一种捕获基因组特定DNA片段的方法与流程
本发明涉及一种捕获基因组特定dna片段,用于后续dna检测的方法。
背景技术:
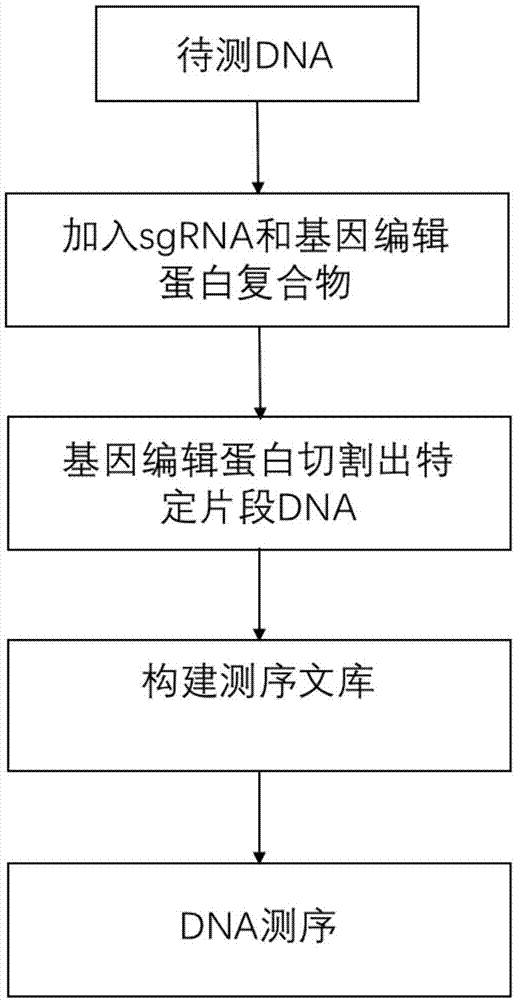
:人的基因组具有30亿个碱基,也就是3gb,其中含有编码区、非编码区和一些调控区域。随着高通量测序技术的发展,对全基因组进行重测序的成本越来越低,人类第一个基因组花费了30亿美金,现在一个人的全基因组重测序已经在几千人民币,近十年测序成本降低了一万倍,成本降低非常快。然而在临床上,比如特定遗传病检测、携带者筛查、耳聋检测、多基因病检测、单基因病检测,并不是每个病例都需要进行全基因组测序,而只需要进行特定区域的检测,从而快速检测该特定区域是否有dna水平的结构异常。目前市场上出现了特定片段测序的方法,基本实现的方法有以下三个方向:1、pcr:pcr反应的核心是通过特定引物和目标dna在合适温度下的结合,从而在聚合酶的存在下进行扩增。采用pcr技术,对需要检测的片段进行扩增,然后对扩增产物进行测序检测。pcr方法简单方便,价格便宜,快速实现区域富集。该技术适用于几十个位点富集,当需要检测位点比较多的时候,要调整优化每一个pcr反应的引物在一个近似的温度,同时进行反应就会非常困难,因为每个引物区域由特定位点决定,而这些区域的gc含量不尽相同,因此并不适用于大规模的片段检测。2、rna探针富集:美国安捷伦公司发布了通过合成rna探针和基因组dna杂交从而一次性获得全基因组所有基因外显子区域的技术。该方法的原理是,首先将基因组dna进行随机破碎,补平、加a、链接接头、扩增6~7个循环,得到一个全基因组小片段的扩增文库,然后将文库dna变性成单链dna和全外显子区域互补的rna探针进行杂交4~16个小时,rna探针上有生物素标记,通过链霉亲和素标记的磁性颗粒进行结合,从而洗去不是靶标区域的dna,剩余的dna进行10~16个循环的pcr扩增得到了最终文库,该文库就可以通过高通量测序进行检测,一般全外显子+调控区域基本在50mb左右,测序一个2g数据量左右,就可以对外显子和调控区域有全面的检测。该技术出现以后,科研和临床范围应用广泛,目前主要的供应商为安捷伦和罗氏公司。这个方法相较于pcr最大的优点,可以一次性对全部外显子区域进行富集,这是常规pcr、多重pcr技术都无法实现的。该技术也可以针对特定区域进行定制,灵活性也不错。但是该技术操作相较于pcr技术复杂很多,耗时长,需要3~4天,整个实验过程有两次pcr过程,虽然现在基本都采用高保障pcr聚合酶,但是也不排除多次pcr扩增过程中会人为引入碱基错配。3、dna探针富集:市场上也有用双链dna探针和目标区域杂交富集的技术。该技术的实现路线和rna探针的基本一致,也是首先将基因组dna进行随机破碎,补平、加a、链接接头、扩增6~7个循环,得到一个全基因组小片段的扩增文库,然后将文库dna变性成单链dna和全外显子区域互补的dna探针进行杂交4~16个小时,探针上有生物素标记,通过链霉亲和素标记的磁性颗粒进行结合,从而洗去不是靶标区域dna,剩余的dna进行10~16个循环的pcr扩增得到了最终文库,该文库就可以通过高通量测序进行检测,一般全外显子+调控区域基本在50mb左右,测序一个2g数据量左右,就可以对外显子和调控区域有全面的检测。相较于pcr技术的优缺点和rna探针方法一致。相较于rna探针,具有探针本身稳定较佳,但是和目标片段dna结合能力上不如rna。pcr直接扩增技术通量较低,一般来说基因组捕获都指以探针方式杂交获得的形式。这种技术有几个常见的缺点:1.对于临床开展技术难度大,操作复杂,需要进行严格的分区,不利于该技术的大规模开展;2.当样本量大的时候,样本之间容易交叉污染;3.探针的方法由于需要在65度的温度下探针和目标dna进行长时间的杂交结合,才能成功获得目标片段,因此耗时长久。近年来,基于crispr/cas的第三代基因编辑系统获得了长足的发展,该系统原本为细菌的获得性免疫系统,当细菌受到外源的侵袭时,细菌把特定片段序列记录下来,如果下次再受到这个外源侵袭,细菌会立即启动免疫系统快速识别外源侵袭,切割外源序列,从而保证自身安全。这套crispr/cas9系统在这几年发展非常迅速,被广泛应用于基因治疗、针对遗传病、恶性肿瘤、胚胎治疗、核酸检测。技术实现要素:为了克服现有中的上述问题,本发明提供了一种捕获基因组中特定dna片段的方法,无需常规捕获技术的杂交结合,快速地实现了特定目标区域dna富集,避免了常规方法杂交富集步骤多、流程繁琐等缺点,检测结果也更精准。一种捕获基因组特定dna片段方法,其特征在于,包括以下步骤:(1)待检dna与含有特定区域sgrna探针/基因编辑蛋白复合物结合,温浴切割dna;(2)特定片段dna构建测序文库;(3)通过测序确定特定区域dna序列。进一步地,所述的sgrna探针为基因编辑蛋白识别特定片段所需的探针,探针序列与特定片段互补,特定片段是一个或是多个靶标,每一个特定片段至少需要前后两个探针,中间间隔距离根据后期的检测要求调整。进一步地,sgrna探针/基因编辑蛋白复合物结合dna后,37℃温浴30分钟,实现特定片段dna切割。进一步地,所述方法还包括通过清洗、过柱去除非特定片段dna的步骤。进一步地,构建测序文库的方法是:切割下来的dna,通过t4dna聚合酶、klenowexo-、t4dna磷酸激酶(t4dnapnk)、taq聚合酶或klenow酶中的一种或多种补平和增加碱基a,37℃反应20分钟;加入细菌碱性磷酸酶(bap)1分钟后,75℃变性5~10分钟,待温度降低后加入dna连接酶、dna接头,20℃连接20分钟;纯化去除反应体系中的蛋白和缓冲液,构建dna测序文库,直接用于后续检测;若测序文库浓度过低,通过pcr方法扩增富集。本发明首次采用crispr/cas家族蛋白对于特定序列dna的识别和切割能力,对于特定片段dna同时设计多条~数百万条探针,sgrna探针和cas蛋白复合物快速识别特定片段dna,37℃孵育30分钟,即可把待测dna片段全部切割下来;从基因组切割下来的dna,末端进行补平、加a,随后连接dna接头,构建完成的dna文库可以通过测序仪进行测序。测序仪可以是以边合成边测序为主要技术代表的二代测序仪(illumina、life),也可以是pacbio、oxfordnanopore等为代表的第三代单分子测序仪。常规探针富集杂交技术中,杂交步骤需要65度4~16个小时,一般以过夜杂交为多,费时费力。本方法首次采用了基因编辑方法去识别dna并从待测dna上把需要检测的区域dna切割下来,构建测序文库,从而首次实现了多重靶标dna富集,无需常规的探针过夜杂交过程,最快可在两个小时内完成了特定片段富集及测序文库构建,比常规的探针富集方法要节约90%的时间。常规探针捕获方法是通过重叠的过量探针和一个dna混合文库进行杂交,把结合在目标区域的dna最后洗脱下来,不能控制洗脱dna的原始长度,从而在后期检测的时候就需要用尽可能长的dna测序方法,把洗脱下来的dna进行全长测序,才能准确的获得待检测区域的dna变异情况。而本发明由于特定的切割位点已定,可以定制出符合后续检测要求的dna长度,从而可以大大降低dna测序的成本和时间,精准定向对于目标区域进行深度检测。本发明具有以下技术特点:(1)快速:多重、全基因组外显子通过本发明进行捕获,可以在2个小时内结束,和常规pcr反应实验一致,大大快于常规的探针捕获方法。(2)无需杂交:创新的采用cripsr/cas9系统识别目标片段并切割,切割下来构建测序文库,整个流程在业界首创无需杂交即可进行目标区域富集。(3)扩展性好:cripsr/cas9的识别区域是20个碱基,没有特别的识别区域要求,并且多位点同时工作时,和每个区域的gc含量不同而造成的常规pcrtm温度不同问题在这个系统不存在,因此在全基因组范围内设计难度不高,无需像常规多重pcr反复调整引物位置和浓度即可实现多重的捕获。(4)可塑性强:由于切割捕获下来的dna长度可以自行设计决定,可以根据后续的检测平台不同调整,比如可将特定位点放在不同长短的中间。而常规的捕获方法由于通过探针和随机文库进行杂交,并不明确在捕获下来的片段中特定位点的位置以及捕获dna的长度。(5)特异性好:常规的探针捕获方法,由于有海量重叠覆盖探针,也就是重叠探针放入反应体系,因此会有不少非特异性吸附,造成最终测序结果中一般会有40~50%的数据为非目标区域的dna。而本发明由于不需要探针海量覆盖区域,只需要特定片段左右两端两个探针即可,因此非特异富集区域会大大减少,从而不需要海量的测序数据。(6)长片段dna直接测序:以往所有的高通量测序方法基本都基于鸟枪法的原理,即首先将待测dna打碎,构建随机测序文库,随后测序所得数据要进行拼接分析。当目标片段区域dna是一整段dna的改变时,鸟枪法的方法就不适用于检测该种疾病,比如脆性x综合征、假肥大性肌营养不良(dmd)等等,而本方法可以直接切割待测区域前后的dna,采用新型长片段测序仪pacbio、oxfordnanopore等仪器平台直接进行长片段测序,即可直接检测疾病区域,从而快速让患者得到诊断。附图说明图1是本发明流程图。图2快速富集egfr基因外显子区域原理示意图。图3是快速富集egfr基因外显子区域结果示意图。图4是富集fmr1重复序列区域用于检测脆性x综合征示意图。具体实施方式借助于下述具体实施方式可更好地理解本发明,然而,这些具体实施方式仅用于举例说明本发明,不应被解释为对本发明的限制。如图1所示的流程图,本发明的捕获基因组特定dna片段方法,主要包括包括以下步骤:(1)待检dna与含有特定片段sgrna探针/基因编辑蛋白复合物结合,温浴切割dna;(2)特定片段dna构建测序文库;(3)通过测序确定特定片段dna序列。实施例1:本实施例用于说明本发明用于快速富集egfr基因外显子区域的应用。图2所示是快速富集egfr基因外显子区域原理示意图。针对egfr四个外显子设计了四对探针,cas蛋白切割出整个外显子,通过文库构建以后,采用高通量测序仪进行检测,从而发现外显子上的基因突变情况。步骤主要如下:1.从人外周血白细胞中抽提基因组dna,200ul全血和200ul裂解液混合;2.取5ul总量为2ug基因组dna和20ulegfr互补探针sgrna/cas9蛋白复合物,加25ulcas9缓冲液(tris-hclph8.0),37℃温浴30分钟;3.加入链霉亲和素磁珠5ul(thermofisher),室温混合5分钟;4.用磁力架分离磁珠,去除上清,用pbs冲洗磁珠两次,用磁力架分离,并弃上清;5.3nnaoh40ul加入磁珠混匀5分钟,用磁力架分离,取上清;6.上清中加入40ul0.1n醋酸中和上清;7.加入2单位/ul末端补平酶(t4dna聚合酶或和klenowexo-)、490um的dntps,55mm的tris缓冲液组成,37℃反应20分钟;8.直接在试管中加入1ulbap,37℃1分钟,75℃变性5分钟;9.直接在试管中加入dna连接试剂,连接试剂由100单位/ul的dna连接酶,3.5mm的atp,55mm的tris缓冲液,30mm的二硫苏糖醇及dna接头组成。20℃反应20分钟,反应结束后直接由硅胶柱或磁性颗粒纯化即可直接上高通量测序仪检测。图3所示是快速富集egfr基因外显子区域结果示意图。egfr18、19、20、21四个外显子,通过四对探针直接切割下来,然后构建测序文库,由于起始位置明确,测序数据量要求不高,如图3所示,下方为测序所得序列,和模板dna有出入的碱基用字母表示。locationsgrnatargetingsequencesgrna1intron18ccagatcactgggcagcatgsgrna2intron19ggtccatggctctgaacctcsgrna3intron19ggtccatgtgcccctccttcsgrna4intron20ggagccaggatcctcacatgsgrna5intron20catgaacatgaccctgaattsgrna6intron21gccagcattttcctgacaccsgrna7intron21gagttaactttttccaacagsgrna8intron22agtttgtactgaggccaagc实施例2:本实施例用于说明本发明用于快速富集全基因组外显子区域的应用。1.从人外周血白细胞中抽提基因组dna,200ul全血和200ul裂解液混合;2.取5ul总量为2ug基因组dna和20ul全外互补探针sgrna/cas9蛋白复合物,加25ulcas9缓冲液(tris-hclph8.0),37℃温浴30分钟;3.加入链霉亲和素磁珠5ul(thermofisher),室温混合5分钟;4.用磁力架分离磁珠,去除上清,用pbs冲洗磁珠两次,用磁力架分离,并弃上清;5.3nnaoh40ul加入磁珠混匀5分钟,用磁力架分离,取上清;6.上清中加入40ul0.1n醋酸中和上清;7.加入2单位/ul末端补平酶(t4dna聚合酶或和klenowexo-)、490um的dntps,55mm的tris缓冲液组成,37℃反应20分钟;8.直接在试管中加入1ulbap,37℃1分钟,75℃变性5分钟;9.直接在试管中加入dna连接试剂,连接试剂由100单位/ul的dna连接酶,3.5mm的atp,55mm的tris缓冲液,30mm的二硫苏糖醇及dna接头组成。20℃反应20分钟,反应结束后直接由硅胶柱或磁性颗粒纯化即可直接上高通量测序仪检测。实施例3:本实施例用于说明本发明用于快速富集fmr1基因重复序列区域,用于检测脆性x综合征。图4是富集fmr1重复序列区域用于检测脆性x综合征示意图。通过cas蛋白把导致脆性x综合征的前段重复序列区域切割下来,通过长片段三代测序(oxfordnanopore或pacbio)进行直接测序,可以准确检测是否有脆性x综合征。常规pcr方法由于该区域gc含量太高,很难进行直接扩增检测。1.从人外周血白细胞中抽提基因组dna,200ul全血和200ul裂解液混合;2.取5ul总量为2ug基因组dna和20ul全外互补探针sgrna/cas9蛋白复合物,加25ulcas9缓冲液(tris-hclph8.0),37℃温浴30分钟;3.加入链霉亲和素磁珠5ul(thermofisher),室温混合5分钟;4.用磁力架分离磁珠,去除上清,用pbs冲洗磁珠两次,用磁力架分离,并弃上清;5.3nnaoh40ul加入磁珠混匀5分钟,用磁力架分离,取上清;6.上清中加入40ul0.1n醋酸中和上清;7.加入2单位/ul末端补平酶(t4dna聚合酶或和klenowexo-)、490um的dntps,55mm的tris缓冲液组成,37℃反应20分钟;8.直接在试管中加入1ulbap,37℃1分钟,75℃变性5分钟;9.直接在试管中加入dna连接试剂,连接试剂由100单位/ul的dna连接酶,3.5mm的atp,55mm的tris缓冲液,30mm的二硫苏糖醇及dna接头组成。20℃反应20分钟,反应结束后直接由硅胶柱或磁性颗粒纯化即可通过oxfordnanopore或pacbio测序仪进行长片段测序。序列表<110>杭州恺思医疗器械有限公司<120>一种捕获基因组特定dna片段的方法<160>8<170>siposequencelisting1.0<210>1<211>20<212>dna<213>人工序列(artificialsequence)<400>1ccagatcactgggcagcatg20<210>2<211>20<212>dna<213>人工序列(artificialsequence)<400>2ggtccatggctctgaacctc20<210>3<211>20<212>dna<213>人工序列(artificialsequence)<400>3ggtccatgtgcccctccttc20<210>4<211>20<212>dna<213>人工序列(artificialsequence)<400>4ggagccaggatcctcacatg20<210>5<211>20<212>dna<213>人工序列(artificialsequence)<400>5catgaacatgaccctgaatt20<210>6<211>20<212>dna<213>人工序列(artificialsequence)<400>6gccagcattttcctgacacc20<210>7<211>20<212>dna<213>人工序列(artificialsequence)<400>7gagttaactttttccaacag20<210>8<211>20<212>dna<213>人工序列(artificialsequence)<400>8agtttgtactgaggccaagc20当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1