一种纳米粒子/炭黑复合析氢电催化剂及其制备方法与流程
本发明属于电催化析氢领域,具体涉及了一种纳米粒子/炭黑复合析氢电催化剂及其制备方法。
背景技术:
近年来,人类所面临的环境污染和能源危机日益严重,当前开发和利用新能源是解决目前危机的重要挑战之一。氢气作为一种理想清洁的可再生能源代替化石燃料,引起了广泛的研究。电解水是一种最简单和环保的方法制备高纯度氢气,电解水制氢是最有希望并能可持续发展的途径,由于该技术的最初反应原料是水,而水是一种地球含量丰富且可再生的资源。但是,需要高效的电催剂来提供低的过电位,使得电解水制氢更加高效和节能。虽然,Pt族金属电催化剂具有最高的电催化析氢活性和稳定性,但由于他们稀缺和昂贵,从而限制了他们广泛应用于电化学析氢。所以,寻找基于储量丰富,廉价,稳定和优良的电催化析氢活性的非Pt族催化剂,具有重要的意义。
目前,过渡金属材料包括过渡金属氮化物,过渡金属硫化物,过渡金属碳化物,以及过渡金属磷化物,已经报道作为最理想的非贵重金属析氢电催化剂。其中,过渡金属磷化物(TMPs),由于电负性的磷原子能够吸引金属的电子并且有利于氢气脱附,同时过渡金属磷化物有一定的机械强度、化学稳定性以及良好的电导率,因此利用这些特性将其应用到电化学析氢中。在过渡金属磷化物当中,MoP最初在工业上加氢脱硫反应中得到广泛的应用,加氢脱硫反应和氢析出反应二者有一些相似之处,如氢的可逆结合与离解方面。根据密度泛函数理论(DFT)计算表明MoP具有贵重金属的性质,MoP在酸性条件中具有高的电催化析氢活性和稳定性,引起了广大研究者的兴趣。
研究者已经做出巨大的努力,通过设计MoP的微观结构及其调控其组分比例来进一步提高HER电催化活性。不同的合成方法制备的MoP的电催化析氢活性和稳定性差别很大,但是合成的MoP活性位点越多和比表面积越大,其电催化析氢活性越好。此外,研究人员发现提高催化剂电导率有助于氢析出反应的高活性,因此将催化剂负载在碳载体(如:碳布、石墨烯、碳纳米管和炭黑等)上提高其电导率从而能够明显增强催化剂析氢活性。但是,在HER测试之前,催化剂需要聚合物粘合剂(例如Nafion)混合来固定,防止在HER测试过程中催化剂脱落,这可能降低导电性并阻断催化活性位点以降低电催化性能。因此,本发明提供一种廉价Mo-W-P纳米粒子/炭黑复合析氢电催化剂制备方法,旨在于利用MoP和WP两者的复合所形成的协同效应来加强复合材料稳定性以及炭黑能够明显提高材料的导电性,从而提高其电催化析氢活性。
技术实现要素:
本发明旨在提供一种廉价Mo-W-P纳米粒子/炭黑复合析氢电催化剂制备方法,该方法通过一甲基咪唑将MoP和WP按照磷钼酸(PMo12)和磷钨酸(PW12)不同的质量比固定在炭黑上。本发明制备的Mo-W-P纳米粒子/炭黑材料可应用于在酸性条件下电解水制氢气。
一种廉价Mo-W-P纳米粒子/炭黑复合析氢电催化剂的制备方法,具体步骤如下:
(1)炭黑(CB)溶解在无水乙醇中超声分散,再加入一甲基咪唑(1-MD)搅拌,再离心分离,水洗醇洗,干燥,得到1-MD改性的炭黑(1-MD/CB)。
(2)1-MD/CB超声分散于无水乙醇中得到溶液1,将磷钼酸,磷钨酸按一定的质量比溶解在无水乙醇中得到溶液2,将溶液2逐滴滴加到溶液1中得到溶液3,将1-MD溶解在无水乙醇中得到溶液4,然后将溶液4滴加到溶液3中,然后分离,水洗醇洗,烘干,煅烧得到前驱体。
(3)将前驱体,红磷于两个瓷舟中放于管式炉中,然后煅烧磷化,得到Mo-W-P纳米粒子/炭黑复合析氢电催化剂。
步骤(1)中,炭黑、无水乙醇和一甲基咪唑的质量体积比为0.1-0.2g:40-50ml:0.1-0.5ml,水洗,醇洗各三遍,干燥指在60度烘箱中烘干。
步骤(2)中,溶液1中,1-MD/CB与无水乙醇的质量体积比为0.1-0.2g:40-50ml,溶液2中,磷钼酸和磷钨酸的质量之和与无水乙醇的质量体积比为0.2-0.3g:10-30ml,磷钼酸和磷钨酸的质量比为2:1-1:2。溶液4中,一甲基咪唑与无水乙醇的体积比为1-5:200;溶液1中的1-MD/CB、溶液2中的磷钼酸和磷钨酸的质量之和与溶液4中的1-MD的质量体积比为:0.1-0.2g:0.2-0.3g:0.1-0.5ml;水洗,醇洗各三遍,烘干指在60度烘箱中烘干;煅烧指于200-300度煅烧2-5小时。
步骤(3)中,前驱体与红磷的质量比为:1-2:2-4;磷化温度为800-950度,磷化时间2-5小时。
本发明的优点为:
(1)本发明制备的Mo-W-P纳米粒子/炭黑复合电催化剂复合材料,其制备简单,成本低,易于大规模工业化生产,该复合材料具有良好的稳定性和持久性,在解决未来能源危机具有重要的意义。
(2)通过钼钨元素复合后对比电催化制氢,明显大幅度的降低了起始过电位,比单纯的MoP与WP表现了更优良的电催化析氢效果,在电催化制氢实际应用领域具有广阔的前景。
附图说明
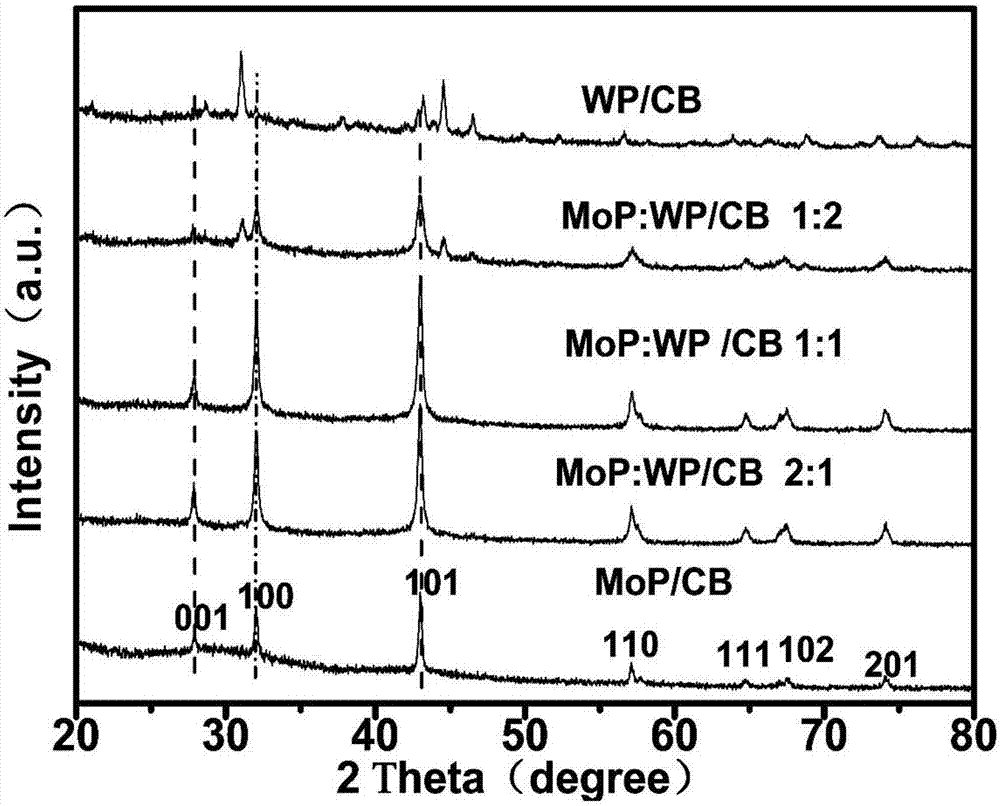
图1为所制备Mo-W-P纳米粒子/炭黑复合析氢电催化剂材料的XRD衍射谱图,包括单纯的MoP,WP及不同磷钼酸和磷钨酸质量比复合的样品。
图2为所制备Mo-W-P纳米粒子/炭黑复合析氢电催化剂材料的微观形貌。(a)Mo-W-P/CB(磷钼酸和磷钨酸的质量比为1:1)的扫描电镜图(FESEM);(b)MoP(TEM);(c,d)Mo-W-P/CB(磷钼酸和磷钨酸的质量比为1:1)高清透射电镜图(HRTEM);(e,f)Mo-W-P/CB(磷钼酸和磷钨酸的质量比为1:1)的map图
图3本发明电催化材料在酸性(0.5M H2SO4)中的析氢活性。(a)Mo-W-P纳米粒子/炭黑复合析氢电催化剂材料在0.5M H2SO4中的极化曲线;(b)为(a)所对应的Tafel斜率。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
实施例1:
制备Mo-W-P纳米粒子/炭黑复合析氢电催化剂材料
(1)合成1-MD改性的CB
称量0.1g的炭黑(CB)溶解在50ml的无水乙醇中超声30min,然后加入0.5ml的一甲基咪唑(1-MD)再搅拌两小时,再离心分离,水洗醇洗,最后60℃干燥,即得到一甲基咪唑改性的炭黑固体粉末(1-MD/CB)。
(2)合成MoP/CB,WP/CB和Mo-W-P/CB
0.1g的1-MD/CB超声分散在50ml的无水乙醇中超声30min,称量0.1g的磷钼酸和0.1g的磷钨酸(这里调控磷钼酸与磷钨酸的质量比1:1)溶解在20ml的无水乙醇中超声10min,将此溶液滴加到一甲基咪唑改性的炭黑溶液中然后超声30min,将0.5ml的一甲基咪唑溶解在20ml的无水乙醇滴加到上述溶液中,然后搅拌10个小时,离心分离,水洗醇洗,烘干研磨成粉末待用。将样品放在马弗炉中在空气氛围中220℃煅烧3个小时(升温速率2℃/min)得到MoO2/CB和WO2/CB(Mo-W-O2/CB)前驱体。称量上述前驱体50mg,红磷100mg于两个瓷舟中放于管式炉中,盛有红磷的瓷舟放于气体上游,盛有样品的瓷舟放于下游,管式炉密封好先通氩气半个小时排出里面的空气,然后在氩气氛围中升温到850℃煅烧3个小时,样品溶解在乙醇中超声分散均匀,离心分离,水洗醇洗,烘干即可。
实施例2:
不同实施例的地方在于:称量0.1g的磷钼酸溶解在20ml的无水乙醇中超声10min。
实施例3:
不同实施例的地方在于:称量0.2g的磷钼酸和0.1g的磷钨酸溶解在20ml的无水乙醇中超声10min。
实施例4:
不同实施例的地方在于:称量0.1g的磷钼酸和0.2g的磷钨酸溶解在20ml的无水乙醇中超声10min。
实施例5:
不同实施例的地方在于:称量0.1g的磷钨酸溶解在20ml的无水乙醇中超声10min。
实施例6:
Mo-W-P纳米粒子/炭黑复合析氢电催化剂的表征分析
如图1所示,从图中可以看出磷化钼与磷化钨不同比例复合的Mo-W-P催化剂具有单纯的磷化钼和磷化钼的特征峰,这说明我们成功的将钼钨复合在一起了。
如图2所示,从图a中可以看出复合的Mo-W-P/CB催化剂是纳米粒子结构。图b,c中可以看出MoP/CB和Mo-W-P/CB纳米球大约50-70nm,d图可以看出Mo-W-P/CB(磷钼酸和磷钨酸的质量比为1:1)中W,Mo的晶格条纹分别为0.32nm和0.28nm。
如图3所示,图中可以清楚看到钼钨复合后的电催化剂在酸性中具有优异的电催化析氢性能。Mo-W-P/CB(磷钼酸和磷钨酸的质量比为1:1)的电催化析氢活性最高。
实施例7:
Mo-W-P纳米粒子/炭黑复合析氢电催化剂的电催化析氢活性实验
用CHI760e电化学工作站进行析氢反应(HER)电化学测试,采用三电极,制备样品滴在玻碳电极,饱和甘汞电极和碳棒分别作为工作电极,参比电极以及对电极。工作电极的制备,称量5mg的样品溶解在1ml去离子水和乙醇混合溶液中(体积比为3:1),加入20μl的萘酚溶液(5wt%),然后超声分散均匀形成墨汁状,用移液枪移取5μl的墨汁状样品滴在处理好的玻碳电极上。极化曲线在0.5M的硫酸(pH=0.40)中以扫描速率为5mV/s测试。
本发明复合电催化析氢材料在酸性中的电催化析氢性能测试结果如图3所示,当电流密度为10mA/cm2时,Mo-W-P/CB(磷钼酸与磷钨酸的质量比为(1:0,2:1,1:1,1:2,0:1)各材料的测试过电位依次为195,182,165,173,204mV,Mo-W-P/CB(磷钼酸与磷钨酸的质量比为1:1)的塔菲斜率也是最低的62mV/decade。
- 还没有人留言评论。精彩留言会获得点赞!