一种建立儿泻停药物制剂的指纹图谱的方法与流程
本发明属于药物分析领域,具体涉及一种建立儿泻停药物制剂的指纹图谱的方法。
背景技术:
儿泻停颗粒是由茜草藤、乌梅和甘草三种药材制成的中药复方制剂,具有清热燥湿、固肠止泻的作用,能够用于治疗湿热内蕴型小儿腹泻。自1999年批准上市销售至今,儿泻停颗粒由于其疗效确切、质量稳定、使用安全,深受临床医生及消费者好评;2011年儿泻停颗粒的临床再评价表明,其治疗儿童急性腹泻疗效好、且安全性高,可以作为儿童腹泻用药的首要选择。
目前,儿泻停颗粒通过国家药品标准新药转正标准第40册收载的标准号WS3-160(Z-16)-2002(Z)的标准来进行质量控制,即:通过测定原料药之一甘草中的甘草酸含量对儿泻停颗粒进行质量控制。
然而,由于儿泻停颗粒的原料药包括茜草藤、乌梅和甘草三种药材,所含的化学成分的种类众多、且各化学成分的含量差异较大,仅仅通过化学成分中的一种—甘草酸的含量测定来对儿泻停颗粒的质量进行检测和控制,不能从整体上全面反映儿泻停颗粒的质量,进而导致质量检测结果可能缺乏客观性和准确性,从而无法保证儿泻停颗粒临床用药的有效性和安全性。
因而,建立一种能够全面地、快速地检测儿泻停颗粒的质量的方法,对于其全面质量检测和整体质量控制具有重要意义。
技术实现要素:
为此,本发明要解决的技术问题是现有的儿泻停颗粒的质量检测方法不能从整体上全面反映儿泻停颗粒的质量、进而导致质量检测结果可能缺乏客观性和准确性、从而无法保证儿泻停颗粒临床用药的安全性和有效性的问题,进而提供一种建立儿泻停药物制剂的指纹图谱的方法。
为解决上述技术问题,本发明是通过以下技术方案来实现的:
一种建立儿泻停药物制剂的指纹图谱的方法,该方法包括如下步骤:
(1)取待测儿泻停药物制剂1.0~5.0重量份,精密称定,精密加入体积分数为10%~100%的甲醇水溶液或乙醇水溶液25~100体积份,称定重量,超声提取10~60分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1体积份含0.00005~0.00050重量份的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1体积份含0.00005~0.00050重量份的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为235~239nm,柱温25~40℃,流速为0.5~1.5mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液5~20μL,注入高效液相色谱仪,测定,分别得到供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱;
所述重量份与所述体积份的关系为g/mL。
优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,该方法包括如下步骤:
(1)取待测儿泻停药物制剂1.0~2.0重量份,精密称定,精密加入体积分数为40%~80%的甲醇水溶液或乙醇水溶液25~50体积份,称定重量,超声提取20~30分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1体积份含0.00005~0.00015重量份的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1体积份含0.00010~0.00030重量份的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为237nm,柱温为30℃,流速为1.0mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液5~15μL,注入高效液相色谱仪,测定,分别得到供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,该方法包括如下步骤:
(1)取待测儿泻停药物制剂1.0重量份,精密称定,精密加入体积分数为50%~70%的甲醇水溶液或乙醇水溶液25体积份,称定重量,超声提取30分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1体积份含0.00010重量份的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1体积份含0.00020重量份的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为237nm,柱温为30℃,流速为1.0mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液10μL,注入高效液相色谱仪,测定,分别得到供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
进一步优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,以250mm×4.6mm、5μm的Techmate C18-ST或250mm×4.6mm、5μm的岛津VP-ODS为色谱柱。
进一步优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,所述药物制剂为片剂、胶囊剂、丸剂、颗粒剂、蜜炼丸剂、缓释制剂、速释制剂、控释制剂、口服液体制剂或注射制剂。
进一步优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,所述药物制剂为颗粒剂。
进一步优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,所述儿泻停药物制剂的原料药组成为茜草藤、乌梅和甘草。
进一步优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,所述儿泻停药物制剂的指纹图谱中,共有特征峰为:1号峰、2号峰甘草苷、3号峰甘草酸铵、4号峰、5号峰、6号峰、7号峰和8号峰;以2号峰为内参照峰,各峰号的相对保留时间分别为:1号峰0.916±0.046、2号峰1.0、3号峰2.024±0.101、4号峰2.097±0.105、5号峰2.153±0.108、6号峰2.378±0.119、7号峰3.333±0.167和8号峰3.512±0.176。
进一步优选地,所述的建立儿泻停药物制剂的指纹图谱的方法,所述儿泻停药物制剂的指纹图谱中,各峰号的相对保留时间分别为:1号峰0.916、2号峰1.0、3号峰2.024、4号峰2.097、5号峰2.153、6号峰2.378、7号峰3.333和8号峰3.512。
本发明还提供上述的方法在儿泻停药物制剂的质量检测和质量控制中的应用。
与现有技术相比,本发明的上述技术方案具有如下优点:
(1)本发明建立儿泻停药物制剂的指纹图谱的方法,以儿泻停药物制剂为检测对象,建立了针对该药物制剂的指纹图谱的方法,获得了较为全面的图谱信息,确认了共有特征峰1号峰、2号峰甘草苷、3号峰甘草酸铵、4号峰、5号峰、6号峰、7号峰和8号峰;选择2号峰为内参照峰,确定了儿泻停药物制剂的共有特征峰1号峰、3号峰甘草酸铵、4号峰、5号峰、6号峰、7号峰和8号峰的相对保留时间,且结合该指纹图谱中的多个色谱峰的信息,能够全面地、快速地检测儿泻停药物制剂的质量,有利于其全面质量检测和整体质量控制,从而有助于提高该药物临床用药的有效性和安全性。
(2)本发明建立儿泻停药物制剂的指纹图谱的方法,具有稳定性高、精密度高、重复性好等优点,可以用于儿泻停药物制剂的生产过程监测和质量控制中,从而可以确保儿泻停药物制剂质量的稳定性和均一性,从而有助于提高该药物临床用药的有效性和安全性。
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中:
图1为本发明实验例1中对照品A溶液的液相色谱图;
图2为本发明实验例1中儿泻停颗粒供试品溶液(批号:1511481)的液相色谱图;
图3为本发明实验例1中10批儿泻停颗粒供试品溶液导入指纹图谱相似度计算软件后导出的叠加色谱图;
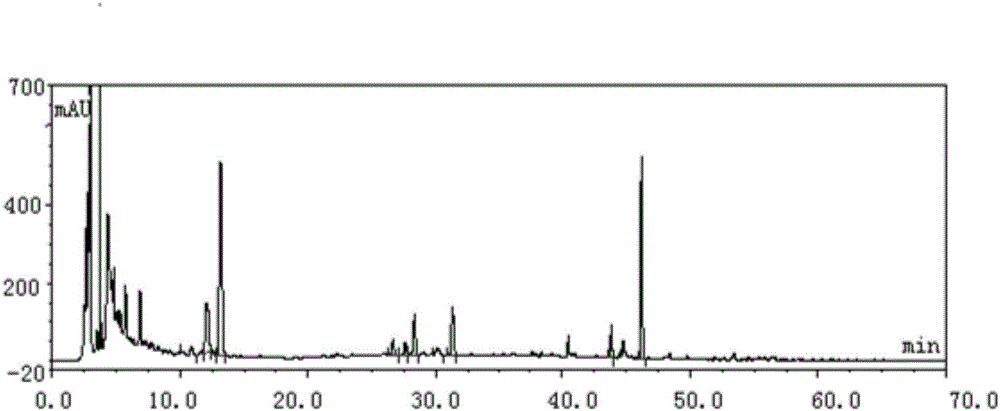
图4为本发明实验例1中建立的儿泻停颗粒的指纹图谱。
具体实施方式
实施例1
本实施例建立儿泻停颗粒的指纹图谱的方法,包括如下步骤:
(1)取待测儿泻停颗粒1.0g,精密称定,精密加入体积分数为70%的乙醇水溶液25mL,称定重量,超声提取30分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1mL含0.00010g的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1mL含0.00020g的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以250mm×4.6mm、5μm的Techmate C18-ST为色谱柱,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为237nm,柱温30℃,流速为1.0mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液10μL,注入高效液相色谱仪,测定,分别得到供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
实施例2
本实施例建立儿泻停颗粒的指纹图谱的方法,包括如下步骤:
(1)取待测儿泻停颗粒1.0g,精密称定,精密加入体积分数为80%的甲醇水溶液或乙醇水溶液25mL,称定重量,超声提取30分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1mL含0.00005g的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1mL含0.00010g的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以250mm×4.6mm、5μm的Techmate C18-ST为色谱柱,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为237nm,柱温为30℃,流速为1.0mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液20μL,注入高效液相色谱仪,测定,分别得供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
实施例3
本实施例建立儿泻停颗粒的指纹图谱的方法,包括如下步骤:
(1)取待测儿泻停颗粒2.0g,精密称定,精密加入体积分数为40%的甲醇水溶液或乙醇水溶液50mL,称定重量,超声提取20分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1mL含0.00015g的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1mL含0.00030g的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以250mm×4.6mm、5μm的Techmate C18-ST为色谱柱,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为237nm,柱温30℃,流速为1.0mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液10μL,注入高效液相色谱仪,测定,分别得到供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
实施例4
本实施例建立儿泻停颗粒的指纹图谱的方法,包括如下步骤:
(1)取待测儿泻停颗粒1.5g,精密称定,精密加入体积分数为60%的甲醇水溶液或乙醇水溶液35mL,称定重量,超声提取25分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1mL含0.00010g的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1mL含0.00020g的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以250mm×4.6mm、5μm的Techmate C18-ST为色谱柱,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为237nm,柱温为30℃,流速为1.0mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液15μL,注入高效液相色谱仪,测定,分别得供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
实施例5
本实施例建立儿泻停颗粒的指纹图谱的方法与实施例1的区别仅在于:供试品溶液的制备步骤中,以体积分数为50%的乙醇水溶液为提取溶剂,其余实验条件和实验操作均与实施例1相同。
实施例6
本实施例建立儿泻停颗粒的指纹图谱的方法与实施例1的区别仅在于:供试品溶液的制备步骤中,以体积分数为60%的乙醇水溶液为提取溶剂,其余实验条件和实验操作均与实施例1相同。
实施例7
本实施例建立儿泻停颗粒的指纹图谱的方法,包括如下步骤:
(1)取待测儿泻停颗粒1.0g,精密称定,精密加入体积分数为100%的甲醇水溶液或乙醇水溶液25mL,称定重量,超声提取60分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1mL含0.00005g的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1mL含0.00005g的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以250mm×4.6mm、5μm的Techmate C18-ST为色谱柱,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为239nm,柱温40℃,流速为1.5mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液20μL,注入高效液相色谱仪,测定,分别得到供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
实施例8
本实施例建立儿泻停颗粒的指纹图谱的方法,包括如下步骤:
(1)取待测儿泻停颗粒5.0g,精密称定,精密加入体积分数为10%的甲醇水溶液或乙醇水溶液100mL,称定重量,超声提取10分钟,放冷,过滤,取续滤液,作为供试品溶液;
(2)精密称取甘草苷对照品,加甲醇制成每1mL含0.00050g的溶液,摇匀,作为对照品A溶液;
精密称取甘草酸铵对照品,加甲醇制成每1mL含0.00050g的溶液,摇匀,作为对照品B溶液;
(3)色谱条件:以十八烷基硅烷键合硅胶为填充剂,以250mm×4.6mm、5μm的岛津VP-ODS为色谱柱,以乙腈为流动相A、以体积分数为0.1%的磷酸水溶液为流动相B按照如下程序进行梯度洗脱:0min,A:B为10%:90%;0~0.1min,A:B为10%:90%→19%:81%;0.1min,A:B为19%:81%;0.1~8min,A:B为19%:81%;8min,A:B为19%:81%;8~15min,A:B为19%:81%→20%:80%;15min,A:B为20%:80%;15~50min,A:B为20%:80%→50%:50%;50min,A:B为50%:50%;50~60min,A:B为50%:50%→80%:20%;60min,A:B为80%:20%;60~65min,A:B为80%:20%;65min,A:B为80%:20%;65~66min,A:B为80%:20%→10%:90%;66min,A:B为10%:90%;66~70min,A:B为10%:90%;70min,A:B为10%:90%;检测波长为235nm,柱温25℃,流速为0.5mL/min;
(4)分别精密吸取供试品溶液、对照品A溶液和对照品B溶液5μL,注入高效液相色谱仪,测定,分别得供试品溶液、对照品A溶液和对照品B溶液的液相色谱;
(5)利用国家药典委员会中药色谱指纹图谱相似度评价系统,对供试品溶液、对照品A溶液和对照品B溶液的液相色谱分别经过数据导入、多点校正和数据匹配,即得指纹图谱。
实验例1
1、仪器与试药
仪器包括:高效液相色谱仪:戴安UItiMate3000高效液相色谱仪;色谱柱:Techmate C18-ST(250mm×4.6mm,5μm)色谱柱;紫外检测器;万分之一天平(Sartorius BS224S),十万分之一天平(Precisa XR205SM DR);超声波发生器:(昆山超声KQ-700DE 40KHz 280W);
试药包括:乙腈为色谱纯(德国Merk公司),水为超纯水(电阻率18.2mΩ.cm),其他试剂均为分析纯;儿泻停药物制剂(序号1-10)由华润三九医药股份有限公司提供。
2、流动相的梯度洗脱程序
表1流动相的梯度洗脱程序
3、儿泻停颗粒指纹图谱的建立
按如下方法建立儿泻停颗粒的指纹图谱:
取儿泻停药物制剂对照品甘草苷溶液(对照品A溶液)和甘草酸铵溶液(对照品B溶液)、以及10个批次经现行标准检测合格的儿泻停颗粒粉末(批号:1312473、11511474、1511475、1511476、1511477、1511478、1511479、1511480、1511481、1511482)制备供试品溶液,按照实施例1的方法建立儿泻停颗粒的指纹图谱。
对照品A溶液的液相色谱图、批号为1511481儿泻停颗粒供试品溶液的液相色谱图、10批儿泻停颗粒供试品溶液导入指纹图谱相似度计算软件后导出的叠加色谱图和建立的儿泻停颗粒的指纹图谱分别如图1-4所示。
采用药典委员会编制的指纹图谱相似度评价软件中“中药色谱指纹图谱相似度评价系统2004版”,生成对照特征图谱。
由图4可知,儿泻停颗粒的指纹图谱中有8个共有特征峰,依次标号为1、2、3、4、5、6、7、8,经过对照品溶液比对,其中2号峰为甘草苷,3号峰为甘草酸铵,选取其中的甘草苷作为内参照峰(S峰),以参照峰的相对保留时间为1,计算其他各峰的相对保留时间,结果见表2。
表2 10批儿泻停颗粒共有峰相对保留时间
由表2可知,1~8号峰的平均相对保留时间为:0.916、1.000、2.024、2.097、2.153、2.378、3.333、3.512,相对标准偏差均≤0.055%。按照保留时间偏差不大于±5%的原则,将保留时间规定为:
1号峰:0.916±0.046;2号峰:1.0;
3号峰:2.024±0.101;4号峰:2.097±0.105;
5号峰:2.153±0.108;6号峰:2.378±0.119;
7号峰:3.333±0.167;8号峰:3.512±0.176。
4、方法学验证
4.1精密度考察
取同一重复性项下的儿泻停颗粒粉末(批号:1511481),按实施例1的供试品溶液制备方法制备供试品溶液,连续进样6次,得到6个色谱图,以参照物甘草苷保留时间为1计算其他特征峰的相对保留时间,记录各共有色谱峰的相对保留时间和相对峰面积,并计算各共有色谱峰的相对保留时间和相对峰面积的RSD值,精密度考察实验结果如表3和表4所示。
表3精密度考察的相对保留时间数据
表4精密度考察的相对峰面积数据
由表3和表4可知,各共有色谱峰的相对保留时间RSD<2%,相对峰面积RSD<2%,说明该方法的精密度良好。
4.2重复性考察
取同一批号的供试品(批号:1511481)6份,分别按实施例1的供试品溶液制备方法制备供试品溶液,并进行高效液相色谱测定,记录各共有色谱峰的相对保留时间和相对峰面积,并计算各共有色谱峰的相对保留时间和相对峰面积的RSD值,重复性考察实验结果如表5和表6所示。
表5重复性考察的相对保留时间数据
表6重复性考察的相对峰面积数据
由表5和表6可知,各共有色谱峰的相对保留时间RSD<2%,相对峰面积RSD<2%,说明该方法的重复性较好。
4.3稳定性考察
取同一重复性项下的供试品溶液(批号:1511481),按实施例1的方法分别在0h、2h、4h、8h、12h、24h进行检测,记录各共有色谱峰的相对保留时间和相对峰面积,并计算各共有色谱峰的相对保留时间和相对峰面积的RSD值,稳定性考察实验结果如表7和表8所示。
表7稳定性考察的相对保留时间数据
表8稳定性考察的相对峰面积数据
由表7和表8可知,24h内儿泻停颗粒各共有色谱峰的相对保留时间RSD<2%,相对峰面积RSD<2%,说明该方法的稳定性较好。
综上,采用本发明的方法建立的儿泻停颗粒的指纹图谱的精密度、重复性和稳定性均较好,结果准确可靠,能较全面的反映儿泻停颗粒的质量。
5、儿泻停颗粒的质量检测
样品批号:1604471、1604473(由合肥华润神鹿药业有限公司提供);
供试品溶液的制备:取1604471、1604473各1.0g,精密称定,精密加入体积分数为70%的乙醇水溶液25mL,称定重量,超声提取30分钟,放冷,过滤,取续滤液,即得供试品溶液;
对照品溶液的制备:精密称取甘草苷对照品5mg至50mL容量瓶中,加甲醇超声溶解并定容至刻度摇匀,即得对照品A溶液;
精密称取甘草酸铵对照品10mg至50mL容量瓶中,加甲醇超声溶解并定容至刻度摇匀,即得对照品B溶液;
色谱条件:同实施例1;
高效液相分析:分别精密吸取等量的供试品溶液和对照品溶液注入高效液相色谱仪,按上述色谱条件记录70min,即得各供试品溶液和对照品溶液的液相色谱图。在待测样品中找到与指纹图谱对应的8个特征峰,以对照品甘草苷保留时间为1计算其他特征峰的相对保留时间,计算结果如表9所示。
表9批号为1604471、1604473样品的特征峰的相对保留时间
由表9可知,批号为1604471、1604473样品的特征峰的相对保留时间均在规定的保留时间范围内,说明该两批儿泻停颗粒的质量检测符合要求。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!