在本体异质结光敏层P/N结的添加剂结晶的制作方法
本申请要求2014年5月7日提交的,题为“在本体异质结光敏层P/N结的添加剂结晶”的美国临时申请第61/989970号的权益。参考专利申请的全部内容通过引用并入本申请。
发明背景
A.发明领域
本发明一般地涉及添加剂在本体异质结光敏层中的用途。具体地,已做出新发现,其使得在本体异质结光敏层的供体/受体界面(或p-n结)增加添加剂的配置或定位。这可以导致这些层的效率提高。
B.相关技术说明
在过去十年,由于对替代能源的永远迫切的需求以及该领域的许多进展,对有机光伏器件(OPV)的兴趣以指数地增长。尽管已经报道了双层或平面异质结OPV器件结构有希望的结果,但是大部分文献侧重于本体异质结(BHJ)OPV的制造,其利用当作为单一功能层沉积时彼此相分离的供体-受体聚合物和/或小分子的混合物共混。最近,已经报道了使用多种独特的合成聚合物的基于BHJ构型的OPV具有接近9%的功率转换效率(ηeff)。
例如,P3HT:PC61BM(聚(3-己基噻吩):苯基-C61-丁酸甲酯)可以说是研究最多的用作BHJ OPV中的活性层的供体-受体对材料,有无数出版物显示这些器件具有2%至5%的neff(Dang等人,Advanced Materials.2011,23:3597–3602)。例如化合物纯度、退火温度和退火时间、使用低沸点溶剂和添加剂以及P3HT与PC61BM的总比例的几个因素均被认为是该效率变化的原因(Dang等人,2011;Dang等人,Chemical Reviews,2013,113:3734–3765)。还被接受的是与P3HT和PC61BM有关的能级不理想,增加的效率可能或可以由修改P3HT或PC61BM的前沿分子轨道的能级造成(Blouin等人,J.Am.Chem.Soc.2008,130:732–742;Boudreault等人,Chem.Mater.,2011,23:456–469)。
替代改变前沿轨道的能级,已显示增加P3HT:PC61BM BHJ OPV器件效率的另一种方法是包含第三种组分(即添加剂),或小分子或聚合物,以充当第二供体或第二受体。该构型被称为级联BHJ,借此通过增加光吸收、激子离解或甚至在P3HT和PC61BM或PC71BM域之间的空穴或电子转移实现器件ηeff提高(Chen等人,ChemSusChem.2013,6:20–35;Khlyabich等人,Journal of the American Chemical Society.2012,134:9074–9077;Khlyabich等人,Journal of the American Chemical Society.2011,133:14534–14537)。例如,Chen等人在2013年利用双极性聚[2,3-双(噻吩-2-基)-丙烯腈-9,9’-二辛基-芴]聚合物作为P3HT:PC61BM级联BHJ OPV器件中的添加剂(Chen等人,2011)。作者注意到当添加相对于P3HT:PC61BM少如~2.5重量%的聚合物时,ηeff最多增加30%。此外,Honda等人探究了含金属的酞菁对形成P3HT:PC61BM级联BHJ的用途(Honda等人,ACS Applied Materials&Interfaces,2009,1:804–810,Honda等人,Chem.Commun.2010,46:6596–6598)。当将双(三-正己基甲硅烷基氧化物)硅酞菁((3HS)2-SiPc)添加到P3HT:PC61BM BHJ OPV器件时,注意到ηeff最高20%的增加(Honda等人,2009)。
然而,在BHJ光敏层中使用添加剂的问题之一是其对改善给定BHJ层效率具有的作用有限或有上限。此外,目前没有太多可用的添加剂提供如在以上段落中讨论的改善效率。
发明概述
已发现增加在BHJ光敏层中所用的添加剂功效的解决方案。具体地,该发现的前提是在BHJ光敏层的p-n结配置或定位添加剂。这使得添加剂更容易起到其增强BHJ光敏层效率的目的。不希望受理论约束,认为在制备BHJ光敏层期间,使添加剂结晶有助于将添加剂配置或定位于电子供体和受体材料之间的界面。本发明的方法提供了增加BHJ光敏层中在p-n结处添加剂的存在的方式。这种配置的益处导致这些BHJ层总效率的增加或增强。该过程还能够使用曾经被认为具有有限价值的已知添加剂,或基于有利于结晶的工艺条件选择添加剂。此外,选择具有高(负)结晶焓(ΔHcryst)的添加剂在本发明全文中是有利的。
在本发明的一个实施方案中,公开了制备本体异质结光敏层的方法,或使添加剂位于本体异质结光敏层界面的方法,或提高本体异质结光敏层效率的方法。这些方法中的每种可以包括:(1)获得包含溶剂、电子供体材料、可接受电子的材料和溶于该溶剂的添加剂的混合物,其中添加剂具有高(负)的结晶焓(ΔHcryst),(2)由混合物形成本体异质结光敏层,其中添加剂的晶体形成并位于本体异质结光敏层的电子供体材料和电子受体材料之间的界面。基于其结晶倾向可以选择所使用的添加剂。例如,可以在本发明全文中使用具有高(负)结晶焓(ΔHcryst)(即至少1微焦每摩尔)的添加剂。在更具体的实施方案中,结晶焓(ΔHcryst)可以为1微焦每摩尔至100焦每摩尔。在更具体的实施方案中,结晶焓(ΔHcryst)可以为至少2微焦每摩尔、至少3微焦每摩尔、至少4微焦每摩尔、至少5微焦每摩尔、至少6微焦每摩尔、至少7微焦每摩尔、至少8微焦每摩尔、至少9微焦每摩尔、至少10微焦每摩尔、至少11微焦每摩尔、至少12微焦每摩尔、至少13微焦每摩尔、至少14微焦每摩尔、至少15微焦每摩尔、至少16微焦每摩尔、至少17微焦每摩尔、至少18微焦每摩尔、至少19微焦每摩尔、至少20微焦每摩尔、至少25微焦每摩尔、至少30微焦每摩尔、至少35微焦每摩尔、至少40微焦每摩尔、至少45微焦每摩尔、至少50微焦每摩尔、至少60微焦每摩尔、至少70微焦每摩尔、至少80微焦每摩尔、至少90微焦每摩尔、至少100微焦每摩尔、至少150微焦每摩尔、至少200微焦每摩尔、至少250微焦每摩尔、至少300微焦每摩尔、至少350微焦每摩尔、至少400微焦每摩尔、至少450微焦每摩尔、至少500微焦每摩尔、至少550微焦每摩尔、至少600微焦每摩尔、至少650微焦每摩尔、至少700微焦每摩尔、至少800微焦每摩尔、至少850微焦每摩尔、至少900微焦每摩尔、至少950微焦每摩尔、至少1焦每摩尔、至少2焦每摩尔、至少3焦每摩尔、至少4焦每摩尔、至少5焦每摩尔、至少6焦每摩尔、至少7焦每摩尔、至少8焦每摩尔、至少9焦每摩尔、至少10焦每摩尔、至少15焦每摩尔、至少20焦每摩尔、至少25焦每摩尔、至少30焦每摩尔、至少35焦每摩尔、至少40焦每摩尔、至少45焦每摩尔、至少50焦每摩尔、至少55焦每摩尔、至少60焦每摩尔、至少65焦每摩尔、至少70焦每摩尔、至少80焦每摩尔、至少85焦每摩尔、至少90焦每摩尔、至少95焦每摩尔、最高100焦每摩尔、或其中任意范围。在其他实施方案中,结晶焓(ΔHcryst)可以大于100焦每摩尔(例如,110焦每摩尔、120焦每摩尔、130焦每摩尔、140焦每摩尔、150焦每摩尔、160焦每摩尔、170焦每摩尔、180焦每摩尔、190焦每摩尔、200焦每摩尔、300焦每摩尔、400焦每摩尔、500焦每摩尔、600焦每摩尔、700焦每摩尔、800焦每摩尔、900焦每摩尔、或更多焦每摩尔、或其中任意范围)。当前用于BHJ层的或将来会发现的所有类型添加剂可以用于本发明全文中。效率提高可能是许多因素的结果,包括在本体异质结光敏层中存在的纳米级区域中的任何一个形态的改善和电子/光敏三元添加剂结晶的增强。可以使用的添加剂的一些非限制性实例包括烷烃二硫醇(例如,1,6-二巯己烷;1,8-二巯辛烷;1,10-二巯癸烷等)、烷基二卤化物(例如,1,6-二氯己烷;1,6-二溴己烷;1,8-二氯辛烷;1,8-二溴辛烷;1,8-二碘辛烷,1,8-二氯癸烷;1,8-二溴癸烷;1,8-二碘癸烷等)、烷基二腈化物(例如,辛二腈;癸二腈;十二烷二腈等)、酞菁、其衍生物(即,经取代的化合物),或其任意组合或混合物。在更优选的方面,添加剂可以是烷烃二硫醇或酞菁或其组合。在具体方面,添加剂可以是双(三-正己基甲硅烷基氧化物)锗酞菁。添加剂(可以包含单一或混合物和添加剂的组合)可以溶于溶剂最多到其饱和点,或者可以在溶剂中过饱和。在一些情况下,混合物还可以包含成核剂以促进添加剂结晶。在一些具体的情况下,可以将混合物加热并在促进添加剂结晶的条件下冷却或干燥(例如,可以使用真空烘箱或热平板的较慢冷却条件)。更进一步地,在此过程中可以添加非溶剂或反溶剂以促进添加剂结晶。本发明的电子受体和电子供体材料可以是那些本领域目前已知的,以及将来可能会发现的。电子供体材料的一些非限制性实例包括聚(三己基噻吩)(P3HT)或聚[2-甲氧基-5-(2-乙基己氧基)-1,4-亚苯基乙烯撑]、或其组合。电子受体材料的一些非限制性实例包括[6,6]苯基-C61-丁酸甲酯(PC61BM)、[6,6]苯基-C71-丁酸甲酯(PC71BM)、或1',1”,4',4”-四氢-二[1,4]亚甲基萘[1,2:2′,3′,56,60:2″,3″][5,6]富勒烯-C60(ICBA)、或其任意组合。在本发明的一个具体方面,供体材料和受体材料是P3HT:PC61BM共混物。可以在本发明全文中使用的溶剂的非限制性实例包括氯苯、氯仿、二氯苯、二氯甲烷、二甲苯、四氢化萘、甲苯、苯、喹诺酮、间甲酚、1,2,4-三甲基苯、甲基萘、或二甲基萘、或其任意组合。可以在衬底上形成本体异质结光敏层。可以将混合物配置在衬底表面上(例如,通过刮刀涂覆、旋涂、弯月面涂覆、转移印刷、喷墨印刷、胶版印刷或丝网印刷方法)。衬底可以是透明的、半透明的、或反射的。在某些情况下,添加剂不是双(三-正己基甲硅烷基氧化物)硅酞菁。本体异质结光敏的功率转换效率(ηeff)可以通过在电子供体材料和电子受体材料之间界面的添加剂结晶定位来提高。本体异质结光敏的短路电流(JSC)可以通过在电子供体材料和电子受体材料之间界面的添加剂结晶定位来增强。
本发明的本体异质结光敏层可以用于电子应用。这些层可以用于电子器件的活性层中。活性层可以是有机层或混合半导电层或导电层。该器件可以包括衬底、光敏层、至少两个电极,其中之一是透明的。将至少一部分或全部光敏层沉积在所述电极之间。透明电极可以是阴极,另一个电极可以是阳极。或者,透明电极可以是阳极,另一个电极可以是阴极。在一些情况下,上述电极都可以是透明的。在其它情况下,电极中的一个可以是透明的,而另一个是非透明的(例如,不透明的)或反射的,使其可以反射电磁辐射如紫外光或可见光或阳光。此外,衬底可以是不透明的、反射的、或透明的。在具体情况下,电子器件可以是光电池或可以包含光电池。所述电池可以不包含电解质。光电池可以包含在有机电子器件中。这些器件的实例包括有机发光二极管(OLED)(例如,聚合的有机发光二极管(PLED)、小分子有机发光二极管(SM-OLED)、有机集成电路(O-IC)、有机场效应晶体管(OFET)、有机薄膜晶体管(OTFT)、有机太阳能电池(O-SC)、和有机激光二极管(O-激光)。
本发明全文还公开了实施方案1至28。实施方案1是制备本体异质结光敏层,使添加剂位于本体异质结光敏层界面,或提高本体异质结光敏层效率的方法,该方法包括以下步骤:(1)获得包含溶剂、电子供体材料、可接受电子的材料和溶于溶剂的添加剂的混合物,其中添加剂具有高(负)的结晶焓(ΔHcryst);(2)由混合物形成本体异质结光敏层,其中添加剂的晶体形成并位于本体异质结光敏层的电子供体材料和电子受体材料之间的界面。实施方案2是实施方案1的方法,其中基于其结晶倾向选择所使用的添加剂。实施方案3是实施方案1的方法,其中将步骤(1)中的添加剂溶于溶剂中最多至其饱和点或在溶剂中过饱和。实施方案4是实施方案1至3中任一项的方法,其中混合物还包含成核剂以促进步骤(2)期间添加剂的结晶。实施方案5是实施方案1至4中任一项的方法,其中将步骤(1)中的混合物加热,将步骤(2)中的混合物在促进添加剂结晶的条件下冷却或干燥。实施方案6是实施方案1至5中任一项的方法,其中向步骤2中的混合物添加非溶剂以促进添加剂的结晶。实施方案7是实施方案1至6中任一项的方法,其中供体材料和受体材料是P3HT:PC61BM共混物。实施方案8是实施方案1至7中任一项的方法,其中电子供体材料是聚(三己基噻吩)(P3HT)或聚[2-甲氧基-5-(2-乙基己氧基)-1,4-亚苯基亚乙烯撑]、或其组合。实施方案9是实施方案1至8中任一项的方法,其中电子受体材料是[6,6]苯基-C61-丁酸甲酯(PC61BM)、[6,6]苯基-C71-丁酸甲酯(PC71BM)或1',1”,4',4”-四氢-二[1,4]亚甲基萘[1,2:2′,3′,56,60:2″,3″][5,6]富勒烯-C60(ICBA)、或其任意组合。实施方案10是实施方案1至9中任一项的方法,其中添加剂是烷烃二巯化物或双(三-正己基甲硅烷基氧化物)锗酞菁或其组合。实施方案11是实施方案1至10中任一项的方法,其中溶剂是氯苯、氯仿、二氯苯、二氯甲烷、二甲苯、四氢化萘、甲苯、苯、喹诺酮、间甲酚、1,2,4-三甲基苯、甲基萘、或二甲基萘、或其任意组合。实施方案12是实施方案1至11中任一项的方法,其中本体异质结光敏层在衬底上形成。实施方案13是实施方案12的方法,其中将来自步骤(1)的混合物配置在衬底表面上。实施方案14是实施方案13的方法,其中混合物通过刮刀涂覆、旋涂、弯月面涂覆、转移印刷、喷墨印刷、胶版印刷或丝网印刷方法配置。实施方案15是实施方案12至14中任一项的方法,其中衬底是电极。实施方案16是实施方案15的方法,其中电极是透明的或半透明的。实施方案17是实施方案15的方法,其中电极是反射的。实施方案18是实施方案17的方法,其中添加剂不是双(三-正己基甲硅烷基氧化物)硅酞菁。实施方案19是实施方案1至18中任一项的方法,其中本体异质结光敏的功率转换效率(ηeff)通过在电子供体材料和电子受体材料之间界面的添加剂结晶定位来提高。实施方案20是实施方案1至19中任一项的方法,其中本体异质结光敏的短路电流(JSC)通过在电子供体材料和电子受体材料之间界面的添加剂结晶定位来增强。实施方案21是光电池,其包含通过实施方案1至20中任一项的方法制备的本体异质结光敏层。实施方案22是实施方案21的光电池,其包含透明衬底、透明电极、本体异质结光敏层、第二电极,其中将光敏层配置于透明电极和第二电极之间。实施方案23是实施方案22的光电池,其中透明电极是阴极,第二电极是阳极。实施方案24是实施方案22的光电池,其中透明电极是阳极,第二电极是阴极。实施方案25是实施方案21至24中任一项的光电池,其中第二电极不是透明的。实施方案26是实施方案21至25中任一项的光电池,其中光电池包含在有机电子器件中。实施方案27是本体异质结光敏层,其通过实施方案1至20中任一项的方法制备。实施方案28是实施方案27的本体异质结光敏层,其包含在光电池中。
在本发明全文中“添加剂”指可以增加本体异质结光敏层的效率或性能的材料(例如,化合物、低聚物、聚合物等)。
术语“约”或“大约”定义如本领域普通技术人员所理解的接近于,在一个非限制性实施方案中,该术语定义为10%以内、优选5%以内、更优选1%以内、最优选0.5%以内。
当在权利要求或说明书中与术语“包含”连用时,指示词前没有数量词可以表示“一个”,但也与“一个或更多个”、“至少一个”和“一个或多于一个”的意思一致。
词语“包含”、“具有”、“包括”、“含有”是包括性的或开放式的,不排除附加的、未列举的要素或方法步骤。
制备本发明的本体异质结光敏层、光电池、和有机电子器件的方法可以“包括”贯穿说明书公开的具体添加剂、材料、成分、化合物、组合物等、“基本由其组成”、或“由其组成”。关于过渡性短语“基本由……组成”,在一个非限制方面,前述方法的基本特征和新特征是实现添加剂在本体异质结光敏层的电子供体材料和电子受体材料之间的界面结晶的能力。
本发明的其他目的、特点和优点由以下附图、详细说明和实施例会变得明显。然而,应理解的是附图、详细说明和实施例尽管表示本发明的具体实施方案,但仅以说明方式给出,不意在限制。此外,预期的是本发明精神和范围内的改变和修改由该详细说明对本领域技术人员会变得明显。
附图说明
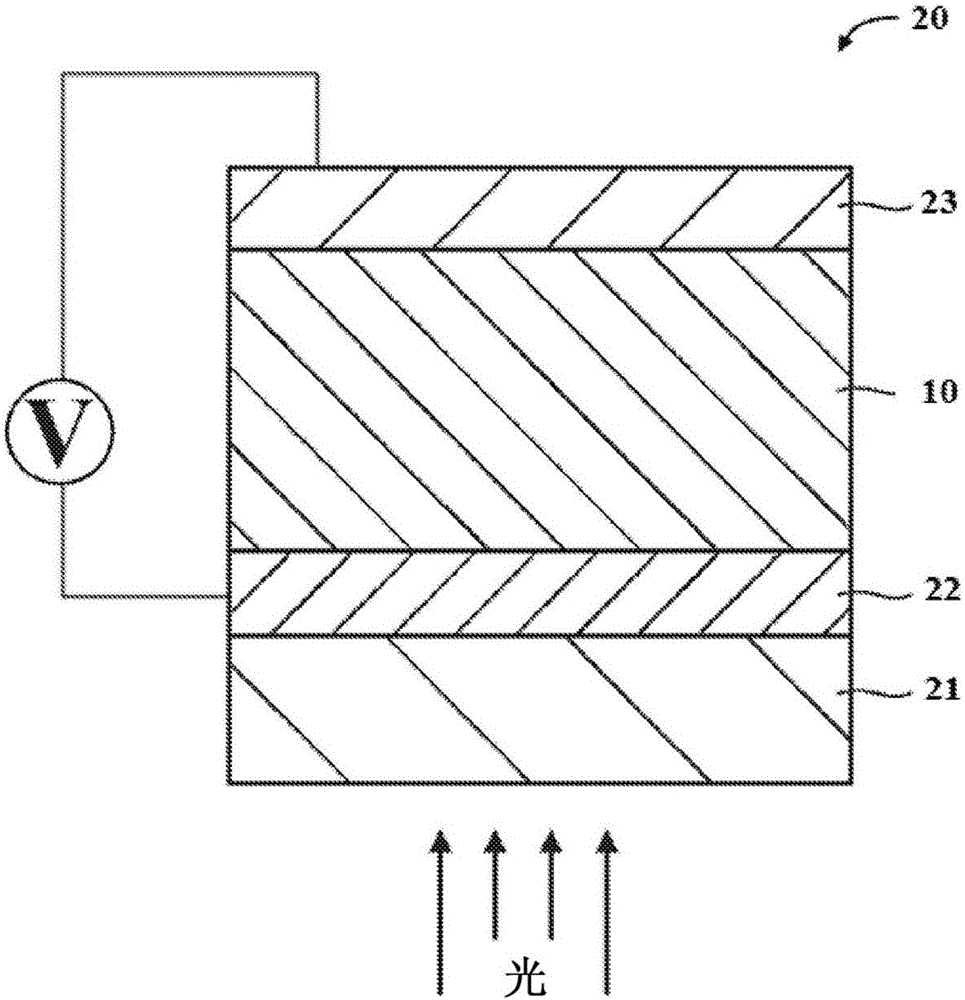
图1:本发明本体异质结光敏层的图解。
图2:包含本发明本体异质结光敏层的有机光电池的图解。
图3:聚(3-己基噻吩)(P3HT)、双(三-正己基甲硅烷基氧化物)硅酞菁((3HS)2-SiPc)、双(3-十五烷基苯氧基)硅酞菁((PDP)2-SiPc)、三-正己基甲硅烷基氧化硼亚酞菁(3HS-BsubPc)、3-十五烷基苯氧基硼亚酞菁(PDP-BsubPc)、3-甲基苯氧基硼亚酞菁(3MP-BsubPc)、五氟代苯氧基硼亚酞菁(F5-BsubPc)、双(三-正己基甲硅烷基氧化物)锗酞菁((3HS)2-GePc)和苯基-C61-丁酸甲酯(PC61BM)的能级图和化学结构。
图4A:P3HT:PC61BM(1.0:0.8,质量比)和P3HT:PC61BM:X(1.0:0.8:Y)的外量子效率(EQE)相对波长的图,其中X=双(三-正己基甲硅烷基氧化物)硅酞菁((3HS)2-SiPc,1)、三-正己基甲硅烷基氧化硼亚酞菁(3HS-BsubPc)和双(三-正己基甲硅烷基氧化物)锗酞菁((3HS)2-GePc),其中Y=0.2(10.6重量%)、0.1(5.3重量%)或0.07(3.7重量%)。显示带有误差条的标准P3HT:PC61BM BHJ太阳能设备数据(无三元添加剂)以说明标准装置设备的空间。
图4B:P3HT:PC61BM(1.0:0.8,质量比)和P3HT:PC61BM:X(1.0:0.8:Y)电流相对电压(IV)的图,其中X=双(三-正己基甲硅烷基氧化物)硅酞菁((3HS)2-SiPc,1)、三-正己基甲硅烷基氧化物硼亚酞菁(3HS-BsubPc)和双(三-正己基甲硅烷基氧化物)锗酞菁((3HS)2-GePc),其中Y=0.2(10.6重量%)、0.1(5.3重量%)或0.07(3.7重量%)。显示带有误差条的标准P3HT:PC61BM BHJ太阳能设备数据(无三元添加剂)以说明标准设备占据的空间。
图5A:双(三-正己基甲硅烷基氧化物)硅酞菁((3HS)2-SiPc)的电化学光谱。
图5B:双(三-正己基甲硅烷基氧化物)锗酞菁((3HS)2-GePc)的电化学光谱,C)三-正己基甲硅烷基氧化物硼亚酞菁(3HS-BsubPc)和D)双(三-十五烷基苯氧基)硅酞菁((PDP)2-SiPc)。
图5C:三-正己基甲硅烷基氧化硼亚酞菁(3HS-BsubPc)的电化学光谱。
图5D:双(三-十五烷基苯氧基)硅酞菁((PDP)2-SiPc)的电化学光谱。
图6A:作为BHJ OPV器件中活性层的P3HT:PC61BM:PDP-BsubPc(1:0.8:X,其中X=0.2(10.6重量%)、0.1(5.3重量%)和0.07(3.7重量%))的外量子效率(EQE)相对波长的图。
图6B:作为BHJ OPV器件中活性层的P3HT:PC61BM:PDP-BsubPc(1:0.8:X,其中X=0.2(10.6重量%)、0.1(5.3重量%)和0.07(3.7重量%))的电流相对电压(IV)的图。
图6C:作为BHJ OPV器件中活性层的P3HT:PC61BM:X(其中X=5.3重量%的3MP-BsubPc添加、5.3重量%的PDP-BsubPc添加、5.3重量%的F5-BsubPc添加、或2.7重量%的3MP-BsubPc和2.7重量%的F5-BsubPc两者)的EQE相对波长的图。P3HT:PC61BM是原始的、标准的BHJ OPV器件(无三元添加剂),其具有由所有P3HT:PC61BM原始BHJ OPV器件的标准偏差得到的误差条。
图6D:作为BHJ OPV器件中活性层的P3HT:PC61BM:X(其中X=5.3重量%的3MP-BsubPc添加、5.3重量%的PDP-BsubPc添加、5.3重量%的F5-BsubPc添加、或2.7重量%的3MP-BsubPc和2.7重量%的F5-BsubPc两者)的IV图。P3HT:PC61BM是原始的、标准的BHJ OPV器件(无三元添加剂),其具有由所有P3HT:PC61BM原始BHJ OPV器件的标准偏差得到的误差条。
图7A:显示(3HS)2-SiPc(CCDC保藏号:988974)的结构和原子编号的椭球图(50%的几率)。
图7B和7C:多个(PDP)2-SiPc分子的晶体结构排列。(3HS)2-SiPc单晶通过从二氯甲烷缓慢蒸发而生长,用x射线晶体表征。立方体形代表晶胞。
图8A:作为BHJ OPV器件中活性层的P3HT:PC61BM(PDP)2-SiPc(1:0.8:X,其中X=0.2(10.6重量%)、0.1(5.3重量%)和0.07(3.7重量%))的外量子效率(EQE)相对波长的图。P3HT:PC61BM是原始的、标准的BHJ OPV器件(无三元添加剂),其具有由如“基线P3HT:PC61BM BHJ器件”章节列出的所有P3HT:PC61BM原始BHJ OPV器件的标准偏差得到的误差条。
图8B:作为BHJ OPV器件中活性层的P3HT:PC61BM:(PDP)2-SiPc(1:0.8:X,其中X=0.2(10.6重量%)、0.1(5.3重量%)和0.07(3.7重量%))的电流相对电压(IV)的图。P3HT:PC61BM是原始的、标准的BHJ OPV器件(无三元添加剂),其具有由如“基线P3HT:PC61BM BHJ装置”章节列出的所有P3HT:PC61BM原始BHJ OPV器件的标准偏差得到的误差条。
图9A:显示(PDP)2-SiPc(CCDC保藏号:9889746)的结构和原子编号的椭球图(50%的几率)。
图9B和9C:多个(PDP)2-SiPc分子的晶体结构排列。(PDP)2-SiPc单晶通过从二氯甲烷缓慢蒸发而生长,用x射线晶体表征。立方体代表晶胞。
发明详述
本发明提供了提高本体异质结光敏层的效率或性能(例如,增加的Jsc或ηeff或两者)的方式。这通过将添加剂经哟所述添加剂的结晶定位在这些光敏层的p-n结而实现的。相比之下,如实施例中非限制性方面所说明的,在这些层的p-n结不结晶(或较难结晶)的添加剂导致层较低的效率和性能。在优选的方面,因为可以最小化实现结晶的加工步骤,使用具有高(负)结晶焓(ΔHcryst)的添加剂。然而,可以使用附加步骤以帮助具有高(负)的结晶焓或具有低(正)的结晶焓的添加剂结晶(例如,成核剂、改变冷却和干燥程序等)。因此,在本发明全文中可以使用所有类型的添加剂。
在以下部分详细讨论本发明的这些和其他非限制性方面。
A.本体异质结层和添加剂结晶
本体异质结(BHJ)光敏层通常利用当沉积为单一功能层时彼此相分离的电子供体-受体聚合物、低聚物、或小分子、或其组合的混合物共混。相分离导致在电子供体材料(即n型材料)和电子受体材料(即p型材料)之间形成界面或结。在本发明全文中可以使用所有类型的供体材料和受体材料。举例来说,供体材料的非限制实例包括P3HT、聚[2-甲氧基-5-(3,7-二甲基辛氧基)-1,4-亚苯基乙烯撑](MDMO-PPV)、或聚(2-甲氧基-5-(2'-乙基-己氧基)-1,4-亚苯基乙烯撑)(MEH-PPV)、或其组合或共混物。电子受体材料的非限制实例包括PCBM或[6,6]-苯基C7i-丁酸甲酯(C70-PCBM)。其他材料如单壁碳纳米管(CNT)和其他n型聚合物也可以用作受体材料。
在本发明全文中可以使用所有类型和种类的添加剂。然而,在优选的方面,具有高(负)结晶焓(ΔHcryst)的添加剂可被用于帮助促进其结晶。可以使用的添加剂的非限制性实例包括烷烃二巯化物(例如,1,6-二巯己烷;1,8-二巯辛烷;1,10-二巯癸烷等)、烷基二卤化物(例如,1,6-二氯己烷;1,6-二溴己烷;1,8-二氯辛烷;1,8-二溴辛烷;1,8-二碘辛烷,1,8-二氯癸烷;1,8-二溴癸烷;1,8-二碘癸烷等)、烷基二腈化物(例如,辛二腈;癸二腈;十二烷二腈等)、酞菁、其衍生物(即,经取代的化合物),或其任意组合或混合物。特别地,本发明全文中可以使用其他添加剂,条件是添加剂或工艺条件导致添加剂结晶并将晶体定位于本发明本体异质结光敏层的p-n结,。
可以将供体材料、受体材料和添加剂混入能够溶解添加剂的溶剂中。溶剂也能够溶解供体材料和受体材料,或供体材料和受体材料可以分散或悬浮在溶剂中。溶剂的非限制性实例包括基于不饱和烃的溶剂(如甲苯、二甲苯、四氢化萘、十氢化萘、均三甲苯、正丁基苯、仲丁基苯和叔丁基苯)、基于卤代芳香烃的溶剂(如氯苯、二氯苯、三氯苯)、基于饱和卤代烃的溶剂(如四氯化碳、氯仿、二氯甲烷、二氯乙烷、氯丁烷、溴丁烷、氯戊烷、氯己烷、溴己烷和氯代环己烷)、醚(如四氢呋喃和四氢吡喃)和极性非质子溶剂(如二氯甲烷(DCM)、四氢呋喃(THF)、乙酸乙酯、乙酸丙酯、乙酸丁酯、乙酸异丁酯(等)、丙酮、二甲基甲酰胺(DMF)、乙腈(MeCN)、苯甲腈、硝基甲烷、二甲基亚砜(DMSO)、碳酸丙烯酯、或N-甲基-2-吡咯烷酮(NMP)、环丁砜(四亚甲基砜、2,3,4,5-四氢噻吩-1,1-二氧化物)、六甲基磷酰胺(HMPA)、甲基乙基酮、甲基异丁基酮、苯乙酮、二苯甲酮等)、或所述溶剂的任意组合。
可以使用以下非限制性方法以制备本发明的本体异质结光敏层:
(1)制备包含溶剂、电子供体材料、可接受电子的材料和溶于溶剂的添加剂的混合物。根据所期望的量可以改变每种这些成分的重量以实现给定的本体异质结层具有所期望的性质。举例来说,量可以是25重量%至75重量%的电子供体材料、75%至25重量%的电子受体材料、0.01重量%至20重量%的添加剂和适量的溶剂。电子供体材料和电子受体材料可以溶解或分散在溶剂中。在优选的方面,添加剂在溶剂中过饱和,有助于促进所述添加剂的结晶。
(2)然后将来自(1)的混合物通过基于溶液的方法(例如,喷涂、卷式涂覆、滴铸、浸涂、Mayer棒涂覆、刮刀涂覆、旋涂、弯月面涂覆、转移印刷、喷墨印刷、胶版印刷、丝网印刷、凹版印刷、柔版印刷、散发涂覆、喷嘴涂覆、毛细管涂覆等)沉积。
(3)(1)和/或(2)的条件可以是其积极地促进添加剂结晶的条件。结晶过程通常包括成核现象以及之后的晶体生长。当添加剂(溶质)溶解在溶剂中,溶质分子开始聚集成簇,从而在小区域内提高溶质浓度时,成核发生。一旦这些簇达到临界尺寸(其可以通过修改工艺条件例如温度、添加剂的饱和等来促进),原子以确定的和周期性的方式排列以生成晶体。不希望受理论约束,认为该结晶现象发生在靠近由供体材料和受体材料形成的p-n结,或这些晶体向p-n结迁移、或在形成p-n结期间向p-n结迁移。过饱和可以是结晶的驱动力。因此,成核和生长的速率可以由来自(2)的溶液中添加剂的存在过饱和驱动。取决于条件,成核和生长中的任一个可以比另一个占优势,结果得到具有不同大小和形状的晶体。此外,或者,沉积在衬底或电极上的混合物的干燥和冷却条件可以修改以进一步促进晶体生长。更进一步,成核剂可以包含在来自(1)的混合物中以进一步加强或促进添加剂的晶体生长。
图1是本发明本体异质结光敏层(10)的非限制性实例的横截面视图,其中添加剂是结晶的,主要存在于层的p-n结处。供体材料(11)和受体材料(12)形成多个界面或p-n结(13)。在以上讨论的处理步骤期间,添加剂的晶体形式(14,用方块表示)位于p-n结(13)旁边或p-n结(13)中。然而,在一些实施方案中,一些添加剂可以分布在供体材料(11)或受体材料(12)中,但不在p-n结(13)处。这种添加剂表示为小圆圈(15),其可以保持溶解的或可以是晶体形式。在本发明的优选实施方案中,以重量计,绝大多数添加剂以结晶形式存在于p-n结中或p-n结旁边。在更优选的实施方案中,所有添加剂存在于p-n结中或p-n结旁边。
B.有机光电池
本发明的本体异质结光敏层(10)可被用于光电应用,如有机光电池。图2是本发明非限制性的有机光电池的横截面视图。有机光电池(20)可以包含透明衬底(21)、前电极(22)、本体异质结光敏层(10)和后电极(23)。可以与光电池(20)一起使用本领域普通技术人员已知的其他材料、层和涂层(未显示),其中一些描述如下。
一般而言,有机光电池(20)可以通过以下步骤将光转换为可用能量:(a)光子吸收以产生激子;(b)激子扩散;(c)电荷转移;(d)电荷分离和转移至电极。关于(a),激子由通过光敏层(10)的光子吸收产生。关于(b),产生的激子扩散到p-n结(13)。然后在(c)中,电荷转移至活性层的其他成分。对于(d),电子和空穴分离和传输到电极(22)和电极(13),用于电路中。
衬底(21)可以用作支撑物。对于有机光电池,其通常是透明的或半透明的,使光能够有效地进入电池。其通常由不容易被热或有机溶剂改变或降解的材料制备而成,如已指出的,其具有优异的光透明度。这种材料的非限制性实例包括无机材料,例如无碱玻璃和石英玻璃、聚合物,例如聚乙烯、PET、PEN、聚酰亚胺、聚酰胺、聚酰胺酰亚胺、聚碳酸酯(例如,LexanTM,其是由SABIC Innovative Plastics提供的聚碳酸酯树脂)、液晶聚合物和环烯烃聚合物、硅和金属。
根据电路的设置,前电极(22)可以用作阴极或阳极。其堆叠在衬底(21)上。前电极(22)可以由透明的或半透明的导电材料制成。或者,前电极(22)可以由不透明的或反光材料制成。通常,前电极(22)通过使用这些材料形成膜而得到(例如,真空沉积、溅射、离子镀、电镀、涂覆等)。透明的或半透明的导电材料的非限制性实例包括金属氧化物膜、金属膜和导电聚合物。可以用于形成膜的金属氧化物的非限制性实例包括氧化铟、氧化锌、氧化锡及其复合物如铟锡酸盐(ITO)、掺杂氟的锡氧化物(FTO)及氧化铟锌膜。可以用于形成膜的金属的非限制性实例包括金、铂、银和铜。导电聚合物的非限制性实例包括聚苯胺和聚噻吩。前电极(22)的膜厚度通常为30nm至300纳米。如果膜厚度小于30纳米,则可以降低导电率和增加电阻,其导致光电转换效率降低。如果膜厚度大于300nm,则光透射率可能会降低。另外,前电极(22)的薄层电阻通常为10Ω/sq或更小。另外,前电极(22)可以是单层或由各自具有不同功函数的材料形成的层压层。
根据电路的设置,后电极(23)可以用作阴极或阳极。该电极(23)可以由透明的或半透明的导电材料制成。或者,其(23)可以由不透明的或反射材料制成。该电极(23)可以堆叠在光敏层(10)上。用于后电极(23)的材料可以是导电的。这些材料的非限制性实例包括金属、金属氧化物、和例如在上文前电极(22)内容中讨论的那些导电聚合物(例如,聚苯胺、聚噻吩等)。当使用具有高功函数的材料形成前电极(22)时,后电极(23)可以由具有低功函数的材料制成。具有低功函数的材料的非限制性实例包括Li、In、Al、Ca、Mg、Sm、Tb、Yb、Zr、Na、K、Rb、Cs、Ba、和其合金。后电极(23)可以是单层或由各自具有不同功函数的材料形成的层压层。进一步地,其可以是具有低功函数的材料中的一种或更多种和选自金、银、铂、铜、锰、钛、钴、镍、钨和锡中的至少一种的合金。合金的实例包括锂-铝合金、锂-镁合金、锂-铟合金、镁-银合金、镁-铟合金、镁-铝合金、铟-银合金、和钙-铝合金。后电极(23)的膜厚度可以是1nm至1000nm、或10nm至500nm。如果膜厚度过小,则电阻可以过大,产生的电荷可能不会充分地传递到外电路。
在一些实施方案中,前电极(22)和后电极(23)还可以用空穴传输层或电子传输层(在图1中未示出)涂覆以提高效率并防止有机光电池(1)短路。可以将空穴传输层和电子传输层插在电极和光敏层(10)之间。可以用于空穴传输层的材料的非限制性实例包括基于聚噻吩的聚合物、例如PEDOT/PSS(聚(3,4-亚乙基二氧噻吩)-聚(苯乙烯磺酸盐))和有机导电聚合物如聚苯胺和聚吡咯。空穴传输层的膜厚度可以为20nm至100nm。如果膜厚度过薄,可以更容易地发生电极短路。如果膜厚度过厚,膜电阻大,产生的电流有限,光转换效率会减小。对于电子传输层,其可以通过阻断空穴并更有效传输电子来运行。可以制备电子传输层的材料类型的非限制实例包括金属氧化物(例如,非晶的氧化钛)。当使用氧化钛时,膜厚度可以为5nm到20nm。如果膜厚度过薄,会降低空穴阻隔效果,从而使生成的激子在激子离解成电子和空穴前失活。通过比较,当膜厚度过厚时,膜电阻大,产生的电流是有限的,导致光转换效率降低。
实施例
以具体实施例的方式会更详细地描述本发明。仅以说明性目的提供以下实施例,不意在以任何方式限制本发明。本领域技术人员会容易地识别可以改变或修改以产生基本相同结果的各种非关键参数。
实施例1(材料、方法和流程)
材料:所有ACS级的溶剂购自Caledon Labs(卡利登,安大略省,加拿大),除非另有说明,不经进一步纯化使用。三-正己基氯硅烷购自Gelest(莫里斯维尔,宾夕法尼亚州,美国),按原样使用。氘代氯仿(CDCl3)和0.05体积/体积%的四甲基硅烷(TMS)购自Cambridge Isotope Laboratories,Inc.(圣伦纳德,魁北克省,加拿大),按原样使用。在涂覆二氧化硅(的孔径)和荧光指示剂的铝板上实施薄层色谱(TLC),从Whatman Ltd获得,在UV光(254nm)下可视化。使用从SiliCycle Inc.(魁北克,魁北克省,加拿大)获得的Silica Gel P60(筛孔尺寸40μm至63μm)实施柱层析。羟基硼亚酞菁(HO-BsubPc)(Fulford等人,2012)、二氯硅酞菁((Cl)2-SiPc)(Lowery等人,Inorg Chem.1965,4:128–128)和二氯锗酞菁((Cl)2-GePc)(Joyner和Kenney,Journal of the American Chemical Society,1960,82:5790–5791)、3-十五烷基苯氧基硼亚酞菁(PDP-BsubPc)(Brisson等人,Industrial and Engineering Chemistry Research 2011,50:10910–10917.)、3-甲基苯氧基硼亚酞菁(3MP-BsubPc)(Paton等人,Industrial and Engineering Chemistry Research,2012,51:6290–6296)和五氟代苯氧基硼亚酞菁(F5-BsubPc)(Morse等人,ACS Applied Materials&Interfaces.2010,2:1934–1944)全部根据文献制备。
方法:所有反应在氩气气氛下、使用烤箱干燥的玻璃器皿进行。用Bruker Avance III光谱仪在23℃下、CDCl3中对于1H NMR在400MHz操作,对于13C NMR在100MHz操作下记录核磁共振(NMR)谱。化学位移(δ)参考四甲基硅烷(0ppm)的1H NMR和CDCl3(77.16ppm)的13C NMR,以百万分之一(ppm)为单位报告。耦合常数(J)以赫兹(Hz)计报告。自旋多重性用以下缩写标记:s(单态)、d(双态)、t(三重态)、q(四重态)、m(多重态)和br(宽)。在GCT Premier TOF质谱计(Waters Corporation,米尔福德,马萨诸塞州,美国)上实施精确的质量测定(HRMS)。在配有Direct Analysis in Real Time(DART)离子源的AccuTOF型JMS T1000LC质谱计(JEOL USA Inc.,皮博迪,马萨诸塞州,美国)或具有EI/CI源的GC Premier TOF质谱仪(Waters Corporation,米尔福德,马萨诸塞州,美国)上获得低分辨率质谱(LRMS)。在PerkinElmer Lambda 1050UV/VIS/NIR光谱仪上使用具有10mm路径长度的PerkinElmer石英吸收池获得紫外-可见(UV-vis)吸收光谱。光致发光(PL)谱在PerkinElmer LS55荧光分光仪上使用具有10mm路径长度的PerkinElmer石英吸收池获得。在具有Waters 2998光电二极管阵列和WatersHR 2THF 4.6×300mm柱的Waters 2695分离模块上实施高压液相色谱(HPLC)分析。使用的流动相为HPLC级的乙腈(80体积%)和N,N-二甲基甲酰胺(20体积%)。使用Bioanalytical Systems C3电化学工作站进行循环伏安法。工作电极是2mm玻璃碳盘,反电极是铂丝,参考电极是Ag/AgCl2饱和盐溶液。在使用光谱级二氯甲烷之前,在室温下用氮气将其净化。在100mV/s的扫描速率下对每个样品测量+1.7V至-1.7V三个周期。将四丁基高氯酸铵(1M)用作支持电解质,将二茂铁癸烷用作内部参考。
三-正己基硅氧基硼亚酞菁(3HS-BsubPc)(图3):在氩气下,向经烘箱干燥的三颈圆底烧瓶添加HO-BsubPc(0.50g,1.21毫摩尔,1当量)、1,2-二氯苯(20mL)和三-正己基氯硅烷(0.90mL、2.46毫摩尔、2当量)。将反应混合物加热至130℃,通过HPLC(乙腈:N,N-二甲基甲酰胺-80:20体积/体积)监测反应HO-BsubPc的消耗。一旦反应停止进行(~5小时),将反应混合物冷却至室温,然后将其在减压下浓缩为深粉色液体。注意向反应混合物进一步添加三-正己基氯硅烷不消耗任何未反应的HO-BsubPc。将粗产物通过硅胶柱色谱纯化,首先使用100%的己烷洗脱未反应的三-正己基氯硅烷和其他硅烷衍生物,然后通过己烷中20%的THF溶液(体积/体积)梯度洗脱亮粉色固体的目标化合物(产率=49%),然后旋转蒸发。1H NMR(400MHz,CDCl3)δ8.87-8.81(m,6H),7.92-7.85(m,6H),1.14-1.04(m,6H),0.96-0.89(m,6H),0.89-0.81(m,6H),0.78(t,J=7.3Hz,9H),0.51-0.42(m,6H),-0.38--0.45(m,6H);13C NMR(100MHz,CDCl3)δ151.0,131.2,129.6,122.2,33.2,31.6,22.9,22.7,14.5,14.3;HRMS(EI)[M]计算为694.3987,得到694.3990。
双(羟基)-硅酞菁((HO)2-SiPc)(图3):在氩气下,向经烘箱干燥的三颈圆底烧瓶添加Cl2-SiPc(2.5g,4.09毫摩尔,1当量)、1,2-二氯苯(25mL)和氢氧化铯(1.50g、10.0毫摩尔、2.5当量)。将反应混合物加热至120℃下4小时。将粗产物在甲醇中沉淀、过滤以得到暗蓝色粉末(粗产率=54%),其未经进一步纯化即使用。LRMS(EI)计算为574.62,得到574.1。
双(羟基)-硅酞菁((HO)2-SiPc)(图3):使用如(HO)2-SiPc同样的方法合成(HO)2-GePc。将粗产物在甲醇中沉淀、过滤以得到深蓝色粉末(粗产率=78%),其未经进一步纯化即使用。因为出现标题化合物的破碎,不能获得质谱数据。
双(三-正己基硅氧基)-硅酞菁((3HS)2-SiPc)(图3):通过改进专利文献(Gessner,等人,美国专利公布第2010/0113767号)由(HO)2-SiPc合成(3HS)2-SiPc。在氩气下,向经烘箱干燥的三颈圆底烧瓶中添加(HO)2-SiPc(1.00g、1.61毫摩尔、1当量)、吡啶(100mL)、三-正己基氯硅烷(4.85g、17.1毫摩尔,每个反应位点5当量)。将反应混合物加热至130℃下5小时,然后将其冷却至室温。将粗产物在水中沉淀、过滤、用水(3次)洗净、在真空烘箱中干燥以得到深蓝色固体(在柱层析前产率=79%,通过1H NMR确定纯度>90%)。将粗产物通过硅胶柱色谱纯化,首先使用100%的己烷洗脱未反应的三-正己基氯硅烷和其他硅烷衍生物,然后通过50%THF的己烷溶液(体积/体积)梯度洗脱为深蓝色固体的目标化合物(产率=47%),然后旋转蒸发。(3HS)2-SiPc单晶通过由热吡啶溶液缓慢冷却而生长。1H NMR(400MHz,CDCl3)δ9.66-9.60(m,8H),8.33-8.28(m,8H),0.87-0.79(m,12H),0.74-0.67(t,J=7.2Hz,18H),0.62-0.55(m,12H),0.40-0.32(m,12H),0.06--0.03(m,12H),-1.21--1.35(m,12H);13C NMR(100MHz,CDCl3)δ148.9,136.4,130.6,123.6,33.6,33.4,32.8,31.8,31.7,31.1,23.4,23.2,22.80,22.75,22.6,21.5,16.0,15.2,14.31,14.29,14.27,12.9;LRMS(EI)m/z[M+H]+计算为1139.78,得到1139.7。
双(三-正己基硅氧基)-锗酞菁((3HS)2-GePc)(图3):以(3HS)2-SiPc的相同方法,由(OH)2-GePc(OH)2合成(3HS)2-GePc。得到的标题化合物为深蓝色固体(产率=49%)。(3HS)2-GePc的单晶从DCM/己烷中通过缓慢冷却而生长。1H NMR(400MHz,CDCl3)δ9.66-9.61(m,8H),8.34-8.29(m,8H),0.86-0.78(m,12H),0.73-0.68(m,18H),0.62-0.55(m,12H),0.41-0.32(m,12H),0.06--0.02(m,12H),-1.07--1.17(m,12H);13C NMR(100MHz,CDCl3)δ149.6,135.9,131.5,124.1,33.4,32.7,31.7,31.1,29.92,29.87,23.2,22.8,22.5,21.7,15.2,14.3,14.3,13.4。因为出现标题化合物的破碎,不能获得质谱数据。
双(3-十五烷基苯氧基)-硅酞菁((PDP)2-SiPc)(图3):以改进自Brisson等人,2011的方法,在氩气下,将Cl2-SiPc(0.5g、0.82毫摩尔、1当量)和3-十五烷基苯酚(0.60g、1.97毫摩尔、2.4当量)添加至氯苯(30mL),加热至120℃下22小时。一经冷却至室温,通过旋转蒸发将反应混合物浓缩至干燥。通过使用配有高真空泵的旋转蒸发仪蒸馏在200℃下去除一些3-十五烷基苯酚。产生的深蓝色固体在硅胶柱上使用DCM作为洗脱剂来洗脱。收集首先洗脱的蓝色带状物,浓缩,在真空烘箱中干燥以得到深蓝色固体(产率=38%)。(PDP)2-SiPc的单晶在二氯甲烷溶液中通过缓慢蒸发生长。1H NMR(400MHz,CDCl3)9.68-9.61(m,8H),8.38-8.30(m,8H),5.55-5.51(m,2H),5.51-5.46(t,J=7.5Hz,2H),2.34-2.29(m,2H),2.26-2.23(m,2H),1.39-1.26(m,40H),1.25-1.16(m,4H),1.14-1.04(m,4H),0.96-0.90(m,6H),0.85-0.75(m,4H),0.61-0.50(m,4H);13C NMR(100MHz,CDCl3)δ149.8,149.3,142.1,135.9,131.1,126.6,123.9,119.5,117.5,114.8,34.8,32.1,30.5,30.0,29.94,29.88,29.8,29.6,29.5,29.3,22.9,14.3;HRMS(DART)m/z[M+H]+计算为1147.61,得到1147.6。
P3HT:PC61BM OPV装置:通过将P3HT、PC61BM和染料在1,2-二氯苯(40mg/mL的溶液)中溶解,并使其在50℃下被搅拌2小时至3小时以确保固体的完全溶解来制备含有P3HT:PC61BM:Dye的装置。将涂覆氧化铟锡(ITO)的玻璃衬底(Colorado Concept Coatings LLC)用含水清洁剂擦拭,然后在含水清洁剂、去离子水、丙酮、和甲醇中各自超声5分钟,然后用氧等离子体处理15分钟。然后将聚(3,4-亚乙基二氧噻吩):聚(苯乙烯磺酸)(PEDOT:PSS)的薄层以3000rpm在ITO玻璃上旋涂30秒,在加热板上、在140℃空气中干燥15分钟。在转涂活性层之前,将P3HT、PC61BM和染料溶液一起搅拌15分钟。然后在氮气下、以600rpm将三元混合物旋涂到经PEDOT:PSS涂覆的衬底上,使其在室温下干燥20小时。在这些设备上没有进行退火。然后在0.7×10-6托至2.0×10-6托下使用Angstrom Engineering(Kitchener,ON)Covap II金属蒸发系统、通过热蒸发用氟化锂(LiF,0.8nm)、铝(Al,100nm)涂覆衬底。通过荫罩限定的器件面积是0.07cm2,在具有100/mWcm的功率密度的模拟AM1.5G条件下使用Keithley 2400源表得到IV曲线。使用硅二极管和KG-5滤波器校准类似光谱的不匹配。使用300W氙灯和Oriel Cornerstone 260 1/4m单色仪记录EQE测量,并与起源于国家标准与技术研究所的硅参考器件对比。
实施例2(结果)
基线P3HT:PC61BM BHJ装置:一系列使用结构ITO/PEDOT:PSS/Active Layer/LiF/Al的基线BHJ器件在整个研究中重复地制备以及不断地分析和比较,其中活性层是P3HT:PC61BM的1.0:0.8混合物。产生的BHJ OPV器件确定具有JSC=8.15±0.78mA/cm-2,VOC=0.62±0.02V,FF=0.55±0.03和ηeff=2.75±0.21(至少40个设备的平均值)。带有误差条的代表性的IV曲线和平均P3HT:PC61BM器件的EQE图如图4A和图4B所示。图4A是EQE%相对波长的特征性外量子效率(EQE)曲线。图4B是P3HT:PC61BM(1.0:0.8,质量比)和P3HT:PC61BM:X(1.0:0.8:Y)的IV曲线(以mA/cm2计的电流相对以伏特计的偏压),其中X=双(三-正己基甲硅烷基氧化物)硅酞菁((3HS)2-SiPc,1)、三-正己基甲硅烷基氧化硼亚酞菁(3HS-BsubPc)和双(三-正己基甲硅烷基氧化物)锗酞菁((3HS)2-GePc),其中Y=0.2(10.6重量%,空心方块,数据线402)、0.1(5.3重量%,实心圆,数据线404)或0.07(3.7重量%,空心三角形,数据线406)。标准的P3HT:PC61BM BHJ太阳能器件数据(无三元添加剂,数据线408)和误差条一起显示以说明标准器件占据的空间。
使用(3HS)2-SiPc的P3HT:PC61BM级联OPV:然后将(3HS)2-SiPc作为P3HT:PC61BM BHJ OPV器件中的添加剂来评估,所述器件具有和基线P3HT:PC61BM OPV器件相同的器件结构。特征性的IV曲线和外量子效率(EQE)曲线在图4A和图4B中示出,可以在表1中找到所有重复的统计。可以观察到归因于EQE中SiPc发色团的≈700nm的峰、JSC增加7%至25%和ηeff增加15%至20%(图4,表1)。添加多如10重量%的(3HS)2-SiPc,观察到FF降低,其导致ηeff略微下降(图4)。使用大致5重量%的(3HS)2-SiPc的AFM和接触角研究确定40%的SiPc分子位于界面。
在所有实验情况下,该值是最少4至5个器件的平均值,在实施例A、B和C的情况下,该值是在每个器件中具有4至5个栅格的最少2至3个器件的平均值,该器件是相同仪器件装配了8至12周。在AM1.5G照射下以100mW/cm-2下得到值。
使用酞菁变体的P3HT:PC61BM级联OPV:将双(三-正己基甲硅烷基氧化物)锗酞菁((3HS)2-GePc)引入(3HS)2-GePc得到相同的P3HT:PC61BM BHJ OPV器件。观察到对(3HS)2-SiPc发现的相反作用。低如3.7重量%的(3HS)2-GePc导致EQE穿过光谱的显著下降以及FF、JSC和ηeff的降低(图4A和图4B,表1)。对(3HS)2-SiPc和(3HS)2-GePc都实施UV-Vis吸收光谱和电化学分析。其各自的循环伏安如图5(A)和图5(B)所示,其计算的HOMO和LUMO能级和吸收最大值在图3和表2中概述。观察到(3HS)2-GePc的双重可逆氧化峰和还原峰,对(3HS)2-SiPc未观察到该发现,这大概是由于(3HS)2-GePc中独特的Si-O-Ge-O-Si序列。相比(3HS)2-SiPc,(3HS)2-GePc具有显著不同的HOMO和LUMO能级(图3)。HOMO和LUMO能级的不同导致与P3HT的跨立的构型,因此可以假定在P3HT:PC61BM OPV中(3HS)2-GePc起电荷捕获的作用,而不是促进级联电子转移作用;主张当(3HS)2-GePc存在时,观察到的显著降低的EQE谱与整体OPV器件的性能相一致。(3HS)-BsubPc和(PDP)2-SiPc的循环伏安如图5(C)和图5(D)示出。
表2(3-正己基甲硅烷氧基硼亚酞菁(3HS-BsubPc)的电化学表征)
1EHOMO=(EOX,1/2)-(4.27)eV(缩放至二茂铁癸烷的内标物)
2ELUMO,电子/ELUMO,Opt,其中ELUMO,电子=(ERed,1/2)-(4.27)eV,ELUMO,Opt=EGap,Opt-EHOMO
3化合物各自的最大吸收λMAX,其中λMAX,溶液/λMAX,膜
4由溶液吸收光谱的起始确定的EGap,Opt。
合成类似于(3HS)2-SiPc的硼亚酞菁(BsubPc)(即,三-正己基甲硅烷基氧化硼亚酞菁)(3HS-BsubPc),并用以上基线P3HT:PC61BM BHJ器件测试。其在普通有机溶剂中特别地可溶。实施3HS-BsubPc的光学及电化学表征,结果在图3、图6、和表2中概述并制表。在所测量的CV行为和经计算的HOMO和LUMO能级的基础上,类似于(3HS)2-SiPc,当与P3HT和PC61BM混合时3HS-BsubPc应该导致级联BHJ(图1)。与(3HS)2-SiPc不同,3HS-BsubPc发色团的吸收在≈545nm,因此由低负载3HS-BsubPc生成的任何光电荷与通过P3HT和PC61BM组合生成的电荷不能区分(图4A)。在增加的负载或当从混合物除去PC61BM并只是用其他的BsubPc衍生物和P3HT制备器件时,观察到来自BsubPc发色团的光生。三个不同质量负载的3HS-BsubPc的添加没有得到BHJ OPV1器件的经测量的JSC或VOC的统计学差异(图4B,表1)。尽管前沿轨道排列是有利的,添加3HS-BsubPc时JSC却没有增长表示P3HT和PC61BM之间通过3HS-BsubPc的媒介作用没有显著的级联效应。在此基础上,结论是3HS-BsubPc分子的重要部分溶于P3HT层或PC61BM层,而不存在于P3HT:PC61BM界面,从而不利于所期望的电子转移现象。
还将以下三个其他的BsubPc衍生物并入以上基线P3HT:PC61BM BHJ器件:3-十五烷基-苯氧基-BsubPc(Brisson等人,2011)(PDP-BsubPc,图3)、3-甲基-苯氧基-BsubPc(Paton等人,2012)(3MP-BsubP,图3)和五氟代苯氧基-BsubPc(Morse等人,2010;Helander等人,ACS Applied Materials&Interfaces,2010,2:3147–3152,2010)(F5-BsubPc,图3)。这三种BsubPc具有与3HS-BsubPc相似的能级,因此满足了促进P3HT和PC61BM之间的级联电子转移的能量标准(图4)。相比3HS-BsubPc,每种具有或相似或不同的物理特性。例如,PDP-BsubPc也高度可溶,十五烷基苯氧片段具有和三己基甲硅烷片段相似的碳原子数。3MP-BsubPc是异常可溶的,且其苯氧基片段具有低碳数的BsubPc的结晶版本(Paton等人,2012)。最后,F5-BsubPc也是相对可溶和晶状的,但具有与其他BsubPc明显不同的HOMO和LUMO能级(Helander等人,2010)(图3)。表1提供了在三元BHJ OPV器件中的这些添加剂的数据。
从PDP-BsubPc开始,在图6A和图4B分别示出P3HT:PC61BM:PDP-BsubPc三元BHJ OPV器件的特征性外量子效率(EQE)和IV曲线和图。当添加3.7重量%的PDP-BsubPc,相比基线P3HT:PC61BM BHJ OPV器件,观察到JSC统计学非显著增加,VOC没有变化,但FF统计学显著改善(从56%到61%)。增加PDP-BsubPc的量使得器件特征的统计学显著减少(图6B,表1)。然而,添加3MP-BsubPc或F5-BsubPc使得JSC的可忽略增加以及VOC和FF中的显著下降(图6B,表1)。
图6C和图6D各自示出向P3HT:PC61BM BHJ OPV器件添加5.3重量%的3MP-BsubPc、F5-BsubPc和PDP-BsubPc,以及在相同的P3HT:PC61BM BHJ OPV器件中使用2.7重量%的3MP-BsubPc和2.7重量%的F5-BsubPc的混合物之间的比较。以P3HT、PC61BM、3MP-BsubPc和F5-BsubPc之间的能量差异会产生具有较低能垒的电子转移活动的有利级联(图3)和增加引出电流的想法,测试了3MP-BsubPc和F5-BsubPc的组合。与单独使用5.3重量%的3MP-BsubPc或5.3重量%的F5-BsubPc相比(表1,图6B),使用2.7重量%的3MP-BsubPc和2.7重量%的F5-BsubPc的组合,P3HT:PC61BM OPV确实显示出JSC统计学的显著增加,但与P3HT:PC61BM基线器件相比,JSC仍然减少。当使用该混合物时,相比单独使用3MP-BsubPc或F5-BsubPc(图6B),测量的VOC不变,观察到EQE谱中610nm处更明显的峰。然而,混合物的FF显著地降低至37%,这在当仅使用3MP-BsubPc或F5-BsubPc时分别得到的47%和33%的FF之间(图6C和图6D)。
这些结果的总和表明,虽然BsubPc发色团具有正确的前沿轨道能量以产生P3HT与PC61BM相之间的级联电子转移,但这些化合物都没有起如3HS-SiPc的作用。结论是,更多需要考虑的是,其并不像具有或高溶解度或低溶解度(以PDP-BsubPc vs F5-BsubPc为例)以及适当前沿轨道能级的发色团那样简单。
在用(3HS)2-SiPc工作期间,观察到其具有高溶解度和强烈结晶趋势的独特组合。通过使粗(3HS)2-SiPc留在热吡啶溶液中并使其冷却至室温过夜,来无意地生长(3HS)2-SiPc的大单晶。使用X射线衍射在低温下分析单晶(CCDC保藏号988974),以非常高的精度确定(3HS)2-SiPc的分子结构和固态排列。产生的热椭球图示于图7A,同时图7B和图7C表示了多重(PDP)2-SiPc分子的晶体结构排列。尽管具有通常会假定为与三己基甲硅烷基有关的高自由度,仍然观察到(3HS)2-SiPc的晶体在己基链中并非是无序的。此外,三己基甲硅烷基的碳原子位置是充分固定的(由紧密的热椭球表示),再次表明晶体内缺乏无序。关于固态排列,(3HS)2-SiPc以良好有序的三维基质排列,其中所有SiPc发色团都指向相同方向,通过相互堆叠的三己基甲硅烷基分离(图5B和图5C)。
为了确定三己基甲硅烷基对BHJ OPV器件中(3HS)2-SiPc的功能及其结晶倾向是否至关重要,合成了替代的SiPc衍生物:双(3-十五烷基苯氧基)-硅酞菁((PDP)2-SiPc)。选择(PDP)2-SiPc作为目标是因为其是(或期望是)高度可溶的SiPc衍生物(Brisson等人,2011)。此外,3-十五烷基苯氧基分子片段具有如三己基甲硅烷基的氧化基团的相似碳数(21个与18个碳原子)。对(PDP)2-SiPc实施电化学和光谱表征,结果列于表2中。HOMO和LUMO能级(计算值)分别与(3HS)2-SiPc相似,因此应该也促进P3HT和PC61BM之间的级联电子转移。因此,使用3.7重量%、5重量%和10重量%的负载,将(PDP)2-SiPc引入到一系列的P3HT:PC61BM BHJ OPV器件。BHJ OPV器件的EQE谱和IV曲线以及相应特征分别在图8A和图8B中示出,且列在表1中。与基线P3HT:PC61BM OPV相比,在低的(PDP)2-SiPc负载(3.7重量%)下JSC显著增加,因此观察到ηeff增加(图8A和图8B,表1)。一旦负载增加到10重量%,观察到JSC的适度增加,然而,FF的显著下降导致ηeff下降。在低负载下这种行为与(3HS)2-SiPc类似,但相似性在高负载偏离。
尽管预期了3-十五烷基苯氧基分子片段的高自由度和(PDP)2-SiPc的高溶解性,仍发现(PDP)2-SiPc具有强烈的结晶倾向。例如,(PDP)2-SiPc的单晶可以或通过使二氯甲烷溶液在乙腈中沉淀或通过简单缓慢蒸发二氯甲烷溶液而获得。使用x射线晶体衍射通过二氯甲烷溶液缓慢蒸发生长的(PDP)2-SiPc的单晶(CCDC保藏号988976)。产生的50%椭球几率图在图9A中示出。(PDP)2-SiPc的固态排列与(3HS)2-SiPc完全不同(图9B和图9C)。例如,SiPc发色团被紧密地填充于交错的十五烷基片段之间,并与其分离(图9B和图9C)。
当在与三元P3HT:PC61BM BHJ OPV器件相似负载下比较(3HS)2-SiPc的添加和(PDP)2-SiPc的添加时,似乎(3HS)2-SiPc是更有效的添加剂(表1,图7)。例如,相比于基线P3HT:PC61BM BHJ OPV器件,添加3.7重量%的(3HS)2-SiPc导致JSC和ηeff分别增加16%和20%,而添加3.7重量%的(PDP)2-SiPc仅导致JSC和ηeff分别增加12%和10%。这些结果表明,当存在(3HS)2-SiPc时,更显著的级联效应在P3HT和PC61BM之间发生。由于在P3HT的正己基链和(PDP)2-SiPc的十五烷基链之间的烃/烃相互作用,(PDP)2-SiPc可能具有对P3HT相更高的溶解度或亲和力,导致P3HT:PC61BM界面上(PDP)2-SiPc减少。由于正己基片段中几何学/位置的差异,这种相互作用可能对(3HS)2-SiPc是不可能的。另一个重要的发现是,3.7重量%或5.3重量%负载的(PDP)2-SiPc导致在700nm处的EQE≈30%,而的5.3重量%负载的(3HS)2-SiPc,导致在700nm处EQE≈55%(图4和8)。该发现进一步表明,相比(3HS)2-SiPc,即使当增加(PDP)2-SiPc的浓度时,较少的(PDP)2-SiPc能够迁移到P3HT:PC61BM界面和结晶以参与级联效应(图4和8)。还发现,在较高的(PDP)2-SiPc负载(10.6重量%)下,EQE谱始终表现出第二个峰≈720nm,其红移自指定为SiPc发色团的峰(≈700nm)(图7A)。由于在高负载存在一些(PDP)2-SiPc分子团聚,该第二个峰可以是吸收扩展的结果,表明形成(PDP)2-SiPc簇。
进一步支持(3HS)2-SiPc具有结晶的非同寻常倾向的想法,尽管多次尝试包括通过蒸汽扩散生长(将己烷引入DCM或将庚烷引入苯)、通过分层法的液体/液体扩散(将己烷引入DCM或将庚烷引入苯)、或从例如氯仿、二氯甲烷或甲苯的热溶剂缓慢冷却,但不能获得3HS-BsubPc的晶体用于X射线衍射分析。值得注意地,虽然比(3HS)2-SiPc更困难,但(3HS)2-GePc的单晶能够生长。使用X射线衍射再次分析产生的分子结构和固态排列(CCDC保藏号988975)。观察到,(3HS)2-GePc具有和(3HS)2-SiPc类似的固态排列。
基于以上数据和观察,(3HS)2-SiPc是P3HT:PC61BM BHJ OPV器件的独特添加剂。其不仅具有适当的前沿分子轨道能级以促进级联电子转移,而且当在沉积过程中(3HS)2-SiPc移动至P3HT:PC61BM界面时(由Honda等人,Adv.Energy Mater.2011,1:588–598确定)其结晶可以导致P3HT和PC61BM相之间更好的电荷传输。对(3HS)2-SiPc结晶的驱动力可能是其完全移动到界面的原因。相反地,3HS-BsubPc和(PDP)2-SiPc不如(3HS)2-SiPc有效的原因是其不容易结晶,或换言之对其存在结晶的有限驱动力,因此显著的部分保持在P3HT和PC61BMC之间的界面外。这些发现代表了为何(3HS)2-SiPc在协助P3HT:PC61BM BHJ OPV器件中是如此好的染料的可能解释,选择三-正己基硅氧基取代基对其性能至关重要。三-正己基硅氧基取代基提供了必要的溶解性质,同时为硅酞菁在固态时结晶提供了方便,导致P3HT和PC61BM之间有利的分散和电荷转移。
- 还没有人留言评论。精彩留言会获得点赞!