一种同时测定活性肽及游离氨基酸的方法与流程
本发明涉及分析检测领域,具体为一种营养及风味物质成分测定方法,尤其涉及一种同时测定活性肽及游离氨基酸含量的方法。
背景技术:
游离氨基酸是水产食品新鲜度和风味的重要评价指标,也是非蛋白氮的重要组成部分;不同的游离氨基酸具有不同的口味,呈现鲜、甜、酸、苦等多种口味,会直接体现出水产品食物的味道以及鲜美程度。如苏氨酸、丝氨酸、脯氨酸具有甜味,而天冬氨酸、谷氨酸则起着重要的鲜味作用。因此游离氨基酸的含量会被用作许多水产品品质评价的重要鲜度的潜在指标。
活性肽是指对生物体的生命活动具有生理活性的肽类化合物,一般是指由2个到20个氨基酸残基组成的特定的蛋白质片段,因为构造的不同也具有不同的生理功能。活性肽除营养功能之外还具有多种生物功能和生理效应,例如:抗氧化、抗菌、降血压、自由基清除剂等等。
贝类是一种重要的水产品种,不同生存环境下的贝类体内活性肽和游离氨基酸的组成以及含量也不尽相同。游离氨基酸的含量是评价水产品品质的重要潜在指标,活性肽具有抗氧化、抗菌、降血压、自由基清除等营养功能,因此活性肽的含量也成为表征贝类等水产品品质的重要参数。
活性肽多为2至10个氨基酸组成的氨基酸分子链,与游离氨基酸分子结构相似,化学性质接近。在营养物质提取液中不容易将二者分离,再分别测定其浓度。因此,现有技术中多采用一步法同时测定贝类中游离氨基酸和活性肽的含量。
目前国内外相关测定活性肽和游离氨基酸的方法有很多,主要有高效液相色谱法和比色法、分光光度法、高效毛细管电泳法。
虽然高效液相色谱法、比色法、分光光度法、高效毛细管电泳法可以对活性肽或者游离氨基酸进行测定,但这些方法存在操作复杂、仪器设备成本较高、容易受到杂质干扰、测定结果不准确以及测试种类有限等缺点,未能得到充分推广。
目前测定游离氨基酸最主要的方法是氨基酸自动分析仪法,现有技术中也有人采用氨基酸自动分析仪测定谷胱甘肽和游离氨基酸,然而氨基酸分析仪相应的标准方法不能够很好的将活性肽和游离氨基酸分开,并且测试时间长。目前还没有一种方法,能够利用氨基酸自动分析仪有效、快速同时测定活性肽及多种游离氨基酸含量。
因此,开发一种利用氨基酸自动分析仪同时测定多种游离氨基酸、谷胱甘肽和肌肽含量的方法,是解决现有技术中问题的关键。
技术实现要素:
本发明旨在提供一种同时测定活性肽及游离氨基酸的方法,以解决现有技术中不能利用氨基酸分析仪准确、快速同时测定活性肽和游离氨基酸含量的问题。
本发明的技术方案为,采用优化后的洗脱程序分离样品中的游离氨基酸和活性肽,绘制标准曲线,然后采用标准曲线法测定待测样品溶液中游离氨基酸和活性肽浓度。
一种同时测定活性肽及游离氨基酸的方法,其步骤包括,将待测样品溶液加入到氨基酸分析仪中,用缓冲液B1~B5按以下程序进行梯度洗脱,采用茚三酮显色液进行显色:
第一阶段:用缓冲液B1洗脱,持续时间20~21min;初始柱温37.5~39℃,第2.0~2.1min时降至34.5~35.5℃,第一阶段结束时将柱温升至55.5~56.5℃;
第二阶段:用缓冲液B1和B2洗脱,持续时间11.5~12.5min;先将柱温升至57.5~58.5℃,瞬时调整为B1 78%~82%、B2 18%~22%;再匀速变化为B1 68%~72%、B2 28%~32%,第二阶段结束时柱温降至55.5~56.5℃;
第三阶段:瞬时调整为缓冲液B1 8%~12%、缓冲液B2 88%~92%,按这一比例洗脱9~10min,并在第2.8~3.1min时柱温降至41.5~42.5℃;
第四阶段:用缓冲液B2洗脱5~6min,并在第四阶段结束时将柱温升至71.5~72.5℃;
第五阶段:用缓冲液B3和B4洗脱,16.5~17.5min内从缓冲液B3 68%~72%、缓冲液B4 28%~32%匀速调整为缓冲液B3 58%~62%、缓冲液B4 38%~42%;柱温为71.5~72.5℃;
第六阶段:用缓冲液B3和B4洗脱,2~3min内匀速调整为B3 54%~56%、B4 44%~46%,柱温为71.5~72.5℃;
第七阶段:用缓冲液B4洗脱17~18min,柱温为71.5~72.5℃;
第八阶段:用缓冲液B5洗脱4~5min。
在第八阶段结束后柱温降至69.5~71℃。
上述比例均为体积比。然后根据同样洗脱条件下活性肽和游离氨基酸的标准工作曲线或回归方程计算待测样品溶液中游离氨基酸和活性肽浓度。
优选的洗脱程序为:
第一阶段为第0~20.8min,用缓冲液B1洗脱,初始柱温37.5~39℃,第2.1min时调整为34.5~35.5℃,第一阶段结束时将柱温升至55.5~56.5℃;
第二阶段为第20.8~32.8min,用缓冲液B1和B2洗脱,将柱温升至57.5~58.5℃,缓冲液B1和B2的比例从B1 78%~82%、B2 18%~22%,匀速调整B168%~72%、B2 28%~32%;第二阶段结束时柱温调整为55.5~56.5℃;
第三阶段为第32.8~42.5min,瞬时调整为缓冲液B1 8%~12%,缓冲液B2 78%~82%,并按这一比例洗脱;第35.8min时柱温调整为41.5~42.5℃;
第四阶段为第42.5~48.0min,用缓冲液B2洗脱,第四阶段结束时柱温调整为71.5~72.5℃;
第五阶段为第48.0~65.0min,从缓冲液B3 68%~72%、缓冲液B4 28%~32%,匀速调整为缓冲液B3 54%~56%、缓冲液B4 44%~46%;
第六阶段为第65.0~67.5min,匀速调整为缓冲液B3为54%~56%,缓冲液B4为44%~46%,柱温为69.5~71℃;
第七阶段为第67.5~84.5min,用缓冲液B4进行洗脱,柱温为71.5~72.5℃;
第八阶段为第84.5~89.0min,用缓冲液B5进行洗脱。
更优选的洗脱程序为:
第一阶段为第0~20.8min,用缓冲液B1洗脱,初始柱温38℃,第2.1min时调整为35℃,第一阶段结束时将柱温升至56℃;
第二阶段为第20.8~32.8min,用缓冲液B1和B2洗脱,将柱温升至58℃,缓冲液B1为80%、B2为20%,并匀速调整为B1为70%、B2为30%,第二阶段结束时柱温调整为56℃;
第三阶段为第32.8~42.5min,调整为缓冲液B1为10%,缓冲液B2为90%,并按这一比例洗脱;第35.8min时柱温调整为42℃;
第四阶段为第42.5~48.0min,用缓冲液B2洗脱,第四阶段结束时柱温调整为72℃;
第五阶段为第48.0~65.0min,瞬时调整为缓冲液B3为70%,缓冲液B4为30%,并匀速调整为B3 60%、B4 40%;
第六阶段为第65.0~67.5min,匀速调整为缓冲液B3为55%,缓冲液B4为45%;
第七阶段为第67.5~84.5min,用缓冲液B4洗脱;
第八阶段为第84.5~89min,用缓冲液B5洗脱。在第八阶段结束后柱温降至70℃。
所述的缓冲液B1~B5为柠檬酸-柠檬酸锂缓冲体系,pH值分别为2.75~2.85、3.65~3.75、3.55~3.65、4.05~4.15;所述的缓冲液B5为氢氧化锂水溶液。
缓冲液B1的pH=2.75~2.85,柠檬酸三锂浓度为5.5~6g/L,氯化锂浓度为1~1.5g/L,柠檬酸浓度为19~21g/L,乙醇体积含量2.5%~3.5%,辛酸体积含量0.005%~0.015%。优选的,B1的pH=2.8,柠檬酸三锂浓度为5.73g/L,氯化锂浓度为1.24g/L,柠檬酸浓度为19.9g/L,乙醇体积含量3.0%,辛酸体积含量0.01%。
所述的缓冲液B2的pH=3.65~3.75,柠檬酸三锂浓度为9.5~11g/L,氯化锂浓度为6~7.5g/L,柠檬酸浓度为11.5~13g/L,乙醇体积含量2.5%~4%,辛酸体积含量0.005%~0.015%。优选的,B2的pH=3.7,柠檬酸三锂浓度9.8g/L,氯化锂浓度为6.36g/L,柠檬酸浓度12.0g/L,乙醇体积含量3%,辛酸体积含量0.01%。
所述的缓冲液B3的pH=3.55~3.65,柠檬酸三锂浓度为8.5~9.6g/L,氯化锂浓度为25~28g/L,柠檬酸浓度为11~12g/L,乙醇体积含量8%~12%,苯乙醇体积含量3.5%~5%,辛酸体积含量0.005%~0.0.15%。优选的,缓冲液B3的pH=3.6,柠檬酸三锂浓度为8.79g/L;氯化锂浓度为26.62g/L、柠檬酸浓度为11.27g/L,乙醇体积含量为10%,苯乙醇体积含量为0.4%,辛酸体积含量为0.01%。
所述的缓冲液B4的pH=4.05~4.15,柠檬酸三锂浓度为9.5~11g/L,氯化锂浓度为37.5~40g/L,柠檬酸浓度为3~4g/L,辛酸体积含量0.005%~0.015%。优选的,缓冲液B4的pH=4.1,柠檬酸三锂浓度为9.8g/L、氯化锂浓度为38.15g/L、柠檬酸浓度为3.3g/L;辛酸体积含量为0.01%。
所述的缓冲液B5中,氢氧化锂浓度为8~10g/L,优选为8.4g/L;乙醇体积含量2.5%~4%,优选为3%;辛酸体积含量0.005%~0.015%,优选为0.01%。
洗脱的产物采用茚三酮衍生试剂显色,显色波长为440nm和570nm。
茚三酮衍生试剂包括茚三酮反应液、反应缓冲液和洗涤液,具体组成如下:
茚三酮反应液(R1)为茚三酮和硼氢化钠的溶液,溶剂选自乙二醇单甲基醚或丙二醇单甲基醚;所述茚三酮浓度为35~42g/L,优选为38~40g/L;所述硼氢化钠浓度为80~85mg/L,优选为80~82mg/L。
反应缓冲液(R2)为乙酸钠、冰醋酸、水与有机溶剂的混合溶液;所述有机溶剂选自乙二醇单甲基醚或丙二醇单甲基醚;所述有机溶剂与冰醋酸、水的体积比为1:0.2~0.4:0.7~1,优选为1:0.25~0.35:0.8~0.85;所述乙酸钠与超纯水的质量比为1:1.5~2.0,优选为1:1.6~1.7。
洗涤液(R3)为乙醇水溶液;所述乙醇与水的体积比为1:18~20,优选为1:18.5~19.5。分离洗脱时用茚三酮反应液和反应缓冲液进行显色,洗脱程序结束时用洗涤液清洗。
本方法中,所述的活性肽为谷胱甘肽和肌肽中的至少一种;所述的游离氨基酸包括GSH(谷胱甘肽)、Car(肌肽)、P-Ser(磷酸丝氨酸)、Tau(牛磺酸)、PEA(磷酸乙醇胺)、Urea(尿素)、Asp(天冬氨酸)、Hypro(羟脯氨酸)、Thr(苏氨酸)、Ser(丝氨酸)、AspNH2(天冬酰胺)、Glu(谷氨酸)、GluNH2(谷氨酰胺)、Sar(肌氨酸)、a-AAA(α-氨基己酸)、Pro(脯氨酸)、Gly(甘氨酸)、Ala(丙氨酸)、Cit(瓜氨酸)、a-ABA(α-氨基丁酸)、Val(缬氨酸)、Cys(胱氨酸)、Met(甲硫氨酸)、Cysthi(胱硫醚)、Ile(异亮氨酸)、Leu(亮氨酸)、Tyr(酪氨酸)、Phe(苯丙氨酸)、b-Ala(β-丙氨酸)、b-AiBA(β-氨基异丁酸)、GABA(γ-氨基丁酸)、Trp(色氨酸)、EOHNH2(乙醇胺)、Hylys(羟基赖氨酸)、Orn(鸟氨酸)、Lys(赖氨酸)、1Mehis(1-甲基组氨酸)、His(组氨酸)、3Mehis(3-甲基组氨酸)、Arg(精氨酸)中的至少一种。
所述待测样品溶液的制备方法为:
a.样品(如水产品、肉类等)与盐酸溶液和抗氧化剂混合,均质后超声处理,固液分离;抗氧化剂与待测水产品样品的质量比为1:50~500;所述抗氧化剂为二硫苏糖醇、连二亚硫酸钠或维生素C,
b.步骤a的固体再次用盐酸溶液提取1~5次;
c. a、b步骤得到的清液合并后用盐酸溶液定容,然后加入磺基水杨酸,离心、过滤,取滤液。
优选的,待测样品溶液的制备方法如下:
a.取待测水产品样品,与盐酸溶液混合,加入抗氧化剂二硫苏糖醇,充分均质后,超声处理3~20min,0~20℃离心后取上清液;二硫苏糖醇与待测水产品样品的质量比为1:100~400;所述待测样品组织与盐酸溶液用量比为1g:1~15mL,优选为1g:5~10mL。
b.按照待测水产品样品与盐酸溶液1g:8~15mL的用量比,取步骤a离心得到的固体与盐酸溶液再次混合搅拌,离心取上清液;并重复上述操作1~5次,合并上清液;
c.合并步骤a和b的上清液,用盐酸溶液定容并加入磺基水杨酸,磺基水杨酸的含量为2wt%~3wt%;离心后采用0.22μm滤膜过滤得到待测液。
以上各步骤所用盐酸浓度为0.005~0.1mol/L,优选为0.01~0.05mol/L。
本方法通过优化洗脱程序、缓冲液组成、洗脱温度,实现了采用氨基酸自动分析仪对食物,尤其是水产品等的营养物质中氨基酸和活性多肽的一步法测定,克服了传统氨基酸分析仪不能够很好的将活性肽和游离氨基酸分开、且测试耗时长的缺点。
本发明的有益效果在于,能够快速、有效的测定样品中各种氨基酸和活性肽的含量;检测时间少于90分钟,并且能够将谷胱甘肽与游离氨基酸完全分离进行测定。采用该方法,混合标准溶液各组分加标平均回收率在84.6%~103.60%之间,日内相对标准偏差在0.31%-0.71%之间,日间相对标准偏差在1.23%~2.38%之间,符合实验室质量控制规范-食品理化检测的相关要求;该测定方法中,色谱峰的面积与浓度之间有着比较好的线性关系,混合标准溶液中的各组分的定量限在0.35~20.12μmol/L之间,最低检测限在0.12~6.25μmol/L之间。表明该全自动氨基酸分析仪方法有良好的灵敏度。
采用本发明的方法,在30min内,可以很好地将谷胱甘肽和肌肽等活性肽与游离氨基酸分离开来,适用于含多种活性肽及游离氨基酸的混合测定,特别是水产品如贝类这些含有多种活性肽和游离氨基酸的样品,缩短了检测时间,并提高灵敏度。
附图说明
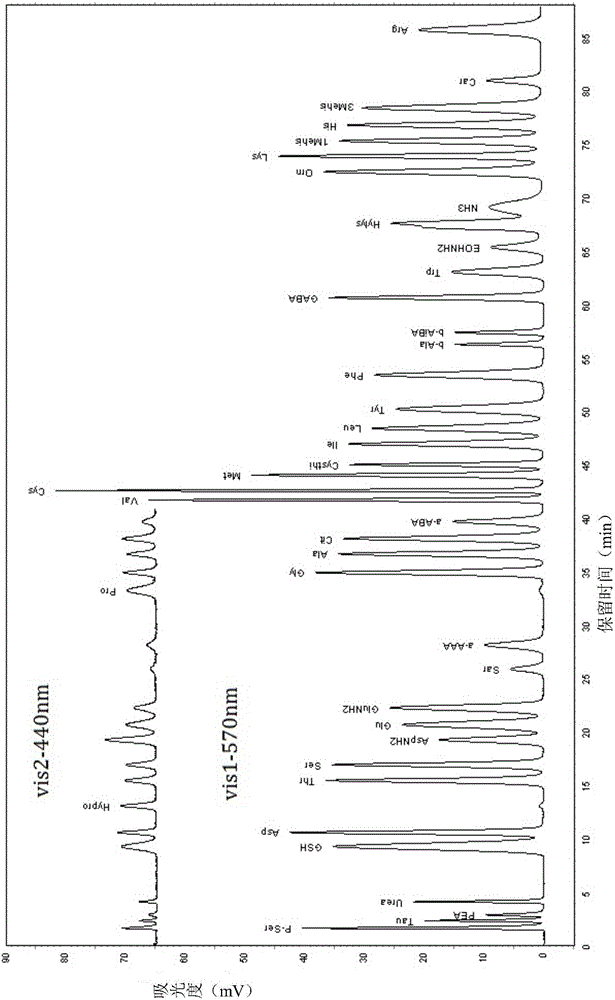
图1为实施例1中,采用现有技术洗脱程序分析活性肽及游离氨基酸混合标准溶液图谱
图2为实施例1中,采用本发明优化后洗脱程序分析活性肽及游离氨基酸混合标准溶液图谱
图3为实施例4中扇贝的检测分离图谱
图4为实施例4中缢蛏的检测分离图谱
图5为实施例4中牡蛎的检测分离图谱
图6为三种抗氧化剂对花蛤中活性肽和氨基酸含量的影响
图7为三种抗氧化剂对白蛤中活性肽和氨基酸含量的影响
具体实施方式
下面结合具体实施例和附图,进一步阐述本发明。以下实施例中,所采用的标准品为活性肽标准品(谷胱甘肽、肌肽)和多种氨基酸混合标液;所采用的仪器型号为L-8800氨基酸全自动分析仪;设定的仪器参数为:分离柱(4.6mm×60mm)树脂为阳离子交换树脂;检测波长:570nm(脯氨酸为440nm);进样量20μL;缓冲液流速为:0.35mL/min;显色液:茚三酮衍生试剂(流量0.35mL/min;单元温度135℃)
实施例1标准溶液的分析测定
(一)洗脱缓冲液和茚三酮衍生试剂的配制
按照表1配制缓冲液B1~B5;按照表2配制茚三酮衍生试剂(茚三酮反应液R1,反应缓冲液R2,洗涤液R3)。
表1 B1~B5缓冲液的组成
表2茚三酮衍生试剂的组成
(二)氨基酸和肽类标准溶液配制
配制GSH(谷胱甘肽)、Car(肌肽)、P-Ser(磷酸丝氨酸)、Tau(牛磺酸)、PEA(磷酸乙醇胺)、Urea(尿素)、Asp(天冬氨酸)、Hypro(羟脯氨酸)、Thr(苏氨酸)、Ser(丝氨酸)、AspNH2(天冬酰胺)、Glu(谷氨酸)、GluNH2(谷氨酰胺)、Sar(肌氨酸)、a-AAA(α-氨基己酸)、Pro(脯氨酸)、Gly(甘氨酸)、Ala(丙氨酸)、Cit(瓜氨酸)、a-ABA(α-氨基丁酸)、Val(缬氨酸)、Cys(胱氨酸)、Met(甲硫氨酸)、Cysthi(胱硫醚)、Ile(异亮氨酸)、Leu(亮氨酸)、Tyr(酪氨酸)、Phe(苯丙氨酸)、b-Ala(β-丙氨酸)、b-AiBA(β-氨基异丁酸)、GABA(γ-氨基丁酸)、Trp(色氨酸)、EOHNH2(乙醇胺)、Hylys(羟基赖氨酸)、Orn(鸟氨酸)、Lys(赖氨酸)、1Mehis(1-甲基组氨酸)、His(组氨酸)、3Mehis(3-甲基组氨酸)、Arg(精氨酸)的标准溶液,并制备混合标准溶液。
标准溶液及混合标准溶液中,各组份含量为:GSH 200μmol/L,Tau50μmol/L,Tau 50μmol/L,AspNH2 200μmol/L,GluNH2 200μmol/L,Sar200μmol/L,a-AAA 50μmol/L,a-ABA 50μmol/L,Cysthi 50μmol/L,其它组份浓度均为100μmol/L。上述溶液均用0.02mol/L稀盐酸配制。
(三)标准溶液及混合标准溶液的检测
将混合标准溶液及各种氨基酸与肽类的标准溶液进样,按照表3的程序洗脱和,根据标准溶液的保留时间确定混合标溶液的各种成分。
被洗脱下来的氨基酸与茚三酮衍生试剂加热反应,产物在570nm和440nm下检测定量。
表3优化后氨基酸分析仪的梯度洗脱以及升温程序
表4氨基酸分析仪的梯度洗脱及升温程序
采用现有技术中洗脱程序(表4)对标准液洗脱得到的谱图如图1所示;采用优化后的洗脱程序(表3)对标准液进行梯度洗脱得到的谱图如图2所示。
根据图1和图2可见,按照现有技术中的梯度洗脱程序不能够将谷胱甘肽与游离氨基酸分离开,无法准确测定溶液中谷胱甘肽的含量。本发明优化后的梯度洗脱程序能在30min内,使谷胱甘肽和肌肽的峰与游离氨基酸较好地分离,实现对谷胱甘肽和肌肽浓度的测定,适用于谷胱甘肽、肌肽和游离氨基酸的混合测定。并且优化后的洗脱程序可缩短分离检测时间。
实施例2线性范围与检测限的测定实验
用0.02mol/L的盐酸溶液将标准混合溶液稀释一定倍数,每个浓度的溶液采用优化后表3的洗脱程序进行三次梯度洗脱测试。
通过出峰的保留时间作为定性的依据,以所测色谱的峰面积和其相对应的浓度作出标准曲线,用线性相关系数(R2)评价;根据信噪比,当所测物质色谱峰的峰高为噪音3倍时(S/N=3),确定其混合标准组分的最低检测限(LOD),当色谱峰的峰高为噪音10倍时(S/N=10),确定其混合标准组分定量限(LOQ),即能够定量该分析物所需的最低浓度值。
具体数据如表5所示:
表5活性肽和多种氨基酸标准混合溶液线性相关系数、检测限和定量限
由表5可见,两种活性肽和游离氨基酸在相应的线性范围内,线性相关系数R2在0.9991~0.9999之间,表明优化过后的全自动氨基酸分析仪法中色谱峰的面积与浓度之间有着比较好的线性关系,混合标准溶液中的各组分的定量限在0.35~20.12μmol/L之间,最低检测限在0.12~6.25μmol/L之间。表明该优化后的全自动氨基酸分析仪方法有良好的灵敏度。
实施例3回收率和精密度的测定实验
取2g贝类的样品组织,样品中分别添加高中低三种不同浓度的混合标准溶液,再加入15mL 0.02mol/L的稀盐酸和1mL浓度为1wt%的二硫苏糖醇,充分均质后超声波清洗5min,然后用冷冻离心机(5000r,4℃)离心10min,收取上清液。将剩余残渣加入10mL浓度为0.02mol/L稀盐酸后搅拌,再次离心(5000r,4℃)5min,合并上清液,定容至50mL。定容后移取2mL,加入2mL 5wt%的磺基水杨酸,再次离心(10000r,4℃)10min,然后用0.22μm水相过滤膜过滤,得到待测液。
每个浓度的待测液采用优化后洗脱程序进行五次梯度洗脱,用外标法计算其实际含量和加标平均回收率。精密度试验对同一待测液在1天内进行5次重复试验分析日内变化,对同一待测液连续测定5天,每天测定1次,分析日间变化,分别确定日内和日间精密度。
回收率计算公式为:回收率=(加入标品后含量-样品含量)/加入标品量*100%,测试结果列于表6。
表6活性肽和氨基酸混合标准溶液加标回收率和精确度实验
根据表6数据可以得出,混合标准溶液各组分加标平均回收率在84.6~103.60%之间,日内相对标准偏差在0.31~0.71%之间,日间相对标准偏差在1.23~2.38%之间,符合实验室质量控制规范-食品理化检测的相关要求。同时也说明该方法回收率以及准确率较高,适用于测定贝类等水产品中的活性肽和多种游离氨基酸。
实施例4新鲜贝类中活性肽和游离氨基酸含量测定
选取市场上比较常见的四种鲜活贝类:扇贝、缢蛏、河蚌、牡蛎,每种水产品各做3个平行样品。每个样品取2g,然后向每个样品中加入15mL0.02mol/L的稀盐酸和1mL浓度为1wt%的二硫苏糖醇,充分均质后超声波清洗5min,然后用冷冻离心机(5000r,4℃)离心10min,收取上清液。将剩余残渣加入10mL浓度为0.02mol/L稀盐酸后搅拌,再次离心(5000r,4℃)5min,合并上清液,定容至50mL。定容后移取2mL,加入2mL 5wt%的磺基水杨酸,再次离心(10000r,4℃)10min,然后用0.22μm水相过滤膜过滤,得到待测液。每份待测液采用优化后洗脱程序进行梯度洗脱,对洗脱检测的数据进行spss分析,计算标准差,并得出显著性。检测结果如表7所示(同一列中相同上标字母表示差异不显著,即P>0.05),谱图如图3~5所示。
表7常见贝类肉组织中活性肽和游离氨基酸的含量(n=3,mg/g)
从表7中可以看出,新鲜贝类肉组织中的游离氨基酸比较丰富,呈味氨基酸含量高,谷氨酸(Glu)、甘氨酸(Gly)、丙氨酸(Ala)在贝类肉组织总游离氨基酸中占有很大比例;牡蛎中总游离氨基酸的含量为5.93mg/g;扇贝中并未检测到肌肽(Car);常见贝类中均含有一定量的谷胱甘肽(GSH);本实施例表明优化后的梯度洗脱程序能快速、有效的测定水产品中多种游离氨基酸和活性肽的含量。
实施例5抗氧化剂对谷胱甘肽和多种游离氨基酸含量的影响
准确称取2g花蛤和白蛤肉组织,分别加入15mL 0.02mol/L的稀盐酸和1mL浓度为1wt%的抗氧剂溶液,抗氧剂为二硫苏糖醇(DTT)、连二亚硫酸钠(Na2S2O4)或维生素C(Vc)。
充分均质后超声波清洗5min,然后用冷冻离心机(5000r,4℃)离心10min,收取上清液。将剩余残渣加入10mL浓度为0.02mol/L稀盐酸后搅拌,再次离心(5000r,4℃)5min,合并上清液,用0.02mol/L的稀盐酸定容至50mL。
定容后移取2mL,加入2mL 5wt%的磺基水杨酸,再次离心(10000r,4℃)10min,然后用0.22μm水相过滤膜过滤,得到待测液。
将待测液分别采用优化后的梯度洗脱程序测定游离氨基酸和活性肽的含量,测试结果如图6和图7所示(Kb为空白对照),抗氧化剂的添加对于贝类肉组织中游离氨基酸的含量基本没有影响,二硫苏糖醇对于贝类肉组织中谷胱甘肽的含量有着更好的保护和抗氧化作用。
需要指出的是,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项目技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!