包含金属纳米颗粒的无机/聚合物的杂化催化材料的制作方法与工艺
本发明涉及新型无机/聚合物杂化催化材料,特别是膜,其在多种化学反应中显示出高活性、稳定性、再利用性和低的金属浸出性。更具体地,本发明涉及聚乙烯醇系杂化催化材料(膜)的制造和它们在化学工艺中的用途。该催化材料(膜)对不饱和化合物的氢化特别有用,但并不局限于此。
背景技术:
由于非常大的表面积,因此金属纳米颗粒(MNPs),尤其是贵金属,如铂、钯、钌、铑和金的金属纳米颗粒作为高效催化剂广泛地用于多种化学工艺中。在大多数情况下,MNPs固定于固体载体材料或作为胶体溶液被稳定化。该载体材料通常基于多孔的无机材料,如碳、氧化硅、氧化钛或氧化铝,以使所有反应物能轻易到达催化剂表面。固定MNPs于载体材料上的常见策略是浸渍法,在该方法中,将载体浸渍于金属前体的溶液中,干燥并焙烧。之后,该金属用某一还原剂通常在苛刻的条件下还原,以形成金属纳米颗粒。但是,很难通过这个方法控制颗粒尺寸,因为对于超过10纳米以上的颗粒,尺寸分布宽。载体材料上的基于MNPs的催化材料只要关注它们的再利用和反应器工艺,就要产生其他难题。在两相液体体系中使用间歇式反应器涉及到在反应完成后通过适当的方法如过滤和离心分离从反应液中回收催化剂。但是,当催化剂为细粉形式时,不易分离催化剂。在有些情况下,分离可能需要超滤。极细的粉末可能还会使在化学反应中使用的反应器或高压釜阻塞或中毒。载体材料也可能在搅拌时粉碎。此外,在载体材料上的催化剂颗粒趋于在使用时聚集,从而形成具有较小表面积并由此具有较低活性,长期使用后最终导致催化剂失活的较大颗粒。金属从催化剂浸出(leaching)至反应溶液在精细化工业(药业,香料业)产品的污染方面也是严重的问题。由于上述理由,大多数承载型MNP系催化剂难以适用于精细化学品大规模生产的高效反应器。本发明的发明者之一在Electrochemistry,72,111-116(2004),JP3889605,US7101638,JP3856699中描述了新的无机/聚合物杂化膜。这些膜由无机氧化物和聚乙烯醇(PVA)的杂化复合物组成,其中无机氧化物通过PVA的羟基与PVA化学结合。这些材料通过简单的方法在水溶液中生产,其中无机氧化物的盐在PVA共存的条件下用酸中和。通过该方法,用中和产生的新生的、活性的无机氧化物与PVA结合并杂化从而形成杂化复合物。杂化复合物与无机氧化物和PVA的混合物不同,即,它们的化学性能与它们的原料相比显著改变。例如,尽管由溶于水的PVA制得,但杂化的材料在任何溶剂中(包括热水)中都不溶解。已经开发这些膜用于作为质子传导固体电解质,尤其是燃料电池中的应用。因此,它们具有对氧化、还原和自由基攻击的高耐化学性以及具有高耐热性。在这种电解质中,由于膜能够吸收水,因此通过使用水分子来承载质子。在这些杂化膜中,由于PVA在杂化复合物的合成过程中防止了无机氧化物长大为大尺寸颗粒,因此无机氧化物以极细(纳米级)颗粒的形式分散。还不知道有上述膜用作MNP系催化剂载体的文献数据。本发明的发明者在PCT/JP2010/056288中公开了这些种类的杂化膜作为分子催化剂的载体材料,其中固定的分子催化剂仅限于金属分子络合物,而不是MNPs。基于纯有机聚合物的嵌入金属纳米颗粒的催化膜之前被记载于文献中,这些催化膜不包含任何无机组分,但是:Adv.Synth.Catal.350,1241–1247(2008),Catal.Today104,305–312(2005)和Ind.Eng.Chem.Res.44,9064–9070(2005)描述了用于氢化和氧化反应中的基于Pd和AuNPs进入聚丙烯酸和聚乙烯吡咯烷酮的催化膜;Chem.Mat.17,301-307(2005)描述了用于烯丙型醇的催化氢化的包含PdNPs的聚乙烯亚胺和聚丙烯酸系膜;WaterRes.42,4656-4664(2008)描述了三氯乙酸催化脱氯的包含Pd/FeNPs的聚偏氟乙烯系膜。
技术实现要素:
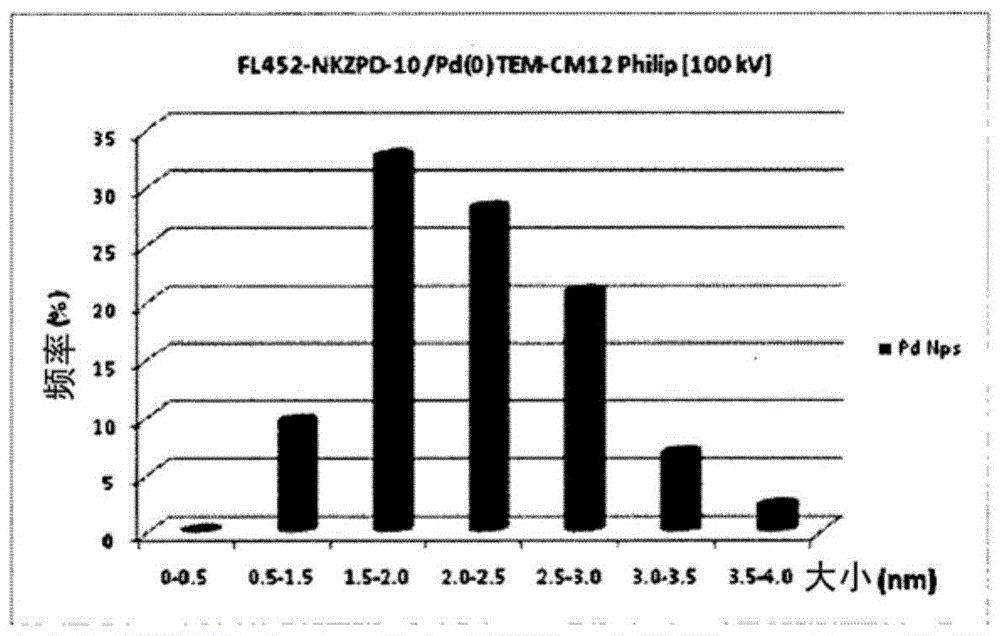
本发明涉及用于化学反应的催化材料尤其是催化膜的制备和用途。在下文中术语“催化材料(膜)”用于表示嵌入金属颗粒并以催化活性为特征的无机/聚合物杂化复合物材料(膜)。无机/聚合物杂化复合物是由金属氧化物和具有羟基的有机聚合物组成,其中该金属氧化物包含选自硅和锆的至少一种,其能通过有机聚合物的羟基与其化学结合。固定在无机/聚合物杂化材料(膜)内的金属颗粒催化剂由大小通常在1μm以下的处于零价态的金属原子的聚集体组成。本发明的一方面涉及到催化材料(催化膜)的制备。本发明的另一方面涉及前述催化材料(膜)在化学工艺中的应用,所述化学工艺例如氢化、脱氢、氢解、加氢甲酰化、羰基化、氧化、二羟化、环氧化、氨基化、次膦酸盐化、羧基化、硅烷基化、异构化、烯丙型烷基化、环丙烷化、烷基化、烯丙基化、芳基化、复分解和其它C-C键形成反应。附图说明图1是包含Pd纳米颗粒的杂化催化膜的截面典型的TEM图像(300K放大)。图2是嵌入杂化催化膜内的PdNPs的典型尺寸分布。图3是嵌入杂化催化膜内的PdNPs的催化前后的典型的XRD图谱。具体实施方式本发明提供用于化学反应的具有高催化活性和高耐久性的新型催化材料,特别是膜,本发明的催化材料(膜)是由包含金属催化剂的无机/聚合物杂化材料(膜)组成。所述无机/聚合物杂化材料(膜)为无机氧化物和具有羟基的聚合物的杂化物。此外,所述无机氧化物优选为硅酸化合物和锆酸化合物。硅酸是指包含SiO2作为其组成单元以及包含水分子的化合物,并且可以由SiO2·xH2O表示。在本发明中,硅酸化合物是指硅酸及其衍生物,或者包含硅酸作为主要组分的任何化合物。锆酸是指包含ZrO2作为其组成单元以及包含水分子的化合物,并且可以由ZrO2·xH2O表示。在本发明中,锆酸化合物是指锆酸及其衍生物,或者包含锆酸作为主要组分的任何化合物。更优选使用锆酸化合物。硅酸化合物和锆酸化合物可包含其他元素,具有非化学计量组成和/或包含一些添加物,条件是维持硅酸和锆酸的原有性能。对于无机/聚合物杂化材料,具有羟基的有机聚合物适合作为有机组分,这是因为羟基可与无机氧化物结合。此外,水溶性聚合物是优选的,这是因为在大多数情况下,杂化过程在水性环境中进行。基于上述理由,聚乙烯醇(PVA)是最适合的组分。纯PVA和/或其改性物(即,用其他基团(部分)取代羟基的PVA衍生物)或部分嵌段共聚化合物可用于该目的。此外,允许将其它聚合物或其它有机和无机添加剂混合至杂化材料(膜),所述其它聚合物例如聚烯烃聚合物如聚乙烯和聚丙烯,聚丙烯酸类聚合物,聚醚聚合物如聚环氧乙烷和聚环氧丙烷,聚酯聚合物如聚对苯二甲酸乙二醇酯和聚对苯二甲酸丁二醇酯,氟聚合物如聚四氟乙烯和聚偏二氟乙烯,含糖聚合物如甲基纤维素,聚乙酸乙烯酯聚合物,聚苯乙烯聚合物,聚碳酸酯聚合物,环氧树脂聚合物。通过简单的含水过程(aqueousprocess)制得有机/无机杂化材料(膜)。在硅酸型的情况下,通过将包含具有羟基的聚合物(如PVA)的硅酸盐水溶液用酸中和来合成杂化物。在这个过程中,硅酸盐通过中和变为硅酸化合物。新生的和初生的化合物如此活泼以致它们具有彼此结合的倾向。然而,在该方法中,聚合物在无机化合物的近旁共存,因此新生的和初生的化合物通过脱水结合而与聚合物的羟基结合。最终杂化材料通过在共存中和过程(co-existentneutralizationprocess)后从上述前体溶液中去除该溶剂(水)来形成。膜可以通过在共存中和过程之后使用上述前体溶液通过通常的流延法来制备。该杂化复合物的纤维可以通过纺粘法、熔喷法或电纺法制备。无机/聚合物杂化材料(膜)显示对于水或者其它具有高极性的溶剂的高亲和性,并且通过吸收这些溶剂而溶胀。膜的溶胀度可以通过醛处理调节(Electrochemistry,72,111-116(2004),JP4041422,US7396616)。在所述醛处理中,通过使膜与包括醛的溶液或气体反应物接触而使残留在无机/聚合物杂化物中的聚合物的自由羟基与醛如戊二醛、苯二醛、乙二醛和丁醛结合。通过上述醛处理,聚合物组分交联或转换为疏水衍生物以便调整膜的溶胀度。为了将金属催化剂固定于硅酸型杂化材料上,将所述材料浸渍于金属盐如硝酸盐或硫酸盐的溶液中,并将这些盐吸收于内部。在金属浸透后,用纯溶剂洗涤所述材料且用合适的还原剂如氢或硼氢化钠还原所吸收的盐以变成金属形式。在锆酸型的情况下,通过在包含具有羟基的聚合物(如PVA)的水溶液中用碱中和锆盐或氧锆盐来合成杂化物。在此过程中,锆盐或氧锆盐通过中和反应变成锆酸化合物,从而如硅酸盐型杂化物那样结合共存的有机聚合物。最终杂化材料可以通过在共存中和过程后从上述前体溶液中去除该溶剂(水)来形成。膜可以通过使用上述前体溶液通过通常的流延法来制备。为了将金属催化剂固定于锆酸型杂化材料上,在中和反应之前添加金属盐至原料溶液中。该盐通过中和反应转化成金属氧化物或氢氧化物。固定的金属氧化物或氢氧化物的尺寸很小(纳米级),这是因为PVA或锆酸化合物防止了在杂化复合物的合成过程中金属氧化物或氢氧化物生长为大尺寸颗粒。此后,金属氧化物或氢氧化物用合适的还原剂如氢或硼氢化钠还原而转变为金属形式。可使用另一种制备方法以合成锆酸型杂化材料。在此方法中,锆盐和/或氧锆盐在包含具有羟基的聚合物如PVA的水溶液中,通过在合适的温度(例如40-60℃)下加热该溶液而部分水解。在此步骤中,杂化反应不完全且仍存在一些锆盐和/或氧锆盐。通过从溶液中去除溶剂,例如通过流延法,形成杂化材料的前体。然后,使如此形成的固体混合物与碱接触,从而中和残留的锆盐和/或氧锆盐并使杂化反应完全。在此过程中,在水解过程前,金属催化剂可通过向原料溶液中添加金属盐而引入至杂化材料中。通过水解和中和过程将该盐转化成金属氧化物或氢氧化物。此后,固定的金属氧化物或氢氧化物用合适的还原剂如氢或硼氢化钠还原而转变为金属颗粒形式。将通过上述方法使用锆型杂化材料获得的催化金属颗粒嵌入所述材料(膜)中,所以当所述材料在催化中使用时,催化金属颗粒特别难以被去除或难以浸出至溶液中。为了补强有机/无机杂化膜,可以使用某些多孔基片如布、无纺布或纸。只要显示足够的耐久性,任何材料如聚酯、聚丙烯、聚乙烯、聚苯乙烯和尼龙都可以用作基体。本发明的催化材料中的金属颗粒催化剂的典型含量的范围为0.2至10重量%。根据本发明,具有催化活性的金属颗粒是指由任何金属组成,优选选自下述组中的至少一种:铁、钴、镍、铜、钌、铑、钯、银、锇、铱、铂和金,且直径大小为0.5-500nm。其中,优选钌、铑、钯、银、铂和金,这是由于它们的更高的稳定性。根据本发明,催化活性金属颗粒由其固定于无机/聚合物杂化材料中的相应的氧化物或金属盐通过还原过程生成,从而杂化材料控制着金属颗粒的生长。本发明的无机/聚合物杂化材料对于溶剂和气体是可渗透的。根据此性能,经固定的金属颗粒催化的化学反应可在杂化材料的表面和其内部发生,因此导致高催化活性。该杂化材料在催化期间还阻碍了催化金属颗粒的聚集,最终导致再利用时恒定的催化活性。在杂化材料中催化活性金属颗粒的强固定化强烈地限制了其在使用时浸出至溶液。与常规的有机聚合物载体材料相比,本发明所描述的杂化催化材料在热稳定性、机械稳定性和化学稳定性(例如对酸和碱,氧化剂,自由基和溶剂的耐久性)方面表现更好的性能。特别地,由于它们与无机氧化物交联,因此,与PVA相比,本发明的杂化材料对于极性和非极性溶剂以及对于高于200℃的温度表现更好的稳定性。尽管杂化材料具有无机氧化物的性能,但是他们也具有有机聚合物的柔韧性且不易碎。在通常的液体体系的化学反应中,搅拌反应液,但是在搅拌期间通过撞击将通常的载体材料,如碳或氧化硅,破碎成更小的粉末。分离由于粉碎而变得更加困难且催化活性显著改变。本发明的杂化材料由于其柔韧性可使其避免此问题。在PVA用做无机/聚合物杂化物的聚合物组分的情况下,相应的催化材料的性能可通过PVA的皂化度(乙酰基的浓度)进行调整:高皂化度在低极性溶剂中提高了催化活性。催化材料(膜)可适用于固定床(在搅拌的反应溶液的情况下)或者旋转膜组件反应器中。在两种情况下,催化材料(膜)可以通过以下方式容易地和直接地再利用:在适当的气体气氛下,例如去除前一反应周期的反应溶液(例如通过简单的倾析),并且添加新一批包含底物的溶液。通过反应溶液无任何催化活性和通过可忽略的金属损失而确保的催化材料(膜)的非均质性,使得在包含期望的产物的反应溶剂中浸出的任何杂质最小化,因此在其回收中无需任何进一步的纯化步骤。根据本发明,如上制备的催化材料(膜)可用于催化多种化学反应,其包含但不仅限于:氢化、脱氢、氢解、加氢甲酰化、羰基化、氧化、二羟化、环氧化、氨基化、次膦酸盐化、羧基化、硅烷基化、异构化、烯丙型烷基化、环丙烷化、烷基化、烯丙基化、芳基化、复分解和其他C-C键形成反应。这些反应可以在溶液中或者在液-气两相体系中进行。此外,对于本领域的熟练技术人员,可使催化膜适用于在固定床或在旋转膜模式中工作的间歇反应器、或连续式流动反应器中的工艺。当在间歇模式中使用时,在包含底物和反应物的溶液存在下,典型地将催化材料(膜)引入反应器中。当要使用气体反应物时,将其在0.1bar至80bar范围内的期望压力下引入反应器中。合适的溶剂包括但并不限于:醇类(优选甲醇)、二醇类、水、醚类、酮类、酯类、脂肪烃和芳香烃、烷基卤化物。典型的底物浓度范围为1×10-2M至10M。基于催化膜中的测定金属含量,底物:催化剂比例可从10:1至100,000:1变化。反应可在-40℃至150℃范围内的温度和在搅拌下进行。由于催化材料(膜)是不溶固体且固定于其上或其中的催化剂是非均质的,反应液可在任何时候通过简单倾析得以轻易地回收,并且催化材料(膜)经过简单添加包含底物和反应物的新溶液而再循环。水作为溶剂使用的可行性也是值得强调的,这是由于其环境相容性。在本发明的优选实施方案中,本发明的催化膜用于底物的氢化,所述底物包括但不限于:烯烃类、炔烃类、亚胺类、烯胺类、酮类、α,β-不饱和醇类、酮类、酯类或酸类。优选的固定化金属颗粒为,但不限于Ir、Rh、Ru、Pd、Au或其混合物的那些。根据本发明的一方面,具有下式的烯烃通过本发明的催化膜氢化:其中R是氢、包含1至约30个碳原子的烷基、包含约从6至18个碳原子的芳基,R1、R2和R3相同或不同并且包括氢、包含1至约30个碳原子的烷基、包含1至约30个碳原子的烯基、包含1至约30个碳原子的炔基、包含约6至18个碳原子的芳基、酰胺、胺、包含1至约30个碳原子的醇盐(alkoxide)、包含1至约30个碳原子的酯、包含1至约30个碳原子的酮。芳基的取代基也可以为双环稠合类或者包含杂原子如硫、氧、氮、磷。将烯烃作为在适当溶剂中的溶液引入包含催化膜的反应器中,所述适当的溶剂优选为,但不限于甲醇。氢化反应在-40℃至150℃的温度范围中并且在0.1bar至50bar的氢气压力范围内进行0.5-48小时。优选的烯类为,但不限于:2-乙酰氨基丙烯酸甲酯、2-乙酰氨基丙烯酸、衣康酸二甲酯、衣康酸、2-乙酰氨基肉桂酸甲酯、2-乙酰氨基肉桂酸。根据本发明的另一方面,具有下式的炔烃通过本发明的催化膜氢化,得到相应的顺式-烯烃产物:其中R1是氢、包含1至约30个碳原子的烷基、约6至18个碳原子的芳基、酰胺、胺、包括1至约30个碳原子的醇盐(alkoxide)、包括1至约30个碳原子的酯。芳基的取代基也可以为双环稠合类或者包含杂原子如硫、氧、氮、磷。将炔烃作为在适当溶剂中的溶液引入包含催化膜的反应器中,所述适当的溶剂优选为,但不限于甲醇。氢化反应在-40℃至150℃的温度范围中并且在0.1bar至50bar的氢气压力范围中进行0.5-48小时。优选的炔烃为,但并不限于:3-己炔-1-醇。总而言之,本发明描述了基于包含金属颗粒的无机/聚合物杂化材料的催化材料(膜)的制备和使用,所述催化材料(膜)在温和的反应条件中并且在低的金属浸出的情况下,催化多种化学反应,特别是高选择性反应。所述催化材料(膜)适用于反应器的工艺并且可以容易地和有效地再利用。给出以下实施例以说明本发明的范围。此外,本发明的实施方案不限于以下给出的实施例。实施例I该实施例说明了依照上述本发明的方法制备催化材料尤其是膜的通常步骤。通过将预定量的硅酸钠和100ml的10重量%聚乙烯醇(PVA)溶液混合获得原料水溶液。PVA具有3100-3900的平均聚合度和86-90%的皂化度。在搅拌下将浓度为2.4M的盐酸溶液滴加至原料水溶液以共存中和,这诱导了杂化反应。在将板加热至温度60-80℃的条件下,将该前体溶液浇注于涂布设备的聚酯膜上。涂布设备为RKPrintCoatInstrumentsLtd.K控制涂布机,所述控制涂布机具有用千分尺调整间隙的刮刀和固定于涂布板上的聚酯膜。在将前体溶液浇注在板上后即刻,为了将前体溶液平铺为预定厚度,将前体溶液通过间隙调节至0.5mm的刮刀以恒定的速度刮扫。在此条件下,水从前体溶液蒸发。在前体溶液的流动性几乎消失之后,将另外的前体溶液再次浇注于其上,通过刮刀刮扫,并且接着将板在110-125℃下加热1-2小时。其后,由此形成的无机/聚合物杂化膜从板剥离掉,用热水洗涤并且干燥。无机/聚合物杂化物的组成如表1所示。尽管这是用于制备膜的方法实例,但是无机/聚合物杂化材料可以由前体溶液形成为任意形状和尺寸。在通过基片补强的情况下,聚酯无纺布或聚丙烯无纺布夹持在前体溶液的第一流延层和第二流延层之间。醛处理通过在室温下将无机/聚合物杂化膜浸入包含对苯二甲醛的1.2M浓度的盐酸溶液进行1小时。在两个特氟隆(Teflon)窗口之间夹紧1cm2的杂化无机/PVA膜样品并将其引入装配有侧向活栓(lateralstopcock)的圆底烧瓶中。转移Pd(NO3)2·2H2O在水中的氮脱气溶液(1.87×10-3M)且在室温下借助定轨振荡器搅拌该悬浮液24小时。此后,在氮气气流下通过从烧瓶中倾析去除该水溶液,用顺次添加的脱气水部分(3×20mL)和甲醇部分(3×20mL)仔细地洗涤该膜并在氮气气流下干燥。在固定后,将该膜转移至不锈钢高压釜中,将新脱气的甲醇转移至反应器中,用5bar的氢气加压该反应器,从而将Pd(II)还原为Pd(0)。在室温下搅拌该溶液2小时。此后,将高压釜减压并且在氢气流下用气密的注射器去除溶液,用顺次添加的脱气甲醇部分(2×20mL)洗涤该膜。因此获得的催化膜组件可在氢气条件下储存且可现成地用于后面的催化氢化反应用高压釜中。出于评价负载在催化膜中的金属的目的,将膜从Teflon固定架去除,在真空下干燥过夜并且通过原子吸收法分析,从而得到Pd含量。表1报道了无机/聚合物杂化催化膜的组成。实施例II该实施例说明了依照上述本发明的方法制备催化材料尤其是膜的另一个通常步骤。通过将预定量的氯氧化锆和氯化钯与100ml的10重量%聚乙烯醇溶液混合获得原料水溶液。PVA具有146,000-186,000的平均分子量和100%的皂化度。在板加热至60-80℃的条件下,将该前体溶液浇注于与实施例1相同的涂布设备的聚酯膜上。在将前体溶液浇注在板上后即刻,为了将前体溶液平滑铺装为预定厚度,将前体溶液通过间隙调节至0.5mm的刮刀以恒定的速度刮扫。在此条件下,水从前体溶液蒸发。在前体溶液的流动性几乎消失之后,将另外的前体溶液再次浇注于其上,通过刮刀刮扫,并且接着将板在110-140℃下加热1-2小时。此后,该固体混合物膜从板上剥离并浸渍于1.7wt%的氨水溶液24小时。在浸渍过程期间,氯氧化锆和氯化钯分别转化为氧化锆(锆酸)和氧化钯(氢氧化物)。将由此制备的杂化膜用热水洗涤并干燥。表1报道了这些膜的组成。在两个特氟隆(Teflon)窗口之间夹紧1cm2的杂化无机/PVA膜样品并将其引入装配有侧向活栓(lateralstopcock)的含氮脱气水(15mL)的圆底烧瓶中。在0℃冷却该混悬液,在氮气气流下,分几份加入大量过量的NaBH4,从而将Pd(II)还原为Pd(0)。在氮气流下,室温下用轨道搅拌器搅拌该溶液24h。此后,在氮气气流下通过倾析去除该水溶液,用顺次添加的脱气水部分(3×20mL)和甲醇部分(3×20mL)仔细地洗涤该膜并在氮气气流下干燥。因此获得的催化膜组件可在氢气条件下储存且可现成地用于后面的催化氢化反应用高压釜中。出于评价负载在催化膜中的金属的目的,将膜从Teflon固定架去除,在真空下干燥过夜并且通过原子吸收法分析,从而得到Pd含量。表1报道了无机/聚合物杂化催化膜的组成。图1、图2和图3分别报道了嵌入杂化催化膜中的Pd纳米颗粒的典型透射电子显微镜图像和柱状图,X射线衍射图谱。实施例III该实施例说明制备包含除钯外其他金属的催化材料,尤其是膜的通常步骤。通过在实施例II描述的制备方法中用氯化钌、氯化铑和氯化金代替氯化钯来制备具有钌颗粒、铑颗粒和金颗粒的杂化膜。如实施例II所述,实施使用NaBH4的还原步骤。表1显示这些催化膜的组成。相同的制备方法通过在实施例II的方法中用铁、钴、镍、铜、银、锇、铱和铂的盐代替氯化钯而适用于具有铁、钴、镍、铜、银、锇、铱和铂的任何催化材料。实施例IV该实施例说明用于依照上述本发明的方法,使用如在实施例I、II和III所述制备的催化杂化PVA/无机膜的多种底物的催化氢化反应的通常步骤。将底物在甲醇中的氢气脱气溶液在氢气流下经由毛细管转移至包含催化膜组件的高压釜中。将高压釜用3个周期的真空/氢气脱气并且接着填充期望的氢气气压。在室温下,用磁力搅拌器搅拌高压釜中的溶液以期望的时间。此后,在氮气流下将高压釜减压且将反应液从底部排液阀去除。该溶液的样品(0.5μL)通过气相色谱法分析以确定转化率和选择性。将剩余的溶液等分部分(aliquot)经由ICP-AES分析以确定浸出至溶液中的金属的量。如下进行再循环实验:将底物的氢气脱气甲醇溶液在氢气流下经由毛细管转移至包含之前氢化反应使用后的催化膜的高压釜中。将高压釜填充期望的氢气气压并且将溶液在室温下搅拌期望的时间。之后,将高压釜减压并且将反应溶液在氢气流下从底部排出阀去除。该溶液的样品(0.5μL)通过气相色谱法分析以确定转化率和选择性。将剩余的溶液等分部分经由ICP-AES分析以确定浸出至溶液中的金属的量。实施例V该实施例说明用于使用包含二氧化硅和钯NPs并且如实施例1所述制备的催化杂化PVA/无机膜NKS-3型的2-乙酰氨基丙烯酸甲酯的氢化反应的步骤。将底物在甲醇(35mL)中的氢气脱气1.5×10-2M溶液在氢气流下经Teflon毛细管转移至包含催化膜组件(9cm2)的高压釜中(摩尔比,底物:Pd=317)。将高压釜用3个周期的真空/氢气脱气并且接着填充5bar氢气气压。在室温下,用轨道搅拌器搅拌高压釜中的溶液1小时。此后,在氮气流下将高压釜减压且将反应液从底部排液阀去除。该溶液样品(0.5μL)使用50m×0.25mmIDLipodex-E(Macherey-Nagel)毛细管柱通过气相色谱法分析以确定转化率。将剩余的溶液等分部分通过ICP-AES分析以测定浸出至溶液中Pd的量(<1ppm)。如下进行再循环实验:将底物的氢气脱气1.5×10-2M甲醇溶液(35mL)在氢气流下经由毛细管转移至包含之前氢化反应使用后的催化膜的高压釜中。将高压釜填充5bar气压并且将溶液在室温下用轨道搅拌器搅拌1小时。之后,将高压釜减压并且将反应溶液在氢气流下从底部排出阀去除。该溶液的样品(0.5μL)通过气相色谱法分析以确定转化率。表2报道了7次再循环试验的代表性数据。实施例VI该实施例说明用于使用依照上述本发明的方法如实施例II所述制备的包含PdNPs的杂化PVA-ZrO2膜的2-乙酰胺丙烯酸甲酯的氢化反应的通常步骤。将底物在甲醇中的氢气脱气1.5×10-2M溶液在氢气流下经由Teflon毛细管转移至包含催化膜组件的高压釜中。将高压釜用3个周期真空/氢气脱气,接着填充期望的氢气气压。在室温下,用磁力搅拌器搅拌高压釜中的溶液以期望的时间。此后,在氮气流下将高压釜减压且将反应液从底部排液阀去除。该溶液样品(0.5μL)使用50m×0.25mmIDLipodex-E(Macherey-Nagel)毛细管柱通过气相色谱法分析以确定转化率。将剩余的溶液等分部分通过ICP-AES分析以测定浸出至溶液中的Pd(<1ppm)。表3报道了使用各种类型的催化膜的代表性结果。实施例VII该实施例说明用于使用依照上述本发明的方法的NKZPD-9型并如实施例II所述制备的包含PdNPs的杂化PVA-ZrO2膜的3-己炔-1-醇的氢化反应的步骤。底物3-己炔-1-醇(0.0529mL,0.484mmol)在甲醇中的氢气脱气溶液(25mL,浓度为0.019M)在氢气气流下经由Teflon毛细管转移至包含催化膜组件的高压釜中。将高压釜用3个周期的真空/氢气脱气,然后填充期望的氢气气压,且将该溶液在期望的温度下搅拌不同的时间。表4报道了不同压力和温度(室温,-10℃,-20℃,-40℃)下代表性结果,并在其中比较了转化率和选择性。此后,在氮气流下将高压釜减压且将反应液从底部排液阀去除。该溶液样品(0.5μL)使用30m×0.25mmIDVF-Waxms毛细管柱通过气相色谱法分析以确定己烯-1-醇的转化率和选择性以及立体选择性(Z/E)。将剩余的溶液等分部分通过ICP-AES分析以测定浸出至溶液中的Pd(<1ppm)。实施例VIII该实施例说明用于使用依照上述本发明的方法,包含NKZPD-11型的PdNPs并且如实施例II所述制备的杂化PVA-ZrO2膜的3-己炔-1-醇的氢化反应的步骤。底物3-己炔-1-醇(0.0529mL,0.484mmol)在甲醇中的氢气脱气溶液(25mL,浓度为0.019M)在氢气流下经由Teflon毛细管转移至包含催化膜组件的高压釜中。将高压釜用三个周期的真空/氢气脱气,然后填充5bar氢气气压且该溶液在室温下搅拌2小时。此后,在氢气流下将高压釜减压且将反应液从底部排液阀去除。该溶液样品(0.5μL)使用30m×0.25mmIDVF-Waxms毛细管柱通过气相色谱法分析以确定3-己烯-1-醇的转化率和选择性以及立体选择性(Z/E)。将剩余的溶液等分部分通过ICP-AES分析以测定浸出至溶液中的Pd(<1ppm)。如下进行再循环实验:将底物(0.0529mL,0.484mmol)在甲醇中的氢气脱气溶液(25mL,0.019M)在氢气流下经由毛细管转移至包含之前氢化反应使用后的催化膜的高压釜中。将高压釜填充5bar气压并且将溶液在室温下用磁力搅拌器搅拌期望的时间。此后,在氢气流下将高压釜减压且将反应液从底部排液阀去除。该溶液样品(0.5μL)使用30m×0.25mmIDVF-Waxms毛细管柱通过气相色谱法分析以确定3-己烯-1-醇的转化率和选择性以及立体选择性(Z/E)。表5报道了6次再循环试验的代表性数据。表1a膜中SiO2对PVA的重量比。b膜中ZrO2对PVA的重量比。c膜中Pd对PVA的重量比。d膜中Ru对PVA的重量比。e膜中Rh对PVA的重量比。f膜中Au对PVA的重量比。g皂化度。*在用硝酸钯浸渍和用氢还原后Pd含量0.26%(w/w)。AAS数据。表2Pd(0)固定在NKS-3膜上的的情况下的2-乙酰氨基丙烯酸甲酯的氢化反应(a)P=5bar,室温,1h,甲醇,浓度=1.5×10-2M,底物/Pd=317。在用硝酸钯浸渍和用氢还原后由原子吸收分析的Pd承载量(0.26%w/w)表32-乙酰氨基丙烯酸甲酯的氢化反应(a)Pd还原后AAS-分析;(b)还原条件:P=5bar,室温,甲醇。表4在NKZPD-9情况下的3-己炔-1-醇的氢化反应(a)(a)Pd含量:3.62%w/w(AAS-分析);(b)还原条件:用NaBH4还原NKZPD-9,底物/Pd=183;甲醇。表5在NKZPD-11情况下的3-己炔-1-醇的氢化反应(a)(a)Pd含量:3.62%w/w(AAS-分析);(b)还原条件:NKZPD-11,用NaBH4还原,催化剂:底物/Pd=183,P=5bar,室温,底物浓度=0.019M。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1