一种双鱼颗粒GC定量指纹图谱的建立方法及其应用与流程
本发明属于化学分析领域,涉及一种双鱼颗粒gc指纹图谱的建立方法。
背景技术:
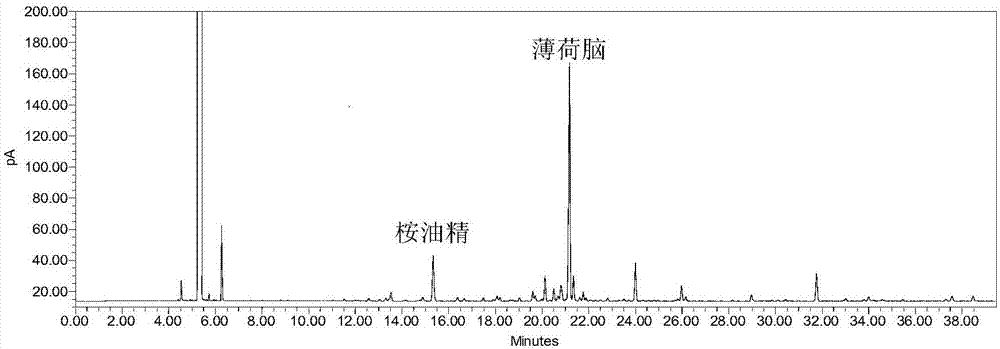
:双鱼颗粒(sg)处方由鱼腥草、金银花、赤芍、艾叶和薄荷5味药组成,为江苏康缘药业股份有限公司研制的中药复方6类新药,具有辛凉解表、清热解毒之功效,用于外感风热型普通感冒,症见发热头痛、全身酸痛、鼻塞、流涕、咳嗽、咳痰、咽喉发痒、口干而渴、咽喉红肿疼痛、舌红、脉数等症。双鱼颗粒为复方制剂,化学成分复杂,有效成分包括苷类、单萜类、酚酸类、挥发油,并含大量辅料如β-环糊精。目前,双鱼颗粒质量标准中对挥发性成分桉油精、薄荷脑和甲基正壬酮进行了气相鉴别,并对绿原酸和芍药苷进行了hplc含量测定。鉴于仅对2个成分进行含量控制,故而进行了基于指纹图谱分析和多成分同时定量的双鱼颗粒质量评价研究,该研究主要是采用hplc对本品中的大中等极性成分进行指纹图谱和含量测定研究。然而对于本品中的挥发性成分仅有鉴别控制,没有含量测定指标和整体性控制。由于双鱼颗粒检测目标成分复杂,且又被辅料β-环糊精包合。以目前的检测方法尚无法对双鱼颗粒中的挥发性有效成分进行有效定量及整体性控制。技术实现要素:本发明的目的是提供一种双鱼颗粒gc定量指纹图谱的建立方法,主要对挥发性成分进行指纹图谱研究,对其中的主要活性成分桉油精和薄荷脑进行了含量测定,以达到整体性控制的目的。本发明的目的是通过以下技术方案实现的:一种双鱼颗粒gc指纹图谱的建立方法,包括以下步骤:步骤(1)、制备供试品溶液:取双鱼颗粒,研细,将双鱼颗粒粉末置于烧瓶中,以水和正己烷为提取溶剂,采用挥发油提取器提取,自沸腾开始计时60~120分钟,冷却,分取正己烷层,用正己烷涮洗挥发油提取器2~3次,合并正己烷,加正己烷定容至25ml,摇匀,过滤,取续滤液,即得供试品溶液;其中,所述的双鱼颗粒粉末、水和正己烷的用量比为10g:250ml:2~5ml;制备对照品溶液:将桉油精、薄荷脑、甲基正壬酮与正己烷混合得到对照品溶液;步骤(2)、分别将对照品溶液与至少10批双鱼颗粒供试品溶液注入气相色谱检测,记录色谱图,对色谱图进行分析,标定出12个共有特征峰,得到双鱼颗粒指纹图谱;其中,气相色谱条件为:agilentdb-1毛细管柱(30m×0.32mm,0.25μm);进样量为1μl;分流比为5:1~50:1;进样口温度为230℃;以氮气为载气,氮气流速0.5~0.7l/min;fid检测器温度为250℃;升温程序为:柱温:78~82℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃。步骤(1)中,优选的,所述的供试品溶液的制备方法为:取装量差异项下的双鱼颗粒,混匀,研细,取10.0g,精密称定,置于500ml烧瓶中,加入玻璃珠数颗,以水和正己烷为提取溶剂;自沸腾开始计时60~90分钟后,冷却,分取正己烷层,用正己烷涮洗挥发油提取器2次,合并正己烷于25ml量瓶内,加正己烷定容至刻度,摇匀,过滤,取续滤液,即得供试品溶液;其中,所述的双鱼颗粒粉末、水和正己烷的用量比为10g:250ml:3ml。进一步优选的,所述的供试品溶液的制备方法为:取装量差异项下的双鱼颗粒,混匀,研细,取10.0g,精密称定,置于500ml烧瓶中,加入玻璃珠数颗,以水和正己烷为提取溶剂;自沸腾开始计时90分钟后,冷却,分取正己烷层,用正己烷涮洗挥发油提取器2次,合并正己烷于25ml量瓶内,加正己烷定容至刻度,摇匀,过滤,取续滤液,即得供试品溶液;其中,所述的双鱼颗粒粉末、水和正己烷的用量比为10g:250ml:3ml。本发明所述的水为去离子水。本发明采用0.22μm微孔滤膜过滤。步骤(2)中,优选的,所述的气相色谱条件为:agilentdb-1毛细管柱(30m×0.32mm,0.25μm);进样量为1μl;分流比为25:1;进样口温度为230℃;以氮气为载气,氮气流速0.6l/min;fid检测器温度为250℃;升温程序为:柱温:80℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃。本发明采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价系统》2012年版对色谱图进行分析,标定出12个共有特征峰。所述的双鱼颗粒指纹图谱的特征峰以桉油精为参照峰,各特征峰及其相对保留时间如下:一种基于本发明所述的双鱼颗粒指纹图谱进行双鱼颗粒gc定量分析的方法,包括以下步骤:步骤(1)、制备供试品溶液:取双鱼颗粒,研细,将双鱼颗粒粉末置于烧瓶中,以水和正己烷为提取溶剂,采用挥发油提取器提取,自沸腾开始计时60~120分钟,冷却,分取正己烷层,用正己烷涮洗挥发油提取器2~3次,合并正己烷,加正己烷定容至25ml,摇匀,过滤,取续滤液,即得供试品溶液;其中,所述的双鱼颗粒粉末、水和正己烷的用量比为10g:250ml:2~5ml;制备对照品溶液:取桉油精对照品、薄荷脑对照品,加正己烷制成桉油精浓度为16.32~326.37μg/ml、薄荷脑浓度为16.32~326.37μg/ml的对照品溶液;步骤(2)、分别将供试品溶液、对照品溶液注入气相色谱检测,记录色谱图,以本发明获得的双鱼颗粒指纹图谱为参照,由相似度软件计算双鱼颗粒供试品相似度;并用外标一点法分别计算双鱼颗粒中桉油精、薄荷脑的含量;其中,气相色谱条件为:agilentdb-1毛细管柱(30m×0.32mm,0.25μm);进样量为1μl;分流比为5:1~50:1;进样口温度为230℃;以氮气为载气,氮气流速0.5~0.7l/min;fid检测器温度为250℃;升温程序为:柱温:78~82℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃。本发明的有效果:本发明方法具有良好的精密度、线性关系、稳定性、重复性,回收率高,耐用性良好;双鱼颗粒挥发性指纹图谱的分离度、重现性均较好,信息全面,标示出12个特征峰,对其中3个成分进行定性,各批次样品相似度在0.90以上。本发明建立的双鱼颗粒挥发性成分指纹图谱及2种成分的含量测定方法为双鱼颗粒的质量控制提供依据;该方法简便可行、快速准确,可以作为评价双鱼颗粒质量的有效方法。附图说明图1为采用升温程序ⅰ的气相色谱图;图2为采用升温程序ⅱ的气相色谱图;图3为采用升温程序ⅲ的气相色谱图;图4为双鱼颗粒水蒸气蒸馏法气相色谱图;图5为双鱼颗粒二氯甲烷-水回流提取法气相色谱图;图6为双鱼颗粒乙醇超声提取法气相色谱图;图7为混合对照品溶液的气相色谱图;图8为双鱼颗粒的供试品溶液的气相色谱图;图9为缺薄荷阴性供试品的气相色谱图;图10为缺艾叶阴性供试品的气相色谱图;图11为缺薄荷和艾叶阴性供试品的气相色谱图;图12为双鱼颗粒对照指纹图谱;图13为双鱼颗粒指纹图谱相关性图,从上往下依次为赤芍、金银花、鱼腥草、薄荷、艾叶、混标(对照品溶液中桉油精81.59μg/ml、薄荷脑208.44μg/ml)、双鱼颗粒样品。具体实施方式下面通过具体实施方式对本发明的技术方案作进一步说明。1仪器与试药agilent7890a气相色谱仪,美国agilent公司;milliporemilli-qcentury纯水仪,美国millipore公司;bsa224s-cw型电子分析天平,德国sartorius公司;mettlertoledox6型电子分析天平,瑞士mettler公司;kq-250db型超声波清洗仪,昆山市超声仪器有限公司;桉油精对照品(中国食品药品检定研究院,批号:110788-201506,含量以98.4%计);薄荷脑对照品(中国食品药品检定研究院,批号:110728-200506);甲基正壬酮对照品(中国食品药品检定研究院,批号:110834-200502);正己烷(南京化学试剂有限公司,批号15078352e,分析纯);乙酸乙酯(南京化学试剂有限公司,批号14092712072,分析纯);双鱼颗粒(批号:161201、161202、161203、160701、160702、160801、160802、160901、160902、161001,江苏康缘药业股份有限公司生产)。2色谱条件选择2.1对照品溶液配制及参照峰选择分别取桉油精、薄荷脑、甲基正壬酮对照品适量,精密称定,加正己烷制成每1ml分别含80μg桉油精、200μg薄荷脑、50μg甲基正壬酮的混合对照品溶液。选择分离度好的桉油精作为参照峰(s)。2.2供试品溶液制备取装量差异项下的双鱼颗粒(批号:161201),混匀,取适量,研细,取约10.0g,精密称定,置500ml烧瓶中,加入玻璃珠数颗,自挥发油提取器上端加入水250ml,再加入正己烷3ml。自沸腾开始计时60分钟后放置30分钟冷却,分取正己烷层,挥发油提取器用正己烷涮洗2次,合并正己烷于25ml量瓶内,加正己烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,作为供试品溶液。2.3升温程序考察毛细管柱:agilentdb-1(30m×0.32mm,0.25μm);进样量1μl;分流比50:1;进样口温度230℃;fid检测器温度为250℃;以氮气为载气,氮气流速0.6l/min。升温程序ⅰ:柱温:50℃保持0分钟,再以4℃/min升至75℃,保持10分钟,再以1℃/min升至100℃,再以8℃/min升至150℃,保持2分钟,再以3℃/min升至165℃,保持5分钟,再以8℃/min升至250℃,保持3分钟。升温程序ⅱ:柱温:50℃保持2分钟,再以8℃/min升至100℃,保持10分钟,再以8℃/min升至250℃,保持5分钟。升温程序ⅲ:柱温:80℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃。表1三种升温程序分离效果对比结果由表1和图1-3表明,采用升温程序ⅱ和升温程序ⅲ桉油精、薄荷脑的分离度均大于1.5,综合考虑出峰时间,优选采用升温程序ⅲ,各色谱峰的整体分离效果较好。结论:气相色谱条件为:进样量1μl;进样口温度230℃;fid检测器温度为250℃;升温程序为:柱温:80℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃。3系统适用性3.1对照品溶液配制分别取桉油精、薄荷脑对照品适量,精密称定,加正己烷制成每1ml分别含830.50μg桉油精、2099.52μg薄荷脑的混合对照品母液①和每1ml分别含815.93μg桉油精、2084.40μg薄荷脑的混合对照品母液②,取混合对照品母液①1ml至10ml量瓶内,加正己烷稀释定容至刻度,摇匀即得混合对照品溶液①。3.2供试品溶液制备取装量差异项下的双鱼颗粒(批号:161201),混匀,取适量,研细,取约10.0g,精密称定,置500ml烧瓶中,加入玻璃珠数颗,自挥发油提取器上端加入水250ml,再加入正己烷3ml。自沸腾开始计时60分钟后放置30分钟,分取正己烷层,挥发油提取器用正己烷涮洗2次,合并正己烷于25ml量瓶内,加正己烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,作为供试品溶液。3.3气相色谱条件毛细管柱:agilentdb-1(30m×0.32mm,0.25μm)。进样量1μl;分流比25:1;进样口温度230℃;fid检测器温度为250℃;氮气流速0.6l/min;升温程序为:柱温:80℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃。在上述条件下,取3.1项下混合对照品溶液①连续进样5次测定,记录待测成分峰面积,取3.2项下供试品溶液连续进样5次,记录待测成分理论板数、分离度、对称因子。结果:桉油精、薄荷脑分离度均大于1.5,已达到基线分离。为确保定量分析的准确,规定理论板数按薄荷脑峰计算不得低于200000。4耐用性考察按照“3系统适用性”项,取供试品溶液及对照品溶液进行色谱条件的耐用性考察。固定其他条件不变时,单一变动以下各个条件:氮气流速、分流比、起始温度,考察不同氮气流速、不同分流比、不同起始温度的变化对桉油精与薄荷脑含量测定结果的影响。结果见表2,以上条件微小变化对含量测定结果无显著影响。表2耐用性考察结果备注:氮气流速0.6l/min样品,所用分流比为25:1,起始柱温为80℃。4.1气相色谱条件的确定根据耐用性考察结果确定桉油精、薄荷脑含量测定气相色谱条件为:agilentdb-1毛细管柱(30m×0.32mm,0.25μm),采用升温程序为:柱温:80℃保持2分钟,再以8℃/min升至112℃,保持4分钟,再以2℃/min升至120℃,保持6分钟;再以20℃/min升至250℃;进样量1μl;分流比25:1;进样口温度230℃;fid检测器温度为250℃;氮气流速0.6l/min。5供试品溶液的制备方法考察为了使制剂中的桉油精、薄荷脑提取完全,对提取方式、提取时间分别进行了考察。气相色谱条件同“4.1”项。5.1提取方法考察提取方法ⅰ(水蒸气蒸馏法):取装量差异项下的双鱼颗粒(批号:161201),混匀,取适量,研细,称取约10.0g,精密称定,置500ml烧瓶中,加入玻璃珠数颗,自挥发油提取器上端加入水250ml,再加入正己烷3ml。自沸腾开始计时60分钟后放置30分钟,分取正己烷层,挥发油提取器用正己烷涮洗2次,合并正己烷于25ml量瓶内,加正己烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,作为供试品溶液ⅰ。提取方法ⅱ(二氯甲烷-水回流提取法):取装量差异项下的双鱼颗粒(批号:161201),混匀,取适量,研细,称取约10.0g,精密称定于150ml烧瓶内,精密加入去离子水30ml,使样品溶解,再加入二氯甲烷15ml,水浴回流30分钟(99.9℃),放凉后分取二氯甲烷,水层用二氯甲烷萃取三次,每次3ml二氯甲烷,合并二氯甲烷于25ml量瓶中,加二氯甲烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,作为供试品溶液ⅱ。提取方法ⅲ(乙醇超声提取法):取装量差异项下的双鱼颗粒(批号:161201),混匀,取适量,研细,称取约10.0g,精密称定于150ml锥形瓶内,精密加入乙醇25ml,超声30分钟(250w,40khz),放冷后,取上清液离心,过0.22μm微孔滤膜,取续滤液,作为供试品溶液ⅲ。表3提取方法考察结果见表3和图4-6,以提取方法ⅰ制得的供试品溶液中桉油精与薄荷脑含量显著高于其他两种提取方法,且供试品溶液无色,不容易污染毛细管柱。5.2提取时间考察取装量差异项下的双鱼颗粒(批号:161201),混匀,取适量,研细,称取约10.0g,平行4份精密称定,置500ml烧瓶中,加入玻璃珠数颗,自挥发油提取器上端加入水250ml,再加入正己烷3ml。自沸腾开始分别计时30、60、90、120分钟后放置30分钟,分取正己烷层,挥发油提取器用正己烷涮洗2次,合并正己烷,置于25ml量瓶内,加正己烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,得到供试品溶液。进行含量测定,测定结果见表4,显示提取60、90、120min桉油精和薄荷脑含量变化不显著,鉴于提取90min所得桉油精和薄荷脑含量微微高出一些,故选择提取时间为90分钟。表4提取时间选择5.3确定供试品溶液的制备方法综上所述确定供试品溶液的制备方法为:取装量差异项下的本品,混匀,取适量,研细,称取约10.0g,精密称定,置500ml烧瓶中,加入玻璃珠数颗,自挥发油提取器上端加入水250ml,再加入正己烷3ml。自沸腾开始计时90分钟后放置30分钟,分取正己烷层,挥发油提取器用正己烷涮洗2次,合并正己烷,置于25ml量瓶内,加正己烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,作为供试品溶液。6方法学研究6.1专属性考察6.1.1对照品溶液的制备取3.1项下混合对照品母液②。6.1.2供试品溶液的制备同5.3项下供试品溶液的制备方法制得供试品溶液。6.1.3阴性供试品溶液的制备原方分别去艾叶、薄荷药材及同时去艾叶和薄荷,按制剂工艺分别制得缺艾叶、缺薄荷、缺艾叶和薄荷的阴性制剂,分别取上述三种阴性制剂约10.0g,按供试品溶液的制备方法,同法制得缺艾叶、缺薄荷以及缺艾叶和薄荷的阴性制剂供试品溶液。分别精密吸取上述混合对照品母液②、供试品溶液、缺艾叶、缺薄荷及缺艾叶和薄荷的阴性制剂供试品溶液各1μl,注入气相色谱仪,按照4.1下气相色谱条件进行测定,结果如图7-图11所示,表明,缺薄荷阴性供试品对薄荷脑的测定无干扰,缺艾叶和薄荷的阴性供试品对桉油精的测定无干扰。说明本发明方法对薄荷脑具有良好的专属性。鉴于桉油精为艾叶和薄荷的共有成分,且无其他药味干扰,故桉油精也纳入含量测定研究。6.1线性关系考察取3.1项下混合对照品母液②,精密量取0.1ml、0.2ml、0.5ml、1.0ml、1.5ml、2.0ml分别置于5ml量瓶内,加入正己烷稀释定容至刻度,摇匀即得分别含桉油精16.32μg/ml、薄荷脑41.69μg/ml、分别含桉油精32.64μg/ml、薄荷脑83.38μg/ml、分别含桉油精81.59μg/ml、薄荷脑208.44μg/ml、分别含桉油精163.19μg/ml、薄荷脑416.88μg/ml、分别含桉油精244.78μg/ml、薄荷脑625.32μg/ml、分别含桉油精326.37μg/ml、薄荷脑833.76μg/ml的一系列混合对照品溶液。分别吸取上述各混合对照品溶液1μl,注入气相色谱仪,按照4.1下气相色谱条件进行测定,结果见表5、6,以峰面积平均值为纵坐标(y),对照品浓度(μg/ml)为横坐标(x),绘制标准曲线得回归方程:桉油精:y=0.7185x+1.0899;r=0.9999;薄荷脑:y=0.7234x+2.6501;r=0.9999。表5线性关系考察-桉油精表6线性关系考察-薄荷脑结果表明,桉油精在浓度为16.32~326.37μg/ml之间呈良好的线性关系,薄荷脑在浓度为41.69~833.76μg/ml之间呈良好的线性关系。6.4精密度试验取低、中、高三个不同浓度的混合对照品溶液(低标:分别含桉油精16.32μg/ml、薄荷脑41.69μg/ml的混合对照品溶液,中标:分别含桉油精81.59μg/ml、薄荷脑208.44μg/ml的混合对照品溶液,高标:分别含桉油精326.37μg/ml、薄荷脑833.76μg/ml的混合对照品溶液),分别连续进样6次,进样量1μl,按照4.1下气相色谱条件进行测定,结果见表7,其峰面积rsd(%)均低于2%,结果表明,仪器精密度良好。取供试品溶液连续进样6次,剪切去掉前6分钟及24分钟以后溶剂峰,以第1次进样所得指纹图谱做为对照,计算后5次进样所得指纹图谱的相似度,结果相似度均大于0.99。结果表明该方法精密度良好。表7精密度结果(峰面积)6.5重复性试验取双鱼颗粒(批号:161201),按供试品溶液制备方法制得6份供试品溶液,按照4.1下气相色谱条件进行测定,结果见表8,桉油精含量0.219mg/g,薄荷脑含量0.609mg/g。剪切去掉前6分钟及24分钟以后溶剂峰,以第1份样品所得指纹图谱作为对照计算另5份样品所得指纹图谱的相似度,结果相似度均大于0.99。表明本发明方法重复性良好。表8重复性结果6.6稳定性试验取6.5项下同一供试品溶液分别于0、2、4、8、12小时进样,结果见表9,供试品溶液中桉油精、薄荷脑、含量的rsd分别为1.62%、1.48%,剪切去掉前6分钟及24分钟以后溶剂峰,以0小时进样所得指纹图谱为对照计算相似度,相似度结果均大于0.99,表明供试品溶液在12小时内稳定。表9稳定性结果6.7回收率试验取装量差异项下的双鱼颗粒(批号:161201,桉油精含量0.219mg/g,薄荷脑含量0.609mg/g),混匀,取适量,研细,分别称取6份,每份约5g,精密称定,置500ml烧瓶中,分别精密加入混合对照品母液②1.5ml,待混合对照品母液与供试品充分混合后,加入玻璃珠数颗,自挥发油提取器上端加入水250ml,再加正己烷3ml,密闭连接好冷凝管后,自沸腾开始计时90分钟,放置30分钟分取正己烷层,挥发油提取器用正己烷涮洗2次,合并正己烷,置于25ml量瓶内,加正己烷稀释定容至刻度,摇匀,过0.22μm微孔滤膜,取续滤液,作为供试品溶液。进样进行含量测定,结果见表10,2个成分回收率在95.62~107.84%之间,rsd%均小于2.3%,表明,该方法回收率良好。表10加样回收率结果6.8多批次样品检测及对照指纹图谱的获得收集十批双鱼颗粒样品,按5.3项下供试品溶液的制备方法制备供试品溶液,进样量1μl,按照4.1下气相色谱条件测定桉油精、薄荷脑含量,同时采用国家药典颁布的《中药色谱指纹图谱相似度评价系统》2012年版进行分析,剪切去掉前6分钟及24分钟以后溶剂峰,以平均数法对照指纹图谱生成法,生成对照指纹图谱(见图12、图13)。以对照指纹图谱为参照,由相似度软件计算各批次样品相似度,结果见表12。以桉油精为参照物计算各特征峰的相对保留时间,并规定相对保留时间的范围。结果见表13。表12双鱼颗粒含量测定及指纹相似度结果表13双鱼颗粒指纹图谱定性结果及特征峰相对保留时间峰号物质名称相对保留时间1未知0.893±20%2桉油精13未知1.321±20%4未知1.361±20%5未知1.424±20%6薄荷脑1.451±20%7未知1.473±20%8未知1.512±20%9未知1.767±20%10甲基正壬酮2.037±20%11未知2.375±20%12未知2.535±20%6.9相关性考察取金银花药材、赤芍药材、鱼腥草药材、薄荷药材、艾叶药材各约3g,精密称定于圆底烧瓶内,按5.3项下供试品制备方法,制备供试品溶液,按4.1项下色谱条件进样分析,见图13,12个特征峰中1、10、11、12号峰主要来自于鱼腥草,2号峰主要来自于艾叶和薄荷,3、5、7号峰主要来自于艾叶,4、6、8号主要来自于薄荷。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1