一种大环化合物、制备方法及其应用与流程
本发明涉及有机化学合成领域,特别是涉及一种大环化合物、制备方法及其应用。
背景技术:
过氧化氢,化学式为h2o2,其水溶液俗称为双氧水,是一种强氧化剂,最大的用途是纸及棉织物的漂白。它还能用于化学合成、水处理、金属抛光、制药、消毒、杀菌、食品加工和火箭助燃剂等(无机盐工业,2013,45(9):1),同时,过氧化氢也是一种潜在能源载体,一种环境友好型的氧化剂。广泛的应用使得过氧化氢成为当今世界上最重要的一百种物质之一(j.chem.edu.,2007,86(5):1182-1182)。然而,目前我国市售过氧化氢含量最高为30%,现有技术限制我们对高浓度过氧化氢的生产,完全依赖国外进口途径,极大的阻碍了我们对过氧化氢的有效利用。现有技术中,工业中过氧化氢生产方式主要为蒽醌氧化还原过程,该过程因工艺条件限制,效率低且耗费大量蒽醌及有机溶剂和无机盐。虽氢气和氧气直接合成简便,但该途径具有潜在的爆炸危险。因而发展可替代的简便、安全、高效的且廉价的过氧化氢生产方式是必不可少的。
近些年来,通过氧阴极还原法(oxygenreductionreaction,orr)制过氧化氢引起了众多科研工作者的注意,该方法简便,经济,现制现用,新鲜高效,很好的解决了对于合成一定浓度过氧化氢的研究所存在的问题。该过程中,阴极材料是高效产生过氧化氢的关键,决定着电流效率及电合成成本的高低。铂、钯、金及其合金等贵金属,因其高效率、高选择性和低过电位使科研工作者对其大量研究,但高昂的成本限制了它们的广泛使用。碳基材料凭借其特殊的性质已成为炙手可热的orr催化剂,尤其是杂原子掺杂的碳基材料,如n、p、b等元素,因其独特的电荷密度在过氧化氢生产过程中表现出优异的电化学性能。然而,大部分已报道的碳基材料,作为电催化剂需要在高温(400-1000℃)下热解,该过程会使具有活性的杂原子含量损失和活性部位微环境的分布不均匀,从而造成催化剂电化学活性的下降。因而发展高活性的碳基材料是必要的。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种大环化合物、制备方法及其在电化学制备过氧化氢中的应用。
为实现上述目的及其他相关目的,本发明一方面提供一种大环化合物的制备方法,包括:将单体和具有对称结构的苄溴化合物反应制备获得第一大环化合物;
其中,所述单体具有式ⅰ所示结构:
本发明另一方面提供一种大环化合物,由本发明所述的大环化合物的制备方法制备获得。
本发明另一方面提供本发明所述的大环化合物在电化学制备过氧化氢中的应用。
本发明具有以下有益效果:
(1)本发明材料在常温下合成,反应条件温和,可以容易放大合成,从而大量制备;
(2)本发明材料未经高温热解且无金属掺杂,将其直接用于燃料电池氧阴极还原,具有优异的过氧化氢产生性能;在o2饱和的0.1mkoh溶液和磷酸缓冲液溶液中,过氧化氢选择性分别可达92%和85%。
(3)本发明材料为离子型碳材料,可通过简单的阴离子交换反应实现对电化学氧阴极还原产生过氧化氢性能的调控,从而明显的提高了过氧化氢的选择性。其中第一大环化合物对过氧化氢的选择性可达93%。第二大环化合物,在碱性电解液中,过氧化氢选择性最高为98.5%。
(4)本发明材料与碳粉等掺杂可制成三明治结构的膜电极,具有较大的电流和良好的稳定性,具有高浓度过氧化氢生产潜力。
附图说明
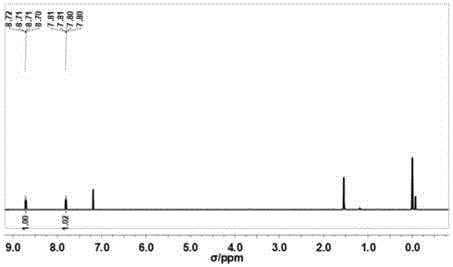
图1为本发明实施例1中2,5-二(4-吡啶)噻唑[5,4-d]并噻唑单体的核磁共振氢谱;
图2为本发明实施例1中2,5-二(4-吡啶)噻唑[5,4-d]并噻唑单体的高分辨质谱;
图3为本发明实施例1中bpyttz-cop:br的核磁共振碳谱。
图4为本发明实施例1中bpyttz-cop:br的红外光谱。
图5为本发明实施例1中bpyttz-cop:br的热重分析图谱。
图6为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)的粉末x射线衍射光谱。
图7为本发明实施例2中bpyttz-cop:br的扫描电子显微镜和透射电子显微镜图谱。
图8为本发明实施例2中bpyttz-cop:f的能量色散x射线吸收图谱。
图9为本发明实施例2中bpyttz-cop:cl的能量色散x射线吸收图谱。
图10为本发明实施例2中bpyttz-cop:br的能量色散x射线吸收图谱。
图11为本发明实施例2中bpyttz-cop:i的能量色散x射线吸收图谱。
图12为本发明实施例2中bpyttz-cop:f的x射线光电子能谱。
图13为本发明实施例2中bpyttz-cop:f中f-的x射线光电子能谱。
图14为本发明实施例2中bpyttz-cop:cl的x射线光电子能谱。
图15为本发明实施例2中bpyttz-cop:cl中cl-的能量色散x射线吸收图谱。
图16为本发明实施例2中bpyttz-cop:i的能量色散x射线吸收图谱。
图17为本发明实施例2中bpyttz-cop:i中i-的能量色散x射线吸收图谱。
图18为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)的氮气吸脱附等温线图谱。
图19为本发明对比例1中紫罗碱型碳基材料(bpy-cop:br)的核磁共振碳谱。
图20为本发明对比例1中紫罗碱型碳基材料(bpy-cop:br)的红外光谱。
图21为本发明对比例1中紫罗碱型碳基材料(bpy-cop:br)和实施例1(bpyttz-cop:br)在氮气饱和的0.1mkoh溶液中的cv曲线图。
图22为本发明对比例1中紫罗碱型碳材料(bpy-cop:br)和实施例1中bpyttz-cop:br在氧气饱和的0.1mkoh溶液中的cv曲线图。
图23为本发明对比例1中紫罗碱型碳基材料(bpy-cop:br)和实施例1中bpyttz-cop:br在氧气饱和的0.1mkoh溶液中的旋转环盘电极曲线图。
图24为本发明对比例1中紫罗碱型碳基材料(bpy-cop:br)和实施例1中bpyttz-cop:br在氧气饱和的0.1mkoh溶液中的h2o2选择性曲线图。
图25为本发明实施例1中bpyttz-cop:br在氧气饱和的0.1mkoh溶液中的时间-电流(i-t)曲线。
图26为本发明实施例1中bpyttz-cop:br在氧气饱和的磷酸缓冲液溶液中的时间-电流(i-t)曲线。
图27为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)在氧气饱和的0.1mkoh溶液中的旋转环盘电极曲线图。
图28为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)在氧气饱和的0.1mkoh溶液中的h2o2选择性曲线图。
图29为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)在氧气饱和的磷酸缓冲液溶液中的旋转环盘电极曲线图。
图30为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)在氧气饱和的磷酸缓冲液溶液中的h2o2选择性曲线图。
图31为本发明实施例2中bpyttz-cop:x(x=f,br)在氧气饱和的0.1mkoh溶液中的h2o2含量图。
图32为本发明实施例2中bpyttz-cop:br和bpyttz-cop:f在氧气饱和的磷酸缓冲液溶液中的h2o2含量图。
图33为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)在氧气饱和的0.1mkoh溶液中的电流效率图。
图34为本发明实施例2中bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)在氧气饱和的磷酸缓冲液溶液中的电流效率图。
具体实施方式
为了使本发明的发明目的、技术方案和有益技术效果更加清晰,以下结合实施例对本发明进行进一步详细说明,熟悉此技术的人士可由本说明书所揭露的内容容易地了解本申请发明的其他优点及功效。
紫罗碱具有良好的电化学活性,而发明人经过大量探索实验意外发现,在碳材料框架中融入吸电子基可以促进电荷载流子迁移,从而有助于提高紫罗碱结构电化学活性和过氧化氢的选择性。噻唑[5,4-d]并噻唑(ttz)结构以其优异的吸电子特性和高效的载流子迁移率被广泛用于半导体材料,因此,将其引入紫罗碱结构,在温和条件下合成了一种新型的二维阳离子型大环化合物。
本发明第一方面提供一种大环化合物的制备方法,包括:将单体和具有对称结构的苄溴化合物反应制备获得第一大环化合物;
其中,所述单体具有式ⅰ所示结构:
式ⅰ所示结构为2,5-二(4-吡啶)噻唑[5,4-d]并噻唑,记为py2ttz。
本发明所提供的制备方法中,通常情况下,反应需要在溶剂存在的条件下进行,所述溶剂选自三氯甲烷、二氧六环、乙腈和n-甲基吡咯烷酮中的一种或多种的组合。在一具体实施例中,将单体溶于溶剂中,再与具有对称结构的苄溴化合物混合,在反应温度为30~120℃下反应,反应后过滤,收集沉淀,再用有机溶剂浸洗,有机溶剂并无特殊限定,例如可以是二氯甲烷、甲醇等。在一些实施例中,反应温度也可以是30~50℃,50~80℃,或80~120℃。在另一具体实施例中,反应温度为30℃。干燥后获得第一大环化合物,命名为bpyttz-cop:br,所述第一大环化合物为噻唑[5,4-d]并噻唑紫罗碱型碳基材料。
本发明所提供的制备方法中,所述苄溴化合物选自式ⅱ~式ⅴ所示结构中的一种或多种的组合;
在优选的实施方式中,所述苄溴化合物选自式ⅲ~式ⅴ所示结构中的一种或多种的组合;
本发明所提供的制备方法中,进一步根据不同卤素的电负性差异,通过调节离子型材料的电子结构可能调控过氧化氢选择性,将第一大环化合物与不同含卤族元素的盐进行离子交换反应,得到含不同卤素阴离子的二维材料,即为第二大环化合物,记为bpyttz-cop:x(x=f,cl,i)。
本发明所提供的制备方法中,所述离子交换反应在溶剂存在的条件下进行,所述溶剂选自水和/或醇。在一具体的实施方式中,可以选择含卤族元素的盐的水溶液和醇混合,在温度为25~60℃反应,在一些实施例中,反应温度也可以是25~40℃,或40~60℃,在另一具体实施例中,反应温度为50℃。收集沉淀,制备获得第二大环化合物,命名为bpyttz-cop:x(x=f,cl,i),所述第二大环化合物为噻唑[5,4-d]并噻唑紫罗碱型碳基材料。
进一步的,所述醇可以选自乙醇。
进一步的,所述含卤族元素的盐选自氯化钠、氟化钠、碘化钠中的一种或多种的组合。
本发明所提供的制备方法中,所述苄溴化合物选自式ⅲ所示结构时,制备获得的第一大环化合物的结构如式ⅵ所示:
本发明所提供的制备方法中,所述苄溴化合物选自式ⅳ所示结构时,制备获得的第一大环化合物的结构如式ⅶ所示:
本发明所提供的制备方法中,所述苄溴化合物选自式ⅴ所示结构时,制备获得的第一大环化合物的结构如式ⅷ所示:
本发明所提供的制备方法中,所述苄溴化合物选自式ⅲ所示结构时,制备获得的第二大环化合物的结构如式ⅸ所示:
其中,x选自cl、f、i;
本发明所提供的制备方法中,所述苄溴化合物选自式ⅳ所示结构时,制备获得的第二大环化合物的结构如式ⅹ所示:
其中,x选自cl、f、i。
本发明所提供的大环化合物的制备方法中,所述苄溴化合物选自式ⅴ所示结构时,制备获得的第二大环化合物的结构如式ⅺ所示:
其中,x选自cl、f、i。
本发明第二方面提供一种大环化合物,由本发明第一方面提供的大环化合物的制备方法制备获得。
本发明第三方面提供本发明第二方面所述的大环化合物在电化学制备过氧化氢中的应用。具体的,提供第一大环化合物和第二大环化合物在电化学制备过氧化氢中的应用。第一大环化合物和第二大环化合物都是噻唑[5,4-d]并噻唑紫罗碱型碳基材料。
本发明具有以下有益效果:
(1)本发明材料在常温下合成,反应条件温和,可以容易放大合成,从而大量制备;
(2)本发明材料未经高温热解且无金属掺杂,将其直接用于燃料电池氧阴极还原,具有优异的过氧化氢产生性能;在o2饱和的0.1mkoh溶液和磷酸缓冲液溶液中,过氧化氢选择性分别可达92%和85%。
(3)本发明材料为离子型碳材料,可通过简单的阴离子交换反应实现对电化学氧阴极还原产生过氧化氢性能的调控,从而明显的提高了过氧化氢的选择性。其中第一大环化合物(bpyttz-cop:br)对过氧化氢的选择性可达93%。第二大环化合物(bpyttz-cop:x(x=f,cl,i)),在碱性电解液中,过氧化氢选择性最高为98.5%。
(4)本发明材料可制成三明治结构的膜电极,具有较大的电流和良好的稳定性,具有高浓度过氧化氢生产潜力。
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
须知,下列实施例中未具体注明的工艺设备或装置均采用本领域内的常规设备或装置。
此外应理解,本发明中提到的一个或多个方法步骤并不排斥在所述组合步骤前后还可以存在其他方法步骤或在这些明确提到的步骤之间还可以插入其他方法步骤,除非另有说明;还应理解,本发明中提到的一个或多个设备/装置之间的组合连接关系并不排斥在所述组合设备/装置前后还可以存在其他设备/装置或在这些明确提到的两个设备/装置之间还可以插入其他设备/装置,除非另有说明。而且,除非另有说明,各方法步骤的编号仅为鉴别各方法步骤的便利工具,而非为限制各方法步骤的排列次序或限定本发明可实施的范围,其相对关系的改变或调整,在无实质变更技术内容的情况下,当亦视为本发明可实施的范畴。
实施例1
步骤(1):2,5-二(4-吡啶)噻唑[5,4-d]并噻唑单体的合成
在一反应瓶中,将4-吡啶甲醛(0.58ml,6.15mmol)和二硫代草酰胺(0.25g,2.08mmol)加入并溶于n,n-二甲基甲酰胺(10ml)中,氮气氛围下150℃回流6小时,冷却,过滤,水洗沉淀,得到浅黄色粉末(0.30g,49%),即为2,5-二(4-吡啶)噻唑[5,4-d]并噻唑,记为py2ttz。
如图1所示,产物的核磁共振氢谱表征如下:1h-nmr(500mhz,cdcl3):δ8.71(d,j=6.0hz,4h),7.81(d,j=6.4hz,4h);
如图2所示,产物的高分辨质谱表征数据如下:hrms(esi)m/z:297.0262。
步骤(2)第一大环化合物的制备
在一250ml反应瓶,将py2ttz(0.24g,0.81mmol)和三氯甲烷(17ml)加入,将1,3,5-三(溴甲基)苯(0.19g,0.54mmol)溶于三氯甲烷(7ml)并逐滴加入上述溶液中,氮气保护,30℃搅拌72小时,过滤,收集沉淀。用二氯甲烷浸洗,重复三次,于真空干燥箱60℃干燥12小时,得到黄色粉末(0.35g,产率82%),即为第一大环化合物,噻唑[5,4-d]并噻唑紫罗碱型碳基材料,记为bpyttz-cop:br。
如图3所示,bpyttz-cop:br的核磁共振碳谱表征如下:13ccp-masnmr:δ166,154,146,137,126,121,64。
如图4所示,bpyttz-cop:br的红外表征如下(波数,cm-1):3106(弱,烯烃c–h伸缩),3030(强,芳香环c–h伸缩),2972(中等强度,烷基c–h伸缩),1634(非常强,吡啶盐环振动),1447(强,芳香环c=c弯曲),1148(强,芳香环c–n弯曲),以及825(非常强,芳香环c–c弯曲)。
实施例2
步骤(1):2,5-二(4-吡啶)噻唑[5,4-d]并噻唑单体的合成
在一反应瓶中,将4-吡啶甲醛(0.58ml,6.15mmol)和二硫代草酰胺(0.25g,2.08mmol)加入并溶于n,n-二甲基甲酰胺(10ml)中,氮气氛围下150℃回流6小时,冷却,过滤,水洗沉淀,得到浅黄色粉末(0.30g,49%),即为2,5-二(4-吡啶)噻唑[5,4-d]并噻唑,记为py2ttz。
步骤(2)第一大环化合物的制备
在一250ml反应瓶,将py2ttz(0.24g,0.81mmol)和三氯甲烷(17ml)加入,将1,3,5-三(溴甲基)苯(0.19g,0.54mmol)溶于三氯甲烷(7ml)并逐滴加入上述溶液中,氮气保护,30℃搅拌72小时,过滤,收集沉淀。用二氯甲烷浸洗,重复三次,于真空干燥箱60℃干燥12小时,得到黄色粉末(0.35g,产率82%),即为第一大环化合物,噻唑[5,4-d]并噻唑紫罗碱型碳基材料,记为bpyttz-cop:br。
步骤(3):离子交换反应。
在一反应瓶,加入nax(x=f,cl或i)和水(50ml),将步骤(2)中所述bpyttz-cop:br(0.5g)加入上述溶液,再加入乙醇(10ml),50℃搅拌10小时,过滤,重新加入前述钠盐溶液,反复5次。过滤,水洗,收集沉淀,60℃真空干燥12小时,得到bpyttz-cop:x(x=f,产率79%;x=br,产率82%;x=i,产率93%)。
如图4红外光谱中,2972cm-1为亚甲基“c-h”振动特征峰,1634cm-1和1447cm-1分别为“c=c”振动和吡啶环的伸缩振动峰。如图3,在13ccp-masnmr中,120–150ppm归属于苯基和吡啶环碳化学位移,166和154ppm为并噻唑环碳的特征值,64ppm为亚甲基碳的特征值。
如表1所示,bpyttz-cop:br的元素分析表明,bpyttz-cop:br的c、n、s、h和br含量分别为49.83%、13.15%、15.43%、2.90%和18.69%。理论值为49.34%、9.59%、10.97%、2.76%和27.34%。
表1
如图5,热重分析(tga)表征,可以看出bpyttz-cop:br从360℃开始分解,到达600℃剩余为初始质量的47%。
如图6,粉末x射线衍射光谱(pxrd)中尖锐的峰形,说明bpyttz-cop:br具有良好的晶型。
如图7,从扫描电子显微镜(sem)和透射电子显微镜(tem)图谱可以看出bpyttz-cop:br为层层相叠而堆积。
如图8~图17,通过能量色散x射线吸收(eds)和x射线光电子能谱分析(xps)对bpyttz-cop:x(x=f,cl,i)离子交换程度进行测试,从图13,15,18中可以看出,br-几乎分别被f-和cl-完全交换,而i-可能由于尺寸较大而交换不完全。
如图6,从pxrd图谱,说明bpyttz-cop:x(x=f,cl,i)结构与bpyttz-cop:br稍有不同,可能是由于bpyttz-cop骨架刚性芳香环之间柔性的亚甲基桥存在重排。
如图18,通过氮气吸脱附等温线对bpyttz-cop:br和bpyttz-cop:x(x=f,cl,i)进行分析,该类材料具有低的比表面积,进一步说明bpyttz-cop:x(x=f,cl,i)层之间交错式堆积。
对比例1
不含噻唑[5,4-d]并噻唑单元的紫罗碱碳基材料的制备
在一250ml反应瓶,将4,4’-二联吡啶(0.13g,0.81mmol)和三氯甲烷(17ml)加入,然后将1,3,5-三(溴甲基苯)(0.19g,0.54mmol)溶于三氯甲烷(7ml)中并逐滴加入上述溶液中,氮气保护,30℃搅拌72小时,过滤,收集沉淀。用四氢呋喃浸洗,重复三次,于真空干燥箱60℃干燥12小时,得到绿色粉末(0.35g,产率82%),记为bpy-cop:br。
如表2所示,是bpy-cop:br的元素分析,其中,bpyttz-cop:br的c、n、h和br含量分别为49.02%、7.14%、3.08%和40.76%。理论值为42.84%、5.28%、3.70%、2.76%和48.18%。
表2
如图19所示,bpy-cop:br的固体核磁表征如下:13cnmr:δ149,144,136,132,128,125,61。
如图20所示,bpy-cop:br的红外表征如下(波数,cm-1):3101(弱,烯基c–h伸缩),3015(强,芳香环c–h伸缩),2961(中等强度,烷基lc–h伸缩),1631(非常强,吡啶盐环振动),1446(强,芳香环c=c弯曲),1158(强,芳香环c–n弯曲),806(非常强,芳香环c–c弯曲)。
实施例3
电化学测量
使用标准三电极系统,pt片和ag/agcl分别为对电极和参考电极,旋转圆盘电极(rde,面积0.1963cm2)或带有pt环的旋转环盘电极(rrde,面积0.2475cm2)作为工作电极。将催化剂(6mg),乙醇(0.95ml),萘酚(0.05ml,10%)加入样品瓶中超声2小时,分10次吸取该混合液共20ul滴在预先抛光的rde或rrde,置于空气中干燥,作为工作电极。循环伏安法(cv)分别于饱和的o2或n2电解液测试,扫描速率为50mvs-1;线性扫描伏安法(lsv)测试在不同的转速下进行,扫描速率为10mvs-1。对于rrde测试,为了测量h2o2电流,环电压设为1.4v。电解液为0.1mkoh或0.1m缓冲盐溶液。
电子转移数目根据koutecky–levich(k-l)公式计算:
其中j和jk分别为测试的电流密度和动力学电流密度(macm-2),w为工作电极的旋转速度(rads-1),n每分子氧气转移的电子数目,f为法拉第常数(96500cmol-1),a是工作电极的面积,ν为运动粘度(0.011cm2s-1),co2为电解液本体部分氧气浓度(1.2×10-6molcm-3),do2为氧气的扩散系数。过氧化氢浓度测量使用ce(so4)2滴定法。
h2o2浓度可从乙烯按公式得出:
其中,m是过氧化氢浓度,mce4+为ce4+消耗的浓度。
在一1000ml容量瓶中加入355mgce(so4)2和0.5m硫酸溶液,用草酸钠滴定ce(so4)2溶液作为标准曲线,并用已知浓度的h2o2溶液检验。
如图21-图22,在o2或n2饱和的0.1mkoh电解液中对bpy-cop:br和bpyttz-cop:br的电化学性能进行测试,如图,在n2下,bpyttz-cop:br曲线没有氧化还原峰,而在o2下具有明显的还原峰,表明了bpyttz-cop:br对o2显著的电化学还原性质。而0.51v和0.47v明显的o2还原峰,更加说明了其优异的orr性质。在相同条件下,bpy-cop:br呈现多个不明显小峰相较于bpyttz-cop:br,表现了其较差的orr活性。
如图23,图24和图30,旋转环盘电极0.1mkoh电解液中进行测试,该过程起始电位为0.67v与热力学平衡电位相近,碱性电解液中对于h2o2选择性为92%,中性电解液中h2o2选择性可达85%。如图25和图26,对它进行12h的稳定性测试,环和盘电流几乎没有改变,表明了它良好的稳定性。中性电解液中,环电流略有下降,这可能是因为阴离子的侵蚀而不是效率的下降,用去离子水清洗即可恢复。然而,在相同条件下,bpy-cop:br起始电位和h2o2选择性分别为0.61v和75%,进一步说明噻唑[5,4-d]并噻唑结构对于该材料orr活性独特的优势。
进一步,如图27~图30,在0.1mkoh和中性电解液中,对bpyttz-cop:x(x=f,cl,br,i)的电化学性质分别进行测试,从图27中可以看出,盘电流和环电流的曲线几乎为对称的,表明该过程还原o2到h2o是被阻碍的,因而h2o2为主要产物。如图27和图28,bpyttz-cop:x(x=f,cl,br,i)的起始电位(0.66v~0.71v)和h2o2选择性(81.7~98.5%)随着负离子元素电负性的增加而增加,说明了orr活性和h2o2选择性分别与卤素阴离子呈正相关关系。
如图29和图30,中性电解液测试结果呈现同样的规律。
如图28和图30,在相同测试条件下,bpyttz-cop:f作为电催化剂产生h2o2性能与一些金属掺杂的电催化剂相媲美甚至更优异,测量了反应过程电解液中h2o2产成含量。
如图31-图32,在碱性电解液,bpyttz-cop:f和bpyttz-cop:br在2h内-0.856v产生的h2o2浓度分别为507.3μmoll-1和377.05μmoll-1,中性电解液下有更高的浓度分别为1156μmoll-1和881.5μmoll-1。如图33-图34,bpyttz-cop:x(x=f,cl,br,i)的电流效率呈现相同的规律,在碱性电解液中,bpyttz-cop:br的电流效率69.1%(0.53v)增加到82.2%(0.37v),bpyttz-cop:f表现出最优的电流效率。中性电解液下bpyttz-cop:x(x=f,cl,br,i)规律与碱性相同。
以上所述,仅为本发明的较佳实施例,并非对本发明任何形式上和实质上的限制,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还将可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。凡熟悉本专业的技术人员,在不脱离本发明的精神和范围的情况下,当可利用以上所揭示的技术内容而做出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对上述实施例所作的任何等同变化的更动、修饰与演变,均仍属于本发明的技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!