针对Rb阳性异常细胞增殖的HSPC节制性治疗的制作方法
本申请涉及并要求2013年3月15日提交的美国临时申请No.61/798,772、2013年8月1日提交的美国临时申请No.61/861,374、2013年12月3日提交的美国临时申请No.61/911,354和2014年3月7日提出的美国临时申请No.61/949,786的权益。这些申请每一者整体以引用的方式并入本文中以达成所有目的。
政府利益
变态反应与传染病国家研究院(the National Institutes of Allergy and Infectious Disease)所颁予的资助5R44AI084284的支持,故美国政府在本发明中具有权利。
技术领域
本发明涉及改善的化合物和方法,其治疗所选的RB阳性癌症和其它Rb阳性异常细胞增殖病症,同时将与当前治疗模态有关的对例如健康造血干细胞和祖细胞(HSPC)等健康细胞的有害作用减到最少。在一个方面,公开了使用本文公开的特定化合物的改善的对所选RB阳性癌症的治疗。当施用至受试者时,在某些实施方案中,本文中描述的化合物充当高度选择性的,且在某些实施方案中,短暂、瞬时起作用的细胞周期蛋白依赖性激酶4/6(CDK 4/6)抑制剂。
发明背景
细胞周期的调节由特定蛋白质管理和控制,所述蛋白质主要通过磷酸化/脱磷酸化过程以准确定时的方式活化和失活。协调细胞周期程序的开始、进展和完成的关键蛋白质是细胞周期蛋白依赖性激酶(CDK)。细胞周期蛋白依赖性激酶属于丝氨酸-苏氨酸蛋白激酶家族。它们是由催化激酶亚单元和调节细胞周期蛋白亚单元构成的异二聚体复合物。CDK活性由与其对应的调节亚单元(细胞周期蛋白)和CDK抑制剂蛋白质(Cip和Kip蛋白质、INK4s)的缔合、由其磷酸化状态和由泛素介导的蛋白水解降解来控制(参见D.G.Johnson,C.L.Walker,Annu.Rev.Pharmacol.Toxicol 39(1999)295–312;D.O.Morgan,Annu.Rev.Cell Dev.Biol.13(1997)261–291;C.J.Sherr,Science 274(1996)1672–1677;T.Shimamura等人,Bioorg.Med.Chem.Lett.16(2006)3751–3754)。
存在四种显著参与细胞增殖的CDK:CDK1,其主要调节从G2期转变至M期;以及CDK2、CDK4和CDK6,其调节从G1期转变至S期(Malumbres M,Barbacid M.Cell cycle,CDKs and cancer:a changing paradigm.Nat.Rev.Cancer 2009;9(3):153-166)。在G1期初期至中期,当细胞对促有丝分裂刺激物起反应时,CDK4细胞周期蛋白D和CDK6细胞周期蛋白D的活化诱发成视网膜细胞瘤蛋白(pRb)的磷酸化。pRb的磷酸化释放转录因子E2F,其进入核中,活化促进细胞周期的进一步进展的其它细胞周期蛋白的转录(参见J.A.Diehl,Cancer Biol.Ther.1(2002)226–231;C.J.Sherr,Cell 73(1993)1059-1065)。CDK4和CDK6是具有基本上不能区别的生物化学特性的密切相关的蛋白质(参见M.Malumbres,M.Barbacid,Trends Biochem.Sci.30(2005)630–641)。
已鉴别大量CDK 4/6抑制剂,包括特定吡啶并[2,3-d]嘧啶、2-苯胺基嘧啶、二芳基脲、苯甲酰基-2,4-二氨基噻唑、吲哚并[6,7-a]吡咯并[3,4-c]咔唑和羟吲哚(参见P.S.Sharma,R.Sharma,R.Tyagi,Curr.Cancer Drug Targets 8(2008)53-75)。例如,WO 03/062236鉴别用于治疗展示对CDK4/6的选择性的Rb阳性癌症的一系列2-(吡啶-2-基氨基-吡啶并[2,3]嘧啶-7-酮,包括6-乙酰基-8-环戊基-5-甲基-2-(5-哌嗪-1-基-吡啶-2-基氨基)-8H-吡啶并-[2,3-d]-嘧啶-7-酮(PD0332991),此化合物目前正由Pfizer在大型临床试验中作为一种针对雌激素阳性、HER2阴性的乳癌的抗赘生剂进行测试。VanderWel等人描述了含碘的吡啶并[2,3-d]嘧啶-7-酮(CKIA)作为有效和选择性的CDK4抑制剂(参见VanderWel等人,J.Med.Chem.48(2005)2371-2387)。Glaxo Group Ltd提交的WO 99/15500公开了蛋白激酶和丝氨酸/苏氨酸激酶抑制剂。Novartis AG提交的WO 2010/020675描述了吡咯并嘧啶化合物作为CDK抑制剂。Novartis还提出的WO 2011/101409描述了具有CDK 4/6抑制活性的吡咯并嘧啶。Novartis提交的WO 2005/052147和Janssen Pharma提交的WO 2006/074985公开了额外CDK4抑制剂。Tavares提交并受让于G1 Thapeutics的WO 2012/061156描述了CDK抑制剂。Francis Tavares提交并转让给G1 Thapeutics的WO 2013/148748描述了内酰胺激酶抑制剂。
虽然选择性CDK4/6抑制剂一般设计成能靶向CDK4/6复制依赖性癌症,但它们抑制CDK4/6活性的确切事实也可能引起对CDK4/6依赖性健康细胞的有害作用,例如其生长抑制。CDK4/6活性是骨髓产生健康血细胞所必需的,因为健康的造血干细胞和祖细胞(HSPC)需要CDK4/6的活性用于增殖(参见Roberts等人Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy.JNCI 2012;104(6):476-487)。健康的造血干细胞产生祖细胞,祖细胞反过来产生血液的所有分化组分,如图1中所示(例如淋巴细胞、红血球、血小板、粒细胞、单核细胞)。在脊髓/红细胞分化期间健康的造血细胞显示对CDK4/6活性的渐进依赖性用于增殖(参见Johnson等人Mitigation of hematological radiation toxicity in mice through pharmacological quiescence induced by CDK4/6inhibition.J Clin.Invest.2010;120(7):2528-2536)。因此,最小分化的细胞(例如健康的造血干细胞(HSC)、多潜能祖细胞(MPP)和共同脊髓祖细胞(CMP))似乎最依赖于CDK4/6活性用于增殖,因此最受CDK4/6抑制剂用于治疗CDK4/6复制依赖性癌症或其它增生性病症不良影响。
因此,正需要改善的化合物、方法和方案治疗所选Rb阳性癌症和异常细胞增殖病症的患者,同时将治疗对例如HSPC等健康细胞的作用减到最少。
发明概要
提供改善的化合物、方法和组合物,通过施用有效量的本文中描述的化合物,治疗所选Rb阳性异常细胞增殖,包括Rb阳性癌症,同时将治疗对例如健康HSPC和其它CDK4/6复制依赖性健康细胞等健康细胞的有害作用减到最少。
在本发明的一个实施方案中,化合物选自如本文中描述的式I、II、III、IV或V的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在一个非限制性实例中,化合物可以选自下表1的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药。
在一个实施方案中,Rb阳性癌症可以是Rb阳性腺癌。Rb阳性癌症可以是结肠的Rb阳性腺癌。Rb阳性癌症也可以是直肠的Rb阳性腺癌。
或者,Rb阳性癌症可以是Rb阳性间变性星形细胞瘤。
Rb阳性癌症可以是Rb阳性乳癌。在一个实施方案中,Rb阳性癌症是Rb阳性雌激素受体阳性、HER2阴性晚期乳癌。或者,Rb阳性癌症可以是Rb阳性雌激素受体阴性乳癌。Rb阳性癌症可以是Rb阳性雌激素受体阳性乳癌。Rb阳性癌症可以是Rb阳性晚期转移性乳癌。Rb阳性癌症可以是Rb阳性管状A型乳癌。Rb阳性癌症可以是Rb阳性管状B型乳癌。Rb阳性癌症可以是Rb阳性Her2阴性乳癌或Rb阳性HER2阳性乳癌。Rb阳性癌症是Rb阳性男性乳癌。在一个实施方案中,Rb阳性癌症是Rb阳性孕激素受体阴性乳癌。Rb阳性癌症可以是Rb阳性孕激素受体阳性乳癌。Rb阳性癌症可以是Rb阳性复发性乳癌。在一个实施方案中,Rb阳性癌症是Rb阳性IV期乳癌。在一个实施方案中,Rb阳性癌症是Rb阳性晚期HER2阳性乳癌。
Rb阳性癌症可以是Rb阳性支气管癌。Rb阳性癌症可以是Rb阳性结肠癌。Rb阳性癌症可以是Rb阳性复发性结肠癌。Rb阳性癌症可以是Rb阳性IV期结肠癌。在一个实施方案中,Rb阳性癌症是Rb阳性结肠直肠癌。
在一个实施方案中,Rb阳性癌症是Rb阳性子宫内膜癌。
Rb阳性癌症可以是Rb阳性性腺外精原细胞瘤。Rb阳性癌症可以是Rb阳性III期性腺外精原细胞瘤。Rb阳性癌症可以是Rb阳性IV期性腺外精原细胞瘤。
Rb阳性癌症可以是Rb阳性生殖细胞癌。Rb阳性癌症可以是Rb阳性中枢神经系统生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性家族性睾丸生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性复发性性腺生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性复发性性腺外非精原细胞瘤性生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性性腺外精原细胞瘤性生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性复发性恶性睾丸生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性复发性卵巢生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性III期恶性睾丸生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性III期卵巢生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性IV期卵巢生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性III期性腺外非精原细胞瘤性生殖细胞肿瘤。Rb阳性癌症可以是Rb阳性IV期性腺外非精原细胞瘤性生殖细胞肿瘤。在一个实施方案中,Rb阳性癌症是Rb阳性生殖细胞癌。在一个实施方案中,Rb阳性癌症是Rb阳性的顺铂难治的不可切除的生殖细胞癌。
在一个实施方案中,Rb阳性癌症是Rb阳性成胶质细胞瘤。
在一个实施方案中,Rb阳性癌症是Rb阳性肝癌。Rb阳性癌症可以是Rb阳性肝细胞癌。
Rb阳性癌症可以是Rb阳性肺癌。在一个实施方案中,Rb阳性癌症是Rb阳性非小细胞肺癌。在一个实施方案中,Rb阳性癌症是Rb阳性KRAS突变体非小细胞肺癌。
Rb阳性癌症可以是Rb阳性黑色素瘤。在一个实施方案中,Rb阳性癌症是Rb阳性复发性黑色素瘤。在一个实施方案中,Rb阳性癌症是Rb阳性IV期黑色素瘤。
Rb阳性癌症可以是Rb阳性卵巢癌。在一个实施方案中,Rb阳性癌症是Rb阳性卵巢上皮癌。
Rb阳性癌症可以是Rb阳性胰腺癌。
Rb阳性癌症可以是Rb阳性前列腺癌。
在一个实施方案中,Rb阳性癌症是Rb阳性直肠癌。Rb阳性癌症可以是Rb阳性复发性直肠癌。Rb阳性癌症可以是Rb阳性IV期直肠癌。
Rb阳性癌症可以是Rb阳性肉瘤。Rb阳性癌症可以是Rb阳性胶质肉瘤。Rb阳性癌症可以是Rb阳性脂肪肉瘤。Rb阳性癌症可以是Rb阳性纤维肉瘤。Rb阳性癌症可以是Rb阳性粘液肉瘤。在一个实施方案中,Rb阳性癌症可以是Rb阳性软骨肉瘤。Rb阳性癌症可以是Rb阳性骨肉瘤。
Rb阳性癌症可以是Rb阳性恶性纤维组织细胞瘤。Rb阳性癌症可以是Rb阳性血管肉瘤。Rb阳性癌症可以是Rb阳性血管肉瘤。Rb阳性癌症可以是Rb阳性淋巴管肉瘤。Rb阳性癌症可以是Rb阳性间皮瘤。Rb阳性癌症可以是Rb阳性平滑肌肉瘤。Rb阳性癌症可以是Rb阳性横纹肌肉瘤。Rb阳性癌症可以是Rb阳性脑膜瘤。Rb阳性癌症可以是Rb阳性神经鞘瘤。
在一个实施方案中,Rb阳性癌症是Rb阳性嗜铬细胞瘤。Rb阳性癌症可以是Rb阳性胰岛细胞癌。Rb阳性癌症可以是Rb阳性类癌瘤。Rb阳性癌症可以是Rb阳性副神经节瘤。
在一个实施方案中,Rb阳性癌症是Rb阳性鳞状细胞癌。Rb阳性癌症可以是Rb阳性腺癌。Rb阳性癌症可以是Rb阳性肝细胞癌。Rb阳性癌症可以是Rb阳性肾细胞癌瘤。Rb阳性癌症可以是Rb阳性肝胆管型肝癌。
Rb阳性癌症可以是Rb阳性难治性实体肿瘤。
Rb阳性癌症可以是Rb阳性神经母细胞瘤。
Rb阳性癌症可以是Rb阳性成神经管细胞瘤。
在一个实施方案中,Rb阳性癌症是畸胎瘤。Rb阳性癌症可以是Rb阳性卵巢未成熟畸胎瘤。Rb阳性癌症可以是Rb阳性卵巢成熟畸胎瘤。Rb阳性癌症可以是Rb阳性卵巢专化畸胎瘤。Rb阳性癌症可以是Rb阳性睾丸未成熟畸胎瘤。Rb阳性癌症可以是Rb阳性睾丸成熟畸胎瘤。Rb阳性癌症可以是Rb阳性畸胎瘤。Rb阳性癌症可以是Rb阳性卵巢单胚层畸胎瘤。
Rb阳性癌症可以是Rb阳性睾丸癌。
在一个实施方案中,Rb阳性癌症是Rb阳性阴道癌。
在一个实施方案中,Rb阳性癌症选自Rb阳性癌瘤、肉瘤,包括(但不限于)肺癌、骨癌、胰腺癌、皮肤癌、头或颈癌、皮肤或眼内黑色素瘤、子宫癌、卵巢癌、直肠癌、肛门部癌、胃癌、结肠癌、乳癌、子宫癌、输卵管癌、子宫内膜癌、宫颈癌、阴道癌、外阴癌、食管癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、前列腺癌、膀胱癌、肾或输尿管癌、肾细胞癌、肾盂癌、中枢神经系统(CNS)的赘生物、原发性CNS淋巴瘤、脊柱肿瘤、脑干神经胶质瘤、垂体腺瘤或上述癌症中一者或多者的组合。
在一个实施方案中,受试者罹患Rb阳性异常细胞增殖病症。在一个实施方案中,Rb阳性异常细胞增殖病症是非癌性的。
在某些实施方案中,本文中描述的化合物当用于治疗例如癌症等所选Rb阳性细胞增殖病症时,允许健康细胞快速再进入正常细胞周期且快速复原破坏的组织和子代细胞,例如血液细胞。在此方面,本文中描述的化合物当用于治疗Rb阳性癌症时将与CDK4/6抑制剂的当前抗赘生性使用有关的药物假期和给药延迟消除、减少和/或减到最少,从而允许破坏的血细胞通过祖细胞和亲本细胞的复制和分化而快速恢复。确切地说,本发明包括向例如Rb阳性癌症等癌症的患者施用有效量的本文中描述的化合物,其中所述化合物具有提供CDK4/6复制依赖性细胞的瞬时可逆的G1停滞的药物动力学和酶半衰期。化合物可以是本申请中描述的那些化合物中的任一者。活性化合物的非限制性实例描述于表1中,或如以下提供的其药学上可接受的组合物、盐、同位素类似物或前药。
在一个实施方案中,本文中描述的化合物可以适用于改善的治疗例如Rb阳性癌症等癌症的方法,其中此类方法具有减少或减到最少的对CDK4/6复制依赖性健康细胞的作用,部分因为其(i)利用显示对CDK4/6复制依赖性健康细胞提供短暂、瞬时和可逆的G1停滞的作用的药物动力学和酶半衰期的化合物,和(ii)在施用停止或受试者中治疗有效水平消散后允许健康细胞快速再进入细胞周期。使用本文中描述的化合物允许例如HSPC因CDK4/6抑制而引起的复制延迟减少,和/或在CDK4/6抑制活性停止后造血细胞谱系恢复加速,和/或血液缺乏减少,因为所利用的化合物短暂起作用,并减少与当前的CDK4/6抑制剂治疗模态有关的非循环期或药物假期的长度,此减少肿瘤药物抗性的促进或减到最少。在某些实施方案中,本文中描述的化合物的使用允许受试者在更长时间段内连续治疗,无需非循环期或药物假期。
CDK4/6复制依赖性健康细胞增殖的及时重新开始是组织修复所必需的,且过度长期的健康细胞的细胞周期停滞,例如HSPC细胞周期停滞,是不希望的。尽管报导指示选择性CDK4/6抑制剂PD0332991是Rb阳性乳癌的有效抑制剂,但已发现此类抑制剂由于化合物的过度骨髓抑制作用而可能不是用作化学治疗剂的最理想化合物。例如,PD0332991具有相对长效的细胞内作用(参见Roberts等人.Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy.JCNI 2012;104(6):476-487(图2A)),延长了例如HSPC等健康细胞的G1停滞的瞬时性,导致剂量限制性骨髓抑制。此类长效作用延迟例如使生长已因可能抑制CDK4/6活性并因此抑制Rb感受态细胞中Rb磷酸化的治疗而受到抑制的血液细胞系复原所需的HSPC细胞谱系的增殖。虽然是其抗赘生作用所需的,但由PD0332991提供的长效G1停滞需要延长的非循环期以使不利地受到急性HSPC G1-停滞影响的红细胞、血小板和骨髓细胞(单核细胞和粒细胞)复原,从而限制骨髓抑制并允许血液复制时期。使用本文中描述的化合物作为抗赘生剂治疗所选Rb阳性癌症可以将非循环期或药物假期的所需长度消除、减少或减到最少,允许在抗赘生性方案的过程中更长的对癌症的有效CDK4/6抑制时间段。
因此在一个实施方案中,本发明在治疗方案中包括向罹患Rb阳性癌症的主体施用有效量的本文中描述的化合物,包括选自表1的一者,其中(单独或其任何组合,每一者视为特定和独立地描述):i)相当大部分的CDK4/6复制依赖性健康细胞(例如至少80%或更大),例如HSPC在离人类中本文中描述的化合物的上次施用不到24小时、30小时或36小时内恢复至或接近治疗前基线细胞周期活性(即再进入细胞周期);ii)相当大部分的CDK4/6复制依赖性健康细胞,例如HSPC在离本文中描述的化合物上次施用不到24小时、30小时或36小时内同步再进入细胞周期;(iii)化合物对CDK4/6复制依赖性健康细胞,例如HSPC的抑制作用的消散在离化合物的施用不到24小时、30小时或36小时内发生;(iv)相当大部分的CDK4/6复制依赖性健康细胞,例如HSPC在离化合物的CDK4/6抑制作用的消散不到24小时、30小时或36小时内恢复至或接近治疗前基线细胞周期活性(即再进入细胞周期);或(vi)相当大部分的CDK4/6复制依赖性健康细胞,例如HSPC在离受试者血液中的施用化合物的浓度水平降到治疗有效浓度的时刻不到约24小时、约30小时或约36小时内恢复至或接近治疗前基线细胞周期活性(即再进入细胞周期)。
在本发明的重要实施方案中,本文中描述的化合物可以与另一剂例如非DNA破坏的靶向抗赘生剂或造血生长因子剂在配合方案中施用,以针对异常细胞增殖实现有益、累加或协同效应。近来已经报导了造血生长因子不合时宜的施用可能具有严重的副作用。例如,生长因子的EPO家族的使用已经与高动脉压、大脑惊厥、高血压性脑病、血栓栓塞、缺铁症、流感类综合症和静脉血栓形成有关。生长因子的G-CSF家族已经与脾扩大和破裂、呼吸窘迫综合症、变态反应和镰刀形红细胞并发症有关。通过将本文中描述的化合物和本发明的方法的施用与造血生长因子的及时施用组合,例如在患病细胞不再处于生长停滞的时间点,保健医师可降低生长因子的量以使不需要的副作用减到最少,同时实现想要的治疗益处。在一个实施方案中,生长因子在化合物对CDK4/6复制依赖性健康细胞,例如HSPC的抑制作用的作用停止后施用。因此,在此实施方案中,在抗赘生性治疗方案中使用本文中描述的选择性CDK4/6抑制剂允许受试者接收减少量的生长因子,因为靶向的造血细胞将比例如PD0332991等其它CDK4/6抑制剂时更快地再进入细胞周期。另外,在使用本文中描述的化合物进行G1停滞后同步再进入细胞周期提供了定时造血生长因子的施用以帮助再造造血细胞系,从而最大化生长因子作用的能力。因而,在一个实施方案中,本文中描述的化合物或方法的使用与造血生长因子的使用组合,所述造血生长因子包括(但不限于)粒细胞群落刺激因子(G-CSF)、粒细胞-巨噬细胞群落刺激因子(GM-CSF)、促血小板生成素、介白素(IL)-12、青灰因子和促红细胞生成素(EPO)或其衍生物。在一个实施方案中,CDK4/6抑制剂在施用造血生长因子前施用。在一个实施方案中,造血生长因子施用时间经安排,使得CDK4/6抑制剂对HSPC的作用已经消散。
在一个实施方案中,本文中描述的化合物的使用在治疗方案内与至少一种其它化学治疗剂组合,且可以是不依赖于增殖或进展通过细胞周期以达到抗增殖活性的化合物。此类剂可以包括(但不限于)他莫昔芬(tamoxifen)、咪达唑仑(midazolam)、来曲唑(letrozole)、硼替佐米(bortezomib)、阿那曲唑(anastrozole)、戈舍瑞林(goserelin)、mTOR抑制剂、PI3激酶抑制剂、双重mTOR-PI3K抑制剂、MEK抑制剂、RAS抑制剂、ALK抑制剂、HSP抑制剂(例如HSP70和HSP 90抑制剂或其组合)。mTOR抑制剂的实例包括(但不限于)雷帕霉素(rapamycin)和其类似物、依维莫司(everolimus)(Afinitor)、坦西莫司(temsirolimus)、雷达莫司(ridaforolimus)、西罗莫司(sirolimus)和德佛莫司(deforolimus)。P13激酶抑制剂的实例包括(但不限于)渥曼青霉素(Wortmannin)、脱甲绿胶酶素(demethoxyviridin)、哌立福新(perifosine)、依德西伯(idelalisib)、PX-866、IPI-145、BAY 80-6946、BEZ235、RP6503、TGR 1202(RP5264)、MLN1117(INK1117)、皮提西伯(Pictilisib)、布帕西伯(Buparlisib)、SAR245408(XL147)、SAR245409(XL765)、帕洛米德(Palomid)529、ZSTK474、PWT33597、RP6530、CUDC-907和AEZS-136。MEK抑制剂的实例包括(但不限于)曲美替尼(Tametinib)、司美替尼(Selumetinib)、MEK162、GDC-0973(XL518)和PD0325901。RAS抑制剂的实例包括(但不限于)瑞赖新(Reolysin)和siG12D LODER。ALK抑制剂的实例包括(但不限于)克唑替尼(Crizotinib)、AP26113和LDK378。HSP抑制剂包括(但不限于)格尔德霉素(Geldanamycin)或17-N-烯丙基氨基-17-脱氧基格尔德霉素(17AAG)和根赤壳菌素(Radicicol)。
在某些实施方案中,本文中描述的化合物在用另一化学治疗剂治疗前,在用另一化学治疗剂治疗期间,在施用另一化学治疗剂后或其组合施用至受试者。在一个实施方案中,本文中描述的化合物在用另一化学治疗剂治疗前不到约24小时、20小时、16小时、12小时、8小时或4小时或更短时间施用至受试者以使Rb阳性癌症对化学治疗剂敏感。在一个实施方案中,化合物在用另一化学治疗剂治疗前至多4小时施用。
在一个实施方案中,本文中描述的化合物以允许药物容易进入血流的方式,例如经由静脉内注射或舌下、大动脉内或其它有效的进入血流的途径施用。在一个实施方案中,本文中描述的化合物在可口服的制剂中施用。在其它实施方案中,本文中描述的化合物经由局部、经皮或其它想要的施用途径施用。
在一个实施方案中,本文中描述的化合物在用造血生长因子治疗前不到约24小时、20小时、16小时、12小时、8小时或4小时或更短时间施用至受试者。在一个实施方案中,化合物在用造血生长因子或其它化学治疗剂治疗前至多4小时施用。
与例如CDK2等其它CKD相比,适用于本发明的化合物展示对抑制CDK4和/或CDK6的显著选择性。例如,适用于本发明的化合物对受试者的Rb阳性癌细胞提供剂量依赖性的G1停滞作用,且本文中提供的方法足以提供RB阳性癌细胞的化学治疗剂治疗和生长抑制,同时不影响CDK4/6复制非依赖性细胞。
在一个实施方案中,本文中描述的化合物的使用引起G1停滞作用消散,使得受试者的CDK4/6复制依赖性健康细胞在不到约12小时、14小时、16小时、18小时、20小时、24小时、30小时、36小时或40小时内恢复至其施用前基线细胞周期活性。
在一个实施方案中,G1停滞作用消散使得受试者的CDK4/6复制依赖性健康细胞在施用停止不到约24小时、30小时、36小时或40小时内或约48小时内恢复至其施用前基线细胞周期活性。在一个实施方案中,CDK4/6复制依赖性健康细胞是HSPC。在一个实施方案中,本文中描述的CDK4/6抑制剂的使用引起G1停滞作用消散,使得受试者的CDK4/6复制依赖性健康细胞在离受试者血液中的CDK4/6抑制剂的浓度水平降到治疗有效浓度以下的时刻不到约24小时、30小时、36小时、40小时内或不到约48小时内恢复至或接近其施用前基线细胞循环活性。在一个实施方案中,G1停滞作用消散,使得受试者的CDK4/6复制依赖性健康细胞在离受试者血液中的CDK4/6抑制剂的浓度水平降到治疗有效浓度以下的时刻不到约24小时、30小时、36小时、40小时内或48小时内恢复至其施用前基线细胞周期活性。
在一个实施方案中,本文中描述并适用于所述方法的化合物在其终止效应上可以是同步的,那就是说,在G1停滞作用消散后,暴露于本文中描述的化合物的CDK4/6复制依赖性健康细胞以类似时间安排方式再进入细胞周期。再进入细胞周期的CDK4/6复制依赖性健康细胞这样做,使得G1和S细胞的正常比例在离受试者血液中的化合物的浓度水平降到治疗有效浓度以下的时刻不到约24小时、30小时、36小时、40小时内或约48小时内快速有效地重建。
与化合物的快速终止效应相关的快速细胞周期再进入有利地使得与例如PD0332991等其它CDK4/6抑制剂相比,更大数目的CDK4/6复制依赖性健康细胞在G1停滞消散时开始复制。因此,例如HSPC等CDK4/6复制依赖性健康细胞可以在非循环期期间或在施用期期间快速开始复制。
在靶向CDK4/6复制依赖性癌症的治疗方案中如本文中描述的化合物的使用可以引起与用目前可用的抗赘生性化学治疗剂治疗后典型预期、常见或与其有关的情况相比,贫血减少、淋巴球减少症减少、血小板减少症减少或中性白细胞减少。如本文中描述的化合物的使用可以使得在CDK4/6抑制剂使用停止后从与长期使用CDK4/6抑制剂有关的骨髓抑制,例如骨髓抑制、贫血、淋巴球减少症、血小板减少症或中性白细胞减少的恢复更快。在一些实施方案中,如本文中描述的化合物的使用使得与长期使用CDK4/6抑制剂有关的骨髓抑制,例如骨髓抑制、贫血、淋巴球减少症、白血球减少症、血小板减少症或粒性白细胞减少症,例如中性白细胞减少减少。
在一些实施方案中,受试者或主体是哺乳动物,包括人类。化合物可以通过任何想要的途径,包括静脉内、舌下、经颊、经口、大动脉内、局部、鼻内、胃肠外、经皮、全身性、肌肉内或经由吸入施用至受试者。
总之,本发明包括以下特征:
A)作为化学治疗剂的最佳化合物、方法和组合物,其将对针对所选Rb阳性癌症进行治疗的受试者中CDK4/6复制依赖性健康细胞,例如造血干细胞和祖细胞(HSPC)的有害作用减到最少,其包括施用有效量的式I、II、III、IV或V的化合物,包括如本文中描述的选自表1的化合物;
B)作为化学治疗剂的最佳化合物、方法和组合物,其将对针对Rb阳性癌症进行治疗的受试者中CDK4/6复制依赖性健康细胞,例如造血干细胞和祖细胞(HSPC)的有害作用减到最少,其包括施用有效量的本文中描述的选择性化合物,其中相当大部分的健康细胞在离CDK4/6抑制剂的上次施用不到约24小时、30小时、36小时或约40小时内恢复至或接近治疗前基线细胞周期活性(即再进入细胞周期)且其中CDK4/6抑制剂对CDK4抑制的IC50浓度比其对CDK2抑制的IC50浓度小超过约1500倍。在某些实施方案中,CDK4/6复制依赖性健康细胞是HSPC。在某些实施方案中,CDK4/6复制依赖性健康细胞是肾上皮细胞;
C)作为化学治疗剂的最佳化合物、方法和组合物,其将对针对Rb阳性癌症进行治疗的受试者中CDK4/6复制依赖性健康细胞的有害作用减到最少,其包括施用有效量的本文中描述的化合物,其中相当大部分的CDK复制依赖性健康细胞在化合物的CDK4/6抑制作用消散后不到约24小时、30小时、36小时或约40小时内同步再进入细胞周期,其中化合物对CDK4抑制的IC50浓度比其对CDK2抑制的IC50浓度小超过约1500倍。在某些实施方案中,CDK4/6复制依赖性健康细胞是HSPC。在某些实施方案中,CDK4/6复制依赖性健康细胞是肾上皮细胞。
D)作为化学治疗剂的最佳化合物、方法和组合物,其将对受试者中CDK4/6复制依赖性健康细胞的有害作用减到最少,所述方法包括向患有Rb阳性异常细胞增殖病症的受试者施用有效量的选自本文中描述的化合物的选择性CDK4/6抑制剂。在某些实施方案中,受试者的健康细胞在离受试者血液中的化合物的浓度水平降到治疗有效浓度以下的时刻不到约24小时、约30小时、约36小时或约40小时内恢复至或接近治疗前基线细胞周期活性(即再进入细胞周期)。在某些实施方案中,CDK4/6复制依赖性健康细胞是HSPC。在某些实施方案中,CDK4/6复制依赖性健康细胞是肾上皮细胞。
E)如本文中描述的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药,其用作治疗Rb阳性异常细胞增殖病症(包括Rb阳性癌症)的化学治疗剂;
F)如本文中描述的化合物,和其药学上可接受的组合物、盐、同位素类似物和前药,其用作治疗Rb阳性异常细胞增殖病症(包括Rb阳性癌症)的化学治疗剂方案,其将对CDK4/6复制依赖性健康细胞,例如HSPC或肾细胞的有害作用减至最少;
G)如本文中描述的化合物,和其药学上可接受的组合物、盐、同位素类似物和前药,其在进行治疗Rb阳性异常细胞增殖病症(包括Rb阳性癌症)的治疗方案的受试者中与造血生长因子组合使用;
H)如本文中描述的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药,其在进行治疗Rb阳性异常细胞增殖病症(包括Rb阳性癌症)的治疗方案的受试者中与第二化学治疗剂组合使用;
I)本文中描述的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药的用途,其用于制造用作治疗Rb阳性异常细胞增殖病症(包括Rb阳性癌症)的化学治疗剂的药剂;
J)本文中描述的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药的用途,其用于制造用作治疗患有当暴露于CDK4/6抑制剂时,生长停滞或生长受到抑制的Rb阳性异常细胞增殖病症(包括Rb阳性癌症)的受试者的化学治疗剂的药剂;
K)用于制备含有有效量的本文中描述的化合物的治疗产品的方法,其用于治疗患有Rb阳性异常细胞增殖病症,例如癌症的受试者,和;
L)一种制造选自本文中描述的化合物的药剂的方法,所述化合物意图用于作为治疗例如癌症等对CDK4/6抑制剂起反应的Rb阳性异常细胞增殖病症的化学治疗剂的治疗用途。
附图简述
图1是展示健康造血干细胞(HSC)和健康造血祖细胞的分层增殖和在增殖后分化增加的造血作用的示意图。
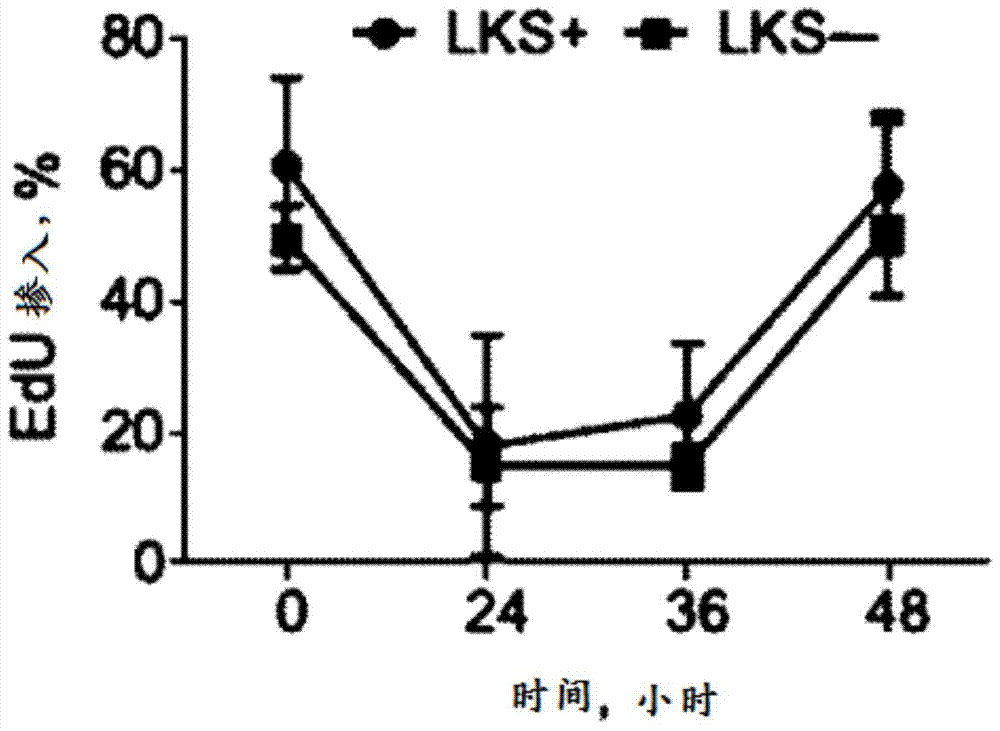
图2是EdU掺入对比PD0332991施用健康小鼠HSPC和健康脊髓祖细胞后的时间(小时)的图。通过口腔管饲法施用PD0332991(150mg/kg)以评估瞬时CDK4/6抑制对骨髓停滞的暂时作用,如Roberts等人Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy.JCNI 2012;104(6):476-487(图2A)中报导。如实施例153中所描述,单一口服剂量的PD0332991引起HSPC EdU掺入(圆形;LKS+)和脊髓祖细胞EdU掺入(正方形;LKS-)持久减少超过36小时。
图3A是血浆药物浓度(ng/ml)对比化合物T施用后时间(小时)的图。图3B是血浆药物浓度(ng/ml)对比化合物Q施用后Q(小时)的图。图3C是血浆药物浓度(ng/ml)对比化合物GG施用后时间(小时)的图。图3D是血浆药物浓度(ng/ml)对比化合物U施用后时间(小时)的图。化合物以30mg/kg通过口腔管饲法(菱形)或10mg/kg通过静脉内注射(正方形)给予小鼠。在给药后0、0.25、0.5、1.0、2.0、4.0和8.0小时取血液样品并通过HPLC测定血浆浓度。
图4A是在人类成纤维细胞(Rb阳性)细胞中细胞周期的G0-G1期中的细胞百分比对比化合物洗脱后时间(小时)的图。图4B是在人类成纤维细胞(Rb阳性)细胞中细胞周期的S期中的细胞百分比对比化合物洗脱后时间(小时)的图。图4C是在人类肾近端小管上皮(Rb阳性)细胞中细胞周期的G0-G1期中的细胞百分比对比化合物洗脱后时间(小时)的图。图4D是在人类肾近端小管上皮(Rb阳性)细胞中细胞周期的S期中的细胞百分比对比化合物洗脱后时间(小时)的图。这些细胞洗脱实验证明本发明的抑制剂化合物在不同的细胞类型中具有短暂、瞬时的G1停滞作用。在24、36、40和48小时确定洗脱化合物后对细胞周期的作用。如实施例155中所描述,结果展示与用化合物T(正方形)、化合物Q(三角形)、化合物GG(X)或化合物U(具有十字形的X)处理的细胞相比,用PD0332991(圆形)处理的细胞到达细胞分裂的基线水平花费(参见仅仅用DMSO(菱形)处理的细胞)显著更长时间。
图5A是在化合物T、Q或GG以150mg/kg口腔管饲后在施用后12或24小时EdU掺入HSPC的比率(与未处理的对照小鼠相比)的图。图5B是在12或24小时用化合物T处理的小鼠的EdU阳性HSPC细胞百分比的图。通过口腔管饲法,给予小鼠50mg/kg(三角形)、100mg/kg(正方形)或150(倒三角形)mg/kg。图5C是在12、24、36和48小时用化合物T(150mg/kg,通过口腔管饲法)处理的小鼠的EdU阳性HSPC细胞百分比的图。如实施例156中所描述,化合物T和GG证明在12小时EdU掺入减少,并至24小时开始恢复至细胞分裂的正常水平。
图6是用PD0332991(三角形)或化合物T(倒三角形)处理的小鼠的EdU阳性HSPC细胞百分比对比施用化合物后的时间(小时)的图。通过口腔管饲法,给予150mg/kg两种化合物,并在12、24、36或48小时,测量EdU阳性HSPC细胞的百分比。如实施例157中所描述,单一口服剂量的PD0332991引起HSPC增殖持久减少,超过36小时。相比之下,单一口服剂量的化合物T引起HSPC增殖在12小时初期减少,但到给予化合物T 24小时,HSPC增殖重新开始。
图7提供人类和动物(猴、狗、大鼠和小鼠)肝微粒体中化合物T和PD0332991的半衰期(分钟)。如实施例158中所描述,在每一测试物种中PD0332991具有超过60分钟的半衰期。在每一测试物种中化合物T的半衰期比PD0332991短。
图8是展示用100mg/kg/d(正方形)或150mg/kg/d(三角形)化合物T处理的载有MMTV-c-neu(Rb阳性)肿瘤的小鼠中肿瘤体积(mm3)对比化合物T施用后时间(天)的图。将载有肿瘤的MMTV-c-neu小鼠(对照,n=9;化合物T,100mg/kg,n=7;化合物T,150mg/kg,n=6)用在食物或标准食物中递送的化合物T处理(圆形)。第0天表示化合物治疗的第一天。将小鼠用化合物T处理28天(如由指示治疗施用天数的x轴上数目周围的方框表示)。28天后,所有小鼠都喂食标准食物。每周记录肿瘤体积(长达56天)并用图表表示为平均值±平均标准误差。如实施例159中所描述,与对照相比,在28天疗法过程期间用化合物T连续处理(100mg/kg/d或150mg/kg/d)引起肿瘤体积显著减少。
图9是用化合物T以100mg/kg(水平线方框)或者150mg/kg(倾斜线方框)处理的每只小鼠的MMTV-c-neu小鼠(Rb阳性)肿瘤体积的变化百分比的瀑布图。在第21天肿瘤体积与未处理的动物的平均肿瘤尺寸相比。用化合物T处理的小鼠的肿瘤体积表示在第28天或超过第28天见到的最佳反应。负值指示肿瘤收缩。
图10是展示在MMTV-c-neu乳腺腺腔乳癌(Rb阳性)模型中用化合物T、GG或U处理的小鼠中MMTV-c-neu(Rb阳性)肿瘤的客观反应率(ORR)的表。所有三种化合物都经由加入药物的饮食(100mg/kg/d)经口施用。加入药物的饮食连续施用28天,接着停止。RECIST准则用以评估客观反应率。客观反应率(ORR)基于肿瘤体积变化百分比,使用以下类别分类:CR(完全反应)=100%反应;PR(部分反应)=至少30%降低;SD(稳定疾病)=无变化(无PR和无PD);和PD(进行性疾病)=20%增加。如实施例160中所描述,在28天疗法过程期间,用化合物T、GG或U连续处理引起肿瘤体积显著减少。
图11是展示用化合物T(空心圆)、化合物GG(菱形)或化合物U(正方形)处理的载有MMTV-c-neu(Rb阳性)肿瘤的小鼠中肿瘤体积(mm3)对比每一化合物施用后时间(天)的图。将载有肿瘤的MMTV-c-neu小鼠(对照,n=9;化合物T,100mg/kg,n=7;化合物GG,100mg/kg,n=7;化合物U,100mg/kg,n=8)用在食物或标准食物中递送的化合物处理(实心圆)。第0天表示化合物治疗的第一天。将小鼠用化合物处理28天(如由指示治疗施用天数的x轴上数目周围的方框表示)。28天后,所有小鼠都喂食标准食物。每周记录肿瘤体积(长达56天)并用图表表示为平均值±平均标准误差。如实施例160中所描述,在28天疗法过程期间,用化合物T、GG或U连续处理引起肿瘤体积显著减少,其中化合物T和U展示100%客观反应率,而化合物GG展示85%客观反应率。
图12是用100mg/kg化合物T(具有对角线的条,n=7)、100mg/kg化合物GG(黑白方框条,n=7)、100mg/kg化合物U(实心条,n=8)处理或无处理(空心条,n=9)的每只小鼠的MMTV-c-neu(Rb阳性)肿瘤体积的变化百分比的瀑布图。在第21天肿瘤体积与未处理的动物的平均肿瘤尺寸相比。用化合物T、GG或U处理的小鼠的肿瘤体积表示在第14天或超过第14天见到的最佳反应。负值指示肿瘤收缩。负值指示肿瘤收缩。如实施例160中所描述,在28天疗法过程期间,用化合物T、GG或U连续处理引起肿瘤体积显著减少。
图13-15说明本发明化合物的R2的若干例示性实施方案。
图16A-16C、17A-17D、18A-18C、19A-19B和20A-20F说明本发明化合物的核心结构的若干例示性实施方案。
图21是MCF7(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用PD0332991(圆形)或化合物T(图1;正方形)处理的可变摩尔浓度(M)的图。MCF7细胞在Costar(Tewksbury,Massachusetts)390396孔经组织培养物处理的白壁/透明底板中接种。进行从10uM到1nM的九点剂量反应连续稀释,并在六天化合物处理后,如所指示,使用发光细胞活力分析(CTG;Promega,Madison,Wisconsin,美国),按照制造商建议,测定细胞活力。在BioTek(Winooski,Vermont)Syngergy2多模板式读数器上读取盘。将相对光单位(RLU)作为可变摩尔浓度的结果绘图,且使用Graphpad(LaJolla,California)Prism 5统计软件分析数据以确定每一化合物的EC50。
图22是MCF7(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用化合物Q(图1;圆形)或者化合物GG(图1;正方形)处理的可变摩尔浓度(M)的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图23是MCF7(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用化合物U(图1;圆形)或者化合物H(图1;正方形)处理的可变摩尔浓度(M)的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图24是MCF7(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用化合物MM(图1;圆形)或者化合物OO(图1;正方形)处理的可变摩尔浓度(M)的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图25是ZR75-1(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用PD0332991或化合物T(图1;正方形)处理的可变摩尔浓度的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图26是ZR75-1(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用化合物Q(表1;圆形)或化合物GG(图1;正方形)处理的可变摩尔浓度的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图27是ZR75-1(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光单位(RLU)测量)对比用化合物U(表1;圆形)或化合物H(图1;正方形)处理的可变摩尔浓度的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图28是ZR75-1(Rb阳性)细胞(乳腺癌)的细胞增殖(如通过相对光毫米(RLU)测量)对比用化合物MM(表1;圆形)或化合物OO(图1;正方形)处理的可变摩尔浓度的图。如图21和实施例152中所描述,使用发光细胞活力分析测定细胞增殖。
图29A是tHS68细胞中处于G2-M期(空心圆)、S期(三角形)、G0-G1期(正方形)、<2N(菱形)中的细胞的百分比对比化合物T的可变浓度(nM)的图。将CDK4/6依赖性细胞系(tHS68)用指示浓度的化合物T处理24小时。化合物T处理后,收获细胞并分析细胞周期分布。如实施例161中所描述,tHS68细胞展示彻底G1停滞,伴随有S期细胞数目对应下降。
图29B是tHS68细胞(CDK4/6依赖性细胞系)的数目对比细胞的DNA含量(如通过碘化丙锭测量)的图。将细胞用DMSO处理24小时,收获并分析细胞周期分布。
图29C是WM2664细胞(CDK4/6依赖性细胞系)的数目对比细胞的DNA含量(如通过碘化丙锭测量)的图。将细胞用DMSO处理24小时,收获并分析细胞周期分布。
图29D是A2058细胞(非CDK4/6依赖性细胞系)的数目对比细胞的DNA含量(如通过碘化丙锭测量)的图。将细胞用DMSO处理24小时,收获并分析细胞周期分布。
图29E是在用化合物T处理后tHS68细胞(CDK4/6依赖性细胞系)的数目对比细胞的DNA含量(如通过碘化丙锭测量)的图。将细胞用化合物T(300nM)处理24小时,收获并分析细胞周期分布。如实施例161中所描述,用化合物T处理tHS68细胞引起S期峰损失(由箭头指示)。
图29F是在用化合物T处理后WM2664细胞(CDK4/6依赖性细胞系)的数目对比细胞的DNA含量(如通过碘化丙锭测量)的图。将细胞用化合物T(300nM)处理24小时,收获并分析细胞周期分布。如实施例161中所描述,用化合物T处理WM2664细胞引起S期峰损失(由箭头指示)。
图29G是在用化合物T处理后A2058细胞(非CDK4/6依赖性细胞系)的数目对比细胞的DNA含量(如通过碘化丙锭测量)的图。将细胞用化合物T(300nM)处理24小时,收获并分析细胞周期分布。如实施例161中所描述,用化合物T处理A2058细胞不引起S期峰损失(由箭头指示)。
图30是展示化合物T处理后Rb在Ser807/811和Ser780的磷酸化水平的蛋白质印迹。CDK4/6依赖性(tHS68或WM2664)和CDK4/6非依赖性细胞系(A2058)用化合物T(300nM)处理所指示的时间(0、4、8、16和24小时)。MAPK水平作为蛋白水平的对照显示。处理后,收获细胞并通过蛋白质印迹分析来分析Rb磷酸化。如实施例162中所描述,化合物T处理引起CDK4/6依赖性细胞系(tHS68和WM2664)中处理后16小时开始Rb磷酸化减少,但CDK4/6非依赖性细胞系(A2058)中则没有。
发明详述
提供改善的化合物、方法和组合物,作为治疗所选RB阳性癌症的化学治疗剂,其将由CDK4/6生长停滞引起的对受试者,典型地人类中CDK4/6复制依赖性健康细胞,例如造血干细胞和/或祖细胞(HSPC)的有害作用减到最少或减少。
定义
除非另有说明,否则用于本申请,包括说明书和权利要求书中的以下术语具有以下给出的定义。除非上下文另外清楚地规定,否则如本说明书和随附权利要求书中所用,单数“一(a/an)”和“所述”包括复数个指示物。标准化学术语的定义可见于参考资料中,包括Carey和Sundberg(2007)Advanced Organic Chemistry第5版A和B卷,Springer Science+Business Media LLC,New York。除非另外指明,否则本发明的实施将采用合成有机化学、质谱分析、色谱的制备和分析法、蛋白质化学、生物化学、重组DNA技术和药理学的常规方法。有机化学的常规方法包括以下中包括的方法:March’s Advanced Organic Chemistry:Reactions,Mechanisms,and Structure,第6版,M.B.Smith和J.March,John Wiley&Sons,Inc.,Hoboken,NJ,2007。
单独或在例如“卤烷基”和“烷基氨基”等其它术语内的术语“烷基”涵盖具有一个至约十二个碳原子的直链或支链基团。“低级烷基”具有一个至约六个碳原子。此类基团的实例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、戊基、异戊基、己基等等。术语“亚烷基”涵盖桥连二价直链和支链烷基。实例包括亚甲基、亚乙基、亚丙基、亚异丙基等等。
术语“烯基”涵盖具有至少一个碳-碳双键和两个至约十二个碳原子的直链或支链基团。“低级烯基”具有两个至约六个碳原子。烯基的实例包括乙烯基、丙烯基、烯丙基、丙烯基、丁烯基和4-甲基丁烯基。术语“烯基”和“低级烯基”涵盖具有“顺式”和“反式”取向或者“E”和“Z”取向的基团。
术语“炔基”表示具有至少一个碳-碳三键并具有两个至约十二个碳原子的直链或支链基团。“低级炔基”具有两个至约六个碳原子。此类基团的实例包括炔丙基、丁炔基等等。
烷基、烯基和炔基可以任选地经一个或多个例如卤基、羟基、硝基、氨基、氰基、卤烷基、芳基、杂芳基、杂环基等官能团取代。
术语“烷基氨基”涵盖“N-烷基氨基”和“N,N-二烷基氨基”,其中氨基对应地独立地经一个烷基和两个烷基取代。“低级烷基氨基”具有一个或两个具有一个至六个碳原子的烷基附接到氮原子。适合的烷基氨基可以是单烷基氨基或二烷基氨基,例如N-甲基氨基、N-乙基氨基、N,N-二甲基氨基、N,N-二乙基氨基等等。
术语“卤基”意指卤素,例如氟、氯、溴或碘原子。
术语“卤烷基”涵盖其中烷基碳原子的任一个或多个经如上定义的一个或多个卤基取代的基团。实例包括单卤烷基、二卤烷基和多卤烷基,包括全卤烷基。举个例子,单卤烷基可以在基团内具有碘基、溴基、氯基或氟基原子。二卤烷基和多卤烷基可以具有两个或超过两个相同卤基原子或不同的卤基的组合。“低级卤烷基”涵盖具有1-6个碳原子的基团。卤烷基的实例包括氟甲基、二氟甲基、三氟甲基、氯甲基、二氯甲基、三氯甲基、五氟乙基、七氟丙基、二氟氯甲基、二氯氟甲基、二氟乙基、二氟丙基、二氯乙基和二氯丙基。“全氟烷基”意指所有氢原子经氟原子置换的烷基。实例包括三氟甲基和五氟乙基。
单独或组合的术语“芳基”意指含有一个或两个环的碳环芳香族系统,其中此类环可以依稠合方式附接在一起。术语“芳基”涵盖芳族基,例如苯基、萘基、茚基、四氢萘基和茚满基。更优选的芳基是苯基。所述“芳基”可以具有1个或更多个取代基,例如低级烷基、羟基、卤基、卤烷基、硝基、氰基、烷氧基、低级烷基氨基等等。芳基可以任选地经例如卤基、羟基、硝基、氨基、氰基、卤烷基、芳基、杂芳基、杂环基等一个或多个官能团取代。
术语“杂环基”(或“杂环”)涵盖饱和和部分饱和含杂原子环基,其中杂原子可以选自氮、硫和氧。杂环包含单环6-8元环以及5-16元双环系统(其可以包括桥连稠合和螺稠合双环系统)。其不包括含有-O-O-、-O-S-或-S-S-部分的环。所述“杂环基”可以具有1个至3个取代基,例如羟基、Boc、卤基、卤烷基、氰基、低级烷基、低级芳烷基、氧代基、低级烷氧基、氨基、低级烷基氨基等等。
饱和杂环基的实例包括含有1至4个氮原子的饱和3至6元杂单环基[例如吡咯烷基、咪唑烷基、哌啶基、吡咯啉基、哌嗪基];含有1至2个氧原子和1至3个氮原子的饱和3至6元杂单环基[例如吗啉基];含有1至2个硫原子和1至3个氮原子的饱和3至6元杂单环基[例如噻唑烷基]。部分饱和杂环基的实例包括二氢噻吩基、二氢吡喃基、二氢呋喃基、二氢噻唑基等等。
部分饱和和饱和杂环基的具体实例包括吡咯烷基、咪唑烷基、哌啶基、吡咯啉基、吡唑烷基、哌嗪基、吗啉基、四氢吡喃基、噻唑烷基、二氢噻吩基、2,3-二氢-苯并[1,4]二噁烷基、吲哚啉基、异吲哚啉基、二氢苯并噻吩基、二氢苯并呋喃基、异色满基、色满基、1,2-二氢喹啉基、1,2,3,4-四氢-异喹啉基、1,2,3,4-四氢-喹啉基、2,3,4,4a,9,9a-六氢-1H-3-氮杂-茀基、5,6,7-三氢-l,2,4-三唑并[3,4-a]异喹啉基、3,4-二氢-2H-苯并[1,4]噁嗪基、苯并[1,4]二噁烷基、2,3-二氢-1H-lλ'-苯并[d]异噻唑-6-基、二氢吡喃基、二氢呋喃基和二氢噻唑基等等。
杂环基还包括杂环基与芳基稠合/缩合的基团:含有1至5个氮原子的缩合杂环基,例如吲哚基、异吲哚基、吲哚嗪基、苯并咪唑基、喹啉基、异喹啉基、吲唑基、苯并三唑基、四氢哒嗪基[例如四唑并[1,5-b]哒嗪基];含有1至2个氧原子和1至3个氮原子的不饱和缩合杂环基[例如苯并噁唑基、苯并噁二唑基];含有1至2个硫原子和1至3个氮原子的不饱和缩合杂环基[例如苯并噻唑基、苯并噻二唑基];和含有1至2个氧或硫原子的饱和、部分不饱和和不饱和缩合杂环基[例如苯并呋喃基、苯并噻吩基、2,3-二氢苯并[1,4]二噁烷基和二氢苯并呋喃基]。
术语“杂芳基”表示含有一个或多个选自O、N和S的群组的杂原子的芳基环系统,其中环氮和硫原子任选地氧化且氮原子任选地季铵化。实例包括含有1至4个氮原子的不饱和5至6元杂单环基,例如吡咯基、咪唑基、吡唑基、2-吡啶基、3-吡啶基、4-吡啶基、嘧啶基、吡嗪基、哒嗪基、三唑基[例如4H-1,2,4-三唑基、1H-1,2,3-三唑基、2H-1,2,3-三唑基];含有氧原子的不饱和5至6元杂单环基,例如吡喃基、2-呋喃基、3-呋喃基等;含有硫原子的不饱和5至6元杂单环基,例如2-噻吩基、3-噻吩基等;含有1至2个氧原子和1至3个氮原子的不饱和5至6元杂单环基,例如噁唑基、异噁唑基、噁二唑基[例如1,2,4-噁二唑基、1,3,4-噁二唑基、1,2,5-噁二唑基];含有1至2个硫原子和1至3个氮原子的不饱和5至6元杂单环基,例如噻唑基、噻二唑基[例如1,2,4-噻二唑基、1,3,4-噻二唑基、1,2,5-噻二唑基]。
术语“杂芳基烷基”表示经杂芳基取代的烷基。实例包括吡啶基甲基和噻吩基乙基。
术语“磺酰基”无论单独使用或与其它术语相联,例如烷基磺酰基,均对应表示二价基-SO2-。
术语“羧基(carboxy或carboxyl)”无论单独使用或与其它术语一起使用,例如“羧基烷基”,均表示-C(O)-OH。
术语“羰基”无论单独使用或与其它术语一起使用,例如“氨基羰基”,均表示-C(O)-。
术语“氨基羰基”表示式-C(O)-NH2的酰胺基。
术语“杂环烷基”涵盖经杂环基取代的烷基。实例包括哌啶基甲基和吗啉基乙基。
术语“芳基烷基”涵盖经芳基取代的烷基。实例包括苯甲基、二苯甲基和苯乙基。所述芳基烷基中的芳基可以另外经卤基、烷基、烷氧基、卤烷基和卤烷氧基取代。
术语“环烷基”包括具有3至10个碳的饱和碳环基团。低级环烷基包括C3-C6环。实例包括环戊基、环丙基和环己基。环烷基可以任选地经例如卤基、羟基、硝基、氨基、氰基、卤烷基、芳基、杂芳基、杂环基等一个或多个官能团取代。
术语“环烷基烷基”涵盖经环烷基取代的烷基。“低级环烷基烷基”是附接到具有一个至六个碳原子的烷基的环烷基。实例包括环己基甲基。所述基团中的环烷基可以另外经卤基、烷基、烷氧基和羟基取代。
术语“环烯基”包括具有一个或多个碳-碳双键的碳环基团,包括“环烷基二烯基”化合物。实例包括环戊烯基、环戊二烯基、环己烯基和环庚二烯基。
术语“包含”意指开放性,包括所指示的组分但不排除其它要素。
如本文所用,术语“氧代基”涵盖用双键附接的氧原子。
如本文所用,术语“硝基”涵盖-NO2。
如本文所用,术语“氰基”涵盖-CN。
如本文所用,术语“前药”意指当体内施用主体时转变为母体药物的化合物。如本文所用,术语“母体药物”意指适用于治疗主体,典型地人类中本文中描述的任何病症,或控制或改善与本文中描述的任何生理性或病理性病症有关的根本原因或症状的任何本发明所述的化合物。前药可以用于实现任何所期望的作用,包括增强母体药物的特性或改善母体药物的药物或药物动力学特性。存在前药策略,其提供了调节母体药物的体内产生的条件的选择,所有都视为包括在本文中。前药策略的非限制性实例包括以下的共价附接:可去除基团或基团的可去除部分,例如(但不限于)酰化、磷酸化、膦酰基化、氨基磷酸酯衍生物、酰胺化、还原、氧化、酯化、烷基化、其它羧基衍生物、硫氧基或砜衍生物、羰化或酸酐。
说明书和权利要求书中,除非另作说明,否则所给出的化学式或名称应该涵盖所有光学和立体异构体以及存在此类异构体和混合物的外消旋混合物。
本发明涉及在Rb阳性增殖病症的治疗期间的HSPC节制性策略。因此,如本文所用,术语“HSPC”意指与患病的HSPC或相关血液来源的细胞相对比,健康的造血干细胞和/或造血祖细胞。HSPC包括造血干细胞,例如长期造血干细胞(LT-HSC)和短期造血干细胞(ST-HSC):和造血祖细胞,包括多潜能祖细胞(MPP)、共同脊髓祖细胞(CMP)、共同淋巴祖细胞(CLP)、粒细胞-单核细胞祖细胞(GMP)和巨核细胞-红细胞祖细胞(MEP)。
在一些实施方案中,CDK4/6复制依赖性健康细胞是造血干细胞祖细胞。在一些实施方案中,CDK4/6复制依赖性健康细胞可以是例如(但不限于)肝、肾、胰腺、脑、肺、肾上腺、肠、肠管、胃、皮肤、听觉系统、骨骼、膀胱、卵巢、子宫、睾丸、胆囊、甲状腺、心脏、胰岛、血管等等非造血组织中的细胞。
本文中描述的化合物的背景下使用的术语“选择性CDK4/6抑制剂”包括在标准磷酸化分析中以IC50摩尔浓度比抑制CDK2活性相同程度所需的IC50摩尔浓度小至少约1/500或1/1000或1/1500或1/1800或1/2000抑制CDK4活性、CDK6活性或CDK4与CDK6活性的化合物。
如本文所用,术语“化学疗法”或“化学治疗剂”是指用细胞生长抑制剂或细胞毒性剂(即化合物)治疗以减少或消除例如癌细胞等不良细胞的生长或增殖。因此,如本文所用,“化学疗法”或“化学治疗剂”是指用于治疗例如癌症等增生病症的细胞毒性剂或细胞生长抑制剂。
“诱发G1停滞”意指抑制剂化合物诱发细胞群体相当大部分处于细胞周期的G1期的静态。
“造血不足”意指血液细胞谱系计数减少或血细胞(即脊髓发育不良)和/或淋巴细胞(即淋巴球减少症、例如B细胞和T细胞等循环淋巴细胞数目减少)产生不足。可以观测到血液不足,例如贫血、血小板数减少(即血小板减少症)或白血球计数减少(即白血球减少症)或粒细胞减少(例如中性白细胞减少)形式的骨髓抑制。
“同步再进入细胞周期”意指由于CDK4/6抑制剂化合物的作用而处于G1停滞的CDK4/6复制依赖性健康细胞,例如HSPC在化合物例如PD0332991作用消散后在相对相同的集中时间框内或以相对相同的速率再进入细胞周期。相对地,“非同步再进入细胞周期”意指由于CDK4/6抑制剂化合物的作用而处于G1停滞的CDK4/6复制依赖性健康细胞,例如HSPC在化合物作用消散后在相对不同的集中时间框内或以相对不同的速率再进入细胞周期。
“非循环期”或“药物假期”意指受试者不施用或暴露于化学治疗剂的时间段。例如,在21天施用至受试者化学治疗剂且7天不施用化学治疗剂且方案大量重复的治疗方案中,未施用的7天时期视为“非循环期”或“药物假期”。脱靶和药物假期也可以指治疗方案的中断,其中受试者由于有害的副作用,例如骨髓抑制而一段时间不施用化学治疗剂。
所治疗的受试者典型地是人类受试者,不过应了解本文中描述的方法对于其它动物,例如哺乳动物和脊椎动物物种也是有效的。更具体地说,术语受试者可以包括用于分析中的动物,例如用于临床前测试中的动物,包括(但不限于)小鼠、大鼠、猴、犬、猪和兔;以及家养猪类(猪和肥猪)、反刍动物、马、家禽、猫科动物、牛科动物、鼠类、犬科动物等等。
活性化合物
在一个实施方案中,本发明是针对式I、II、III、IV或V的化合物或此类化合物或其药学上可接受的盐的用途:
其中:
Z为-(CH2)x-,其中x为1、2、3或4,或-O-(CH2)z-,其中z为2、3或4;
每一X独立地为CH或N;
每一X′独立地为CH或N;
X″独立地为CH2、S或NH,经配置使得所述部分为稳定5元环;
R、R8和R11独立地为H、C1-C3烷基或卤烷基、环烷基或含有一个或多个选自N、O或S的杂原子的环烷基;-(亚烷基)m-C3-C8环烷基、-(亚烷基)m-芳基、-(亚烷基)m-杂环基、-(亚烷基)m-杂芳基、-(亚烷基)m-NR3R4、-(亚烷基)m-C(O)-NR3R4;-(亚烷基)m-O-R5、-(亚烷基)m-S(O)n-R5或-(亚烷基)m-S(O)n-NR3R4,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个R基团取代,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环;
每一R1独立地为芳基、烷基、环烷基或卤烷基,其中所述烷基、环烷基和卤烷基中的每一者任选地在链中包括代替碳的O或N杂原子且相邻环原子上或相同环原子上的两个R1连同其连接的环原子任选地形成3-8元环;
y为0、1、2、3或4;
R2为-(亚烷基)m-杂环基、-(亚烷基)m-杂芳基、-(亚烷基)m-NR3R4、-(亚烷基)m-C(O)-NR3R4;-(亚烷基)m-C(O)-O-烷基;-(亚烷基)m-O-R5、-(亚烷基)m-S(O)n-R5或-(亚烷基)m-S(O)n-NR3R4,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环且其中m为0或1且n为0、1或2;
R3和R4每次出现时独立地为:
(i)氢或
(ii)烷基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环烷基、芳基烷基或杂芳基烷基,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环;或R3和R4连同其连接的氮原子可以组合形成在化合价容许的情况下任选地独立地经一个或多个Rx基团取代的杂环,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环;
R5和R5*每次出现时为
(i)氢或
(ii)烷基、烯基、炔基、环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环烷基、芳基烷基或杂芳基烷基,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代;
Rx每次出现时独立地为卤基、氰基、硝基、氧代基、烷基、卤烷基、烯基、炔基、环烷基、环烯基、杂环基、芳基、杂芳基、芳基烷基、杂芳基烷基、环烷基烷基、杂环烷基、-(亚烷基)m-OR5、-(亚烷基)m-O-亚烷基-OR5、-(亚烷基)m-S(O)n-R5、-(亚烷基)m-NR3R4、-(亚烷基)m-CN、-(亚烷基)m-C(O)-R5、-(亚烷基)m-C(S)-R5、-(亚烷基)m-C(O)-OR5、-(亚烷基)m-O-C(O)-R5、-(亚烷基)m-C(S)-OR5、-(亚烷基)m-C(O)-(亚烷基)m-NR3R4、-(亚烷基)m-C(S)-NR3R4、-(亚烷基)m-N(R3)-C(O)-NR3R4、-(亚烷基)m-N(R3)-C(S)-NR3R4、-(亚烷基)m-N(R3)-C(O)-R5、-(亚烷基)m-N(R3)-C(S)-R5、-(亚烷基)m-O-C(O)-NR3R4、-(亚烷基)m-O-C(S)-NR3R4、-(亚烷基)m-SO2-NR3R4、-(亚烷基)m-N(R3)-SO2-R5、-(亚烷基)m-N(R3)-SO2-NR3R4、-(亚烷基)m-N(R3)-C(O)-OR5)、-(亚烷基)m-N(R3)-C(S)-OR5或-(亚烷基)m-N(R3)-SO2-R5;其中:
所述烷基、卤烷基、烯基、炔基、环烷基、环烯基、杂环基、芳基、杂芳基、芳基烷基、杂芳基烷基、环烷基烷基和杂环烷基可以进一步独立地经一个或多个以下各基取代:
-(亚烷基)m-CN、-(亚烷基)m-OR5*、-(亚烷基)m-S(O)n-R5*、-(亚烷基)m-NR3*R4*、-(亚烷基)m-C(O)-R5*、-(亚烷基)m-C(=S)R5*、-(亚烷基)m-C(=O)O R5*、-(亚烷基)m-OC(=O)R5*、-(亚烷基)m-C(S)-OR5*、-(亚烷基)m-C(O)-NR3*R4*、-(亚烷基)m-C(S)-NR3*R4*、-(亚烷基)m-N(R3*)-C(O)-NR3*R4*、-(亚烷基)m-N(R3*)-C(S)-NR3*R4*、-(亚烷基)m-N(R3*)-C(O)-R5*、-(亚烷基)m-N(R3*)-C(S)-R5*、-(亚烷基)m-O-C(O)-NR3*R4*、-(亚烷基)m-O-C(S)-NR3*R4*、-(亚烷基)m-SO2-NR3*R4*、-(亚烷基)m-N(R3*)-SO2-R5*、-(亚烷基)m-N(R3*)-SO2-NR3*R4*、-(亚烷基)m-N(R3*)-C(O)-OR5*、-(亚烷基)m-N(R3*)-C(S)-OR5*或-(亚烷基)m-N(R3*)-SO2-R5*,
n为0、1或2,且
m为0或1;
R3*和R4*每次出现时独立地为:
(i)氢或
(ii)烷基、烯基、炔基环烷基、杂环基、芳基、杂芳基、环烷基烷基、杂环烷基、芳基烷基或杂芳基烷基,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代;或R3*和R4*连同其连接的氮原子可以组合形成在化合价容许的情况下任选地独立地经一个或多个Rx基团取代的杂环;且
R6为H或低级烷基、-(亚烷基)m-杂环基、-(亚烷基)m-杂芳基、-(亚烷基)m-NR3R4、-(亚烷基)m-C(O)-NR3R4;-(亚烷基)m-O-R5、-(亚烷基)m-S(O)n-R5或-(亚烷基)m-S(O)n-NR3R4,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环;且
R10为(i)NHRA,其中RA为未经取代或经取代的C1-C8烷基、环烷基烷基或-TT-RR、C1-C8环烷基或含有一个或多个选自N、O和S的杂原子的环烷基;TT为未经取代或经取代的C1-C8烷基或C3-C8环烷基连接基团;且RR为羟基、未经取代或经取代的C1-C6烷氧基、氨基、未经取代或经取代的C1-C6烷基氨基、未经取代或经取代的二C1-C6烷基氨基、未经取代或经取代的C6-C10芳基、包含一个或两个5或6元环和1-4个选自N、O和S的杂原子的未经取代或经取代的杂芳基、未经取代或经取代的C3-C10碳环或包含一个或两个5或6元环和1-4个选自N、O和S的杂原子的未经取代或经取代的杂环;或(ii)-C(O)-R12或-C(O)O-R13,其中R12为NHRA或RA且R13为RA;
或其药学上可接受的盐、前药或同位素变体,例如部分或完全氘化形式。
在一些方面,化合物具有式I或式II且R6不存在。
在一些方面,化合物具有式III:
且变数如针对式I和II的化合物和其药学上可接受的盐所定义。
在一些方面,Rx未经进一步取代。
在一些方面,R2为-(亚烷基)m-杂环基、-(亚烷基)m-杂芳基、-(亚烷基)m-NR3R4、-(亚烷基)m-C(O)-NR3R4;-(亚烷基)m-O-R5、-(亚烷基)m-S(O)n-R5或-(亚烷基)m-S(O)n-NR3R4,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环且其中m为0或1且n为0、1或2。
在一些方面,R8为氢或C1-C3烷基。
在一些方面,R为氢或C1-C3烷基。
在一些方面,R2为-(亚烷基)m-杂环基、-(亚烷基)m-NR3R4、-(亚烷基)m-C(O)-NR3R4、-(亚烷基)m-C(O)-O-烷基或-(亚烷基)m-OR5,其中任一者在化合价容许的情况下可以任选地独立地经一个或多个Rx基团取代,且其中两个结合于相同或相邻原子的Rx基团可以任选地组合形成环。
在一些方面,R2为-(亚烷基)m-杂环基、-(亚烷基)m-NR3R4、-(亚烷基)m-C(O)-NR3R4、-(亚烷基)m-C(O)-O-烷基或-(亚烷基)m-OR5,未经进一步取代。
在一些方面,R2中的m为1。在另一方面,R2中的亚烷基为亚甲基。
在一些方面,R2为其中:
R2*为键、亚烷基、-(亚烷基)m-O-(亚烷基)m-、-(亚烷基)m-C(O)-(亚烷基)m-、-(亚烷基)m-S(O)2-(亚烷基)m-和-(亚烷基)m-NH-(亚烷基)m-,其中每一m独立地为0或1;
P为4至8元单环或双环饱和杂环基;
每一Rx1独立地为-(亚烷基)m-(C(O))m-(亚烷基)m-(N(RN))m-(烷基)m,其中每一m独立地为0或1,前提条件为至少一个m为1;-(C(O))-O-烷基;-(亚烷基)m-环烷基,其中m为0或1;-N(RN)-环烷基;-C(O)-环烷基;-(亚烷基)m-杂环基,其中m为0或1;或-N(RN)-杂环基;-C(O)-杂环基;-S(O)2-(亚烷基)m,其中m为1或2,其中:
RN为H、C1至C4烷基或C1至C6杂烷基,且
其中两个Rx1可以连同P上其连接的可以为相同原子的原子形成环;且
t为0、1或2。
在一些方面,每一Rx1仅仅任选地经未经取代的烷基、卤素或羟基取代。
在一些方面,Rx1为氢或未经取代的C1-C4烷基。
在一些方面,至少一个Rx1为-(亚烷基)m-杂环基,其中m为0或1。
在一些方面,R2为其中P*为4至8元单环或双环饱和杂环基。
在一些方面,R2为
在一些方面,R2为
在一些方面,R2为其中:
R2*为键、亚烷基、-(亚烷基)m-O-(亚烷基)m-、-(亚烷基)m-C(O)-(亚烷基)m-、-(亚烷基)m-S(O)2-(亚烷基)m-和-(亚烷基)m-NH-(亚烷基)m-,其中每一m独立地为0或1;
P为4至8元单环或双环饱和杂环基;
P1为4至6元单环饱和杂环基;
每一Rx2独立地为氢或烷基;且
s为0、1或2。
在一些方面,R2为
在一些方面,P1包括至少一个氮。
在一些方面,在任何先前方面的R2*中的任何亚烷基未经进一步取代。
在一些方面,R2选自图13-15中描绘的结构。
在一些方面,R2为
在一些方面,化合物具有通式I且更具体地说,图16-20中通用结构之一,其中变数如先前所定义。
在一些方面,化合物具有通式Ia:
其中R1、R2、R和y如先前所定义。
在一些实施方案中,所述化合物具有式Ia且R为烷基。
在一些实施方案中,所述化合物具有式Ia且R为H。
在一些实施方案中,所述化合物具有式Ia且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ia且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或未经取代的C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ib:
其中R2和R如先前所定义。
在一些实施方案中,所述化合物具有式Ib且R为烷基。
在一些实施方案中,所述化合物具有式Ib且R为H。
在一些实施方案中,所述化合物具有式Ib且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ib且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ic:
其中R2和R如先前所定义。
在一些实施方案中,所述化合物具有式Ic且R为烷基。
在一些实施方案中,所述化合物具有式Ic且R为H。
在一些实施方案中,所述化合物具有式Ic且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ic且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Id:
其中R2和R如先前所定义。
在一些实施方案中,所述化合物具有式Id且R为烷基。
在一些实施方案中,所述化合物具有式Id且R为H。
在一些实施方案中,所述化合物具有式Id且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Id且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ie:
在一些实施方案中,所述化合物具有式Ie且R为烷基。
在一些实施方案中,所述化合物具有式Ie且R为H。
在一些实施方案中,所述化合物具有式Ie且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ie且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式If:
在一些实施方案中,所述化合物具有式If且R为烷基。
在一些实施方案中,所述化合物具有式If且R为H。
在一些实施方案中,所述化合物具有式If且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式If且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ig:
在一些实施方案中,所述化合物具有式Ig且R为烷基。
在一些实施方案中,所述化合物具有式Ig且R为H。
在一些实施方案中,所述化合物具有式Ig且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ig且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ih:
在一些实施方案中,所述化合物具有式Ih且R为烷基。
在一些实施方案中,所述化合物具有式Ih且R为H。
在一些实施方案中,所述化合物具有式Ih且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ih且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ii:
在一些实施方案中,所述化合物具有式Ii且R为烷基。
在一些实施方案中,所述化合物具有式Ii且R为H。
在一些实施方案中,所述化合物具有式Ii且R2为其中P*为4至8元单环或双环饱和杂环基且R2*、Rx1和t如先前所定义。
在一些实施方案中,所述化合物具有式Ii且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基且R2*如先前所定义。
在一些实施方案中,所述化合物具有式Ij:
在一些实施方案中,所述化合物具有式Ij且R为烷基。
在一些实施方案中,所述化合物具有式Ij且R为H。
在一些实施方案中,所述化合物具有式Ij且R2为其中P*为4至8元单环或双环饱和杂环基。
在一些实施方案中,所述化合物具有式Ij且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基。
在一些实施方案中,所述化合物具有式Ij且R为H,且两个X均为N。
在一些实施方案中,所述化合物具有以下结构:
在一些实施方案中,所述化合物具有式Ik且R2为其中P*为4至8元单环或双环饱和杂环基。
在一些实施方案中,所述化合物具有式Ik且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基。
在一些实施方案中,所述化合物具有式Il:
在一些实施方案中,所述化合物具有式Il且R2为其中P*为4至8元单环或双环饱和杂环基。
在一些实施方案中,所述化合物具有式Il且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基。
在一些实施方案中,所述化合物具有式Im:
在一些实施方案中,所述化合物具有式Im且R2为其中P*为4至8元单环或双环饱和杂环基。
在一些实施方案中,所述化合物具有式Im且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基。
在一些实施方案中,所述化合物具有式IIa:
在一些实施方案中,所述化合物具有式IIa且R2为其中P*为4至8元单环或双环饱和杂环基。
在一些实施方案中,所述化合物具有式IIa且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基。
在一些实施方案中,所述化合物具有式IIb:
在一些实施方案中,所述化合物具有式Im且R2为其中P*为4至8元单环或双环饱和杂环基。
在一些实施方案中,所述化合物具有式Im且R2为其中P*为4至8元单环或双环饱和杂环基,Rx1为氢或C1-C4烷基。
在一些方面,活性化合物为:
在本发明内且可以用于所公开的治疗方法和组合物中的其它特定化合物包括下表1中所列出的结构。
表1:抗赘生性和抗增生性剂的结构
同位素取代
本发明包括化合物和具有量超过同位素天然丰度(即富集)的所需原子同位素取代的化合物的使用。同位素是具有相同的原子序数但不同的质量数,即质子数目相同但中子数目不同的原子。通过一般实例且不限于,可以在所述结构中任何地方使用氢的同位素,例如氘(2H)和氚(3H)。或者或另外,可以使用碳的同位素,例如13C和14C。一种优选同位素取代是在分子上一个或多个位置氘取代氢以改善药物的性能。氘可以在代谢期间键断裂的位置中结合(α-氘动力学同位素效应)或紧靠或接近键断裂位点结合(β-氘动力学同位素效应)。
经例如氘的重同位素取代可以提供某些由代谢稳定性更大产生的治疗优点,例如体内半衰期延长或所需剂量减少。在代谢分解位点氘取代氢可以降低该键代谢的速率或消除。在氢原子可以存在的化合物的任何位置,氢原子可以是氢的任何同位素,包括氕(1H)、氘(2H)和氚(3H)。因此,除非上下文清楚地另外规定,否则本文中提及化合物涵盖所有潜在的同位素形式。
术语“同位素标记”类似物是指作为“氘化类似物”、“13C-标记类似物”或“氘化/13C标记类似物”的类似物。术语“氘化类似物”意指本文中描述的化合物,其中H-同位素,即氢/氕(1H)经H同位素(即氘(2H))取代。氘取代可以是部分或完全的。部分氘取代意指至少一个氢经至少一个氘取代。在某些实施方案中,同位素在任何相关位置90%、95%或99%或更多富集同位素。在一些实施方案中,其是在预定位置90%、95%或99%富集的氘。
属于本发明并且可以用于公开的治疗方法和组合物的其它特定化合物包括以下表1中列出的式I、II、III、IV或V的结构。
Rb阳性癌症和增生性病症
具体地说,本文中描述的活性化合物可以用于治疗罹患Rb阳性癌症或其它Rb阳性异常细胞增殖病症的受试者。在一些实施方案中,癌症或细胞增殖病症是CDK4/6复制依赖性癌症或细胞增殖病症,其是指需要CDK4/6活性用于复制或增殖的癌症或细胞增殖病症,或其可以通过选择性CDK4/6抑制剂的活性抑制生长。此类类型的癌症和病症特征可以为(例如细胞显示)存在功能性成视网膜细胞瘤蛋白质。此类癌症和病症分类为Rb阳性。Rb阳性异常细胞增殖病症和如本文所用的此术语的变体是指由不受控制或异常的细胞分裂所引起的病症或疾病,特征为存在功能性成视网膜细胞瘤蛋白质,其可以包括癌症。在本发明的一个方面,本文中描述的化合物和方法可以用于治疗非癌性Rb阳性异常细胞增殖病症。此类病症的实例可以包括非恶性淋巴增生、非恶性乳房赘生物、银屑病、关节炎、皮炎、癌前结肠病变或肉质、血管生成病症、免疫介导和非免疫介导的炎症疾病、关节炎、年龄相关的黄斑变性、糖尿病和其它非癌性或良性细胞增殖病症。
适于施用本文中描述的化合物的靶向癌症可以包括Rb阳性:雌激素受体阳性、HER2阴性晚期乳癌、晚期转移性乳癌、脂肪肉瘤、非小细胞肺癌、肝癌、卵巢癌、成胶质细胞瘤、难治性实体肿瘤、成视网膜细胞瘤阳性乳癌以及成视网膜细胞瘤阳性子宫内膜、阴道和卵巢癌以及肺和支气管癌、结肠腺癌、直肠腺癌、中枢神经系统生殖细胞肿瘤、畸胎瘤、雌激素受体阴性乳癌、雌激素受体阳性乳癌、家族性睾丸生殖细胞肿瘤、HER2阴性乳癌、HER2阳性乳癌、男性乳癌、卵巢未成熟畸胎瘤、卵巢成熟畸胎瘤、卵巢单胚层和高度专化的畸胎瘤、孕激素受体阴性乳癌、孕激素受体阳性乳癌、复发性乳癌、复发性结肠癌、复发性性腺外生殖细胞肿瘤、复发性性腺外非精原细胞瘤性生殖细胞肿瘤、复发性性腺外精原细胞瘤、复发性恶性睾丸生殖细胞肿瘤、复发性黑色素瘤、复发性卵巢生殖细胞肿瘤、复发性直肠癌、III期性腺外非精原细胞瘤性生殖细胞肿瘤、III期性腺外精原细胞瘤、III期恶性睾丸生殖细胞肿瘤、III期卵巢生殖细胞肿瘤、IV期乳癌、IV期结肠癌、IV期性腺外非精原细胞瘤性生殖细胞肿瘤、IV期性腺外精原细胞瘤、IV期黑色素瘤、IV期卵巢生殖细胞肿瘤、IV期直肠癌、睾丸未成熟畸胎瘤、睾丸成熟畸胎瘤。在具体的实施方案中,靶向癌症包括雌激素受体阳性、HER2阴性晚期乳癌、晚期转移性乳癌、脂肪肉瘤、非小细胞肺癌、肝癌、卵巢癌、成胶质细胞瘤、难治性实体肿瘤、成视网膜细胞瘤阳性乳癌以及成视网膜细胞瘤阳性子宫内膜、阴道和卵巢癌以及肺和支气管癌、转移性结肠直肠癌、具有CDK4突变或扩增的转移性黑色素瘤或顺铂难治的不可切除的生殖细胞肿瘤。
在一个实施方案中,Rb阳性癌症选自Rb阳性癌瘤、肉瘤,包括(但不限于)肺癌、骨癌、胰腺癌、皮肤癌、头或颈癌、皮肤或眼内黑色素瘤、子宫癌、卵巢癌、直肠癌、肛门部癌、胃癌、结肠癌、乳癌、子宫癌、输卵管癌、子宫内膜癌、宫颈癌、阴道癌、外阴癌、食管癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、前列腺癌、膀胱癌、肾或输尿管癌、肾细胞癌、肾盂癌、中枢神经系统(CNS)的赘生物、原发性CNS淋巴瘤、脊柱肿瘤、脑干神经胶质瘤、垂体腺瘤或上述癌症中一者或多者的组合。
在一个实施方案中,Rb阳性癌症选自Rb阳性:纤维肉瘤、粘液肉瘤、软骨肉瘤、骨肉瘤、脊索瘤、恶性纤维组织细胞瘤、血管内皮瘤、血管肉瘤、淋巴管肉瘤、间皮瘤、平滑肌肉瘤、横纹肌肉瘤、鳞状细胞癌;表皮样癌、恶性皮肤附属器肿瘤、腺癌、肝癌、肝细胞癌、肾细胞癌瘤、肾上腺样瘤、肝胆管型肝癌、转移细胞癌、恶性合胞体瘤、精原细胞瘤、胚性细菌癌瘤、多形性神经胶质瘤;多形性胶质母细胞瘤、神经母细胞瘤、成神经管细胞瘤、恶性脑膜瘤、恶性神经鞘瘤、神经纤维肉瘤、甲状旁腺腺癌、甲状腺髓样癌、支气管类癌瘤、嗜铬细胞瘤、胰岛细胞癌瘤、恶性类癌瘤、恶性副神经节瘤、黑色素瘤、默克尔细胞赘生物(Merkel cell neoplasm)、分叶状囊性肉瘤、唾液癌、胸腺癌、膀胱癌和威尔姆氏肿瘤(Wilms tumor)。
成视网膜细胞瘤(Rb)肿瘤抑制蛋白的存在或功能正常(Rb阳性)可以通过本领域技术人员已知的任何标准分析,包括(但不限于)蛋白质印迹法、ELISA(酶联免疫吸附分析)、IHC(免疫组织化学)和FACS(荧光活化细胞分选)确定。分析的选择将取决于利用的组织、细胞系或替代组织样品,例如蛋白质印迹法和ELISA可以用于任何或所有类型的组织、细胞系或替代组织,而IHC方法将更适用于本发明的方法中利用的组织是肿瘤活检的情况。FACs分析将大部分可应用于呈单细胞悬浮液的样品,例如细胞系和分离的外周血单核细胞。参见例如US 20070212736“Functional Immunohistochemical Cell Cycle Analysis as a Prognostic Indicator for Cancer”。或者,分子遗传测试可以用于测定成视网膜细胞瘤基因状态。用于成视网膜细胞瘤的分子遗传测试包括如以下中所描述的测试:Lohmann及Gallie“Retinob lastoma.Gene Reviews”(2010)http://www.ncbi.nlm.nih.gov/bookshel f/br.fcgi?book=gene&part=retinoblastoma或Parsam等人“A comprehe nsive,sensitive and economical approach for the detection of mutati ons in the RB1 gene in retinoblastoma”Journal of Genetics,88(4),517-527(2009)。
在一些实施方案中,待治疗的癌症选自雌激素受体阳性、HER2阴性晚期乳癌、晚期转移性乳癌、脂肪肉瘤、非小细胞肺癌、肝癌、卵巢癌、成胶质细胞瘤、难治性实体肿瘤、成视网膜细胞瘤阳性乳癌以及成视网膜细胞瘤阳性子宫内膜、阴道和卵巢癌以及肺和支气管癌。
CDK复制依赖性细胞和细胞周期蛋白依赖性激酶抑制剂
组织特异性干细胞和其它固有增殖细胞子集能够自我更新,意指在成年哺乳动物寿命中其能够通过调控的复制替换本身。另外,干细胞不对称地分裂,产生“子代”或“祖”细胞,这些细胞又产生既定器官的各种组分。例如,在造血系统中,造血干细胞产生祖细胞,祖细胞又产生血液的所有分化组分(例如白血球、红血球和血小板)(参见图1)。
已经发现某些增殖细胞,例如HSPC,需要增生性激酶细胞周期蛋白依赖性激酶4(CDK4)和/或细胞周期蛋白依赖性激酶6(CDK6)的酶活性用于细胞复制。相比之下,成年哺乳动物中的大部分增殖细胞(例如骨髓中更加分化的血液形成细胞)不需要CDK4和/或CDK6(即CDK4/6)的活性。这些分化的细胞可以在缺乏CDK4/6活性下通过使用其它增生性激酶,例如细胞周期蛋白依赖性激酶2(CDK2)或细胞周期蛋白依赖性激酶1(CDK1)增殖。
本发明包括治疗某些癌症,尤其是Rb阳性癌症,同时将对受试者中CDK4/6复制依赖性健康细胞和尤其是造血细胞和/或祖细胞(HSPC)的有害作用减到最少的方法,其通过施用本文中描述的化合物以治疗特定Rb阳性癌症。
在一个实施方案中,与使用例如PD0332991等其它CDK4/6抑制剂相比,使用本文中描述的化合物作为化学治疗剂允许血液恢复加速且因HSPC复制延迟引起的血液不足风险降低。在一个实施方案中,与使用例如PD0332991等其它CDK4/6抑制剂的当前治疗模态相比,使用本文中描述的化合物作为化学治疗剂允许在治疗的过程中非循环期或药物假期减少或减到最少。在一个实施方案中,使用本文中描述的化合物作为化学治疗剂允许消除非循环期或药物假期。在一个实施方案中,与使用例如PD0332991等其它CDK4/6抑制剂的当前治疗模态相比,使用本文中描述的化合物作为化学治疗剂允许施用时期延长且非循环期或药物假期更少。在一个实施方案中,与使用例如PD0332991等其它CDK4/6抑制剂的当前模态相比,使用本文中描述的化合物作为化学治疗剂允许在非循环期或药物假期期间血球数恢复更快。
在某些实施方案中,施用的化合物选自包含式I、式II、式III、式IV或式V或其组合的化合物或组合物。在某些实施方案中,施用的化合物选自从表1选择的化合物。
在某些方面,提供作为化学治疗剂的化合物、方法和组合物,其减少或限制针对Rb阳性癌症进行CDK4/6抑制治疗的受试者中CDK4/6抑制对CDK4/6复制依赖性健康细胞的有害作用,所述方法包括施用有效量的本文中描述的化合物,其中相当大部分的CDK4/6复制依赖性健康细胞在化合物施用不到约24、30、36或40小时内恢复至治疗前基线细胞周期活性(即再进入细胞周期)。在某些实施方案中,其中化合物的IC50CDK4抑制浓度比其对CDK2的IC50抑制浓度小至少1500倍。在某些实施方案中,施用的化合物选自包含式I、式II、式III、式IV或式V的化合物或组合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在某些实施方案中,施用的化合物选自表1中含有的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在一个实施方案中,CDK4/6复制依赖性细胞是造血干细胞和/或祖细胞(HSPC)。
在某些方面,提供用作化学治疗剂的化合物、方法和组合物,其限制针对Rb阳性癌症进行治疗的受试者中CDK4/6抑制对CDK4/6复制依赖性健康细胞的有害作用,所述方法包括施用有效量的本文中描述的化合物,其中相当大部分的CDK4/6复制依赖性健康细胞在化合物抑制作用消散后不到约24、30、36或40小时内同步再进入细胞周期。在某些实施方案中,其中化合物的IC50CDK4抑制浓度比其对CDK2的IC50抑制浓度小至少1500倍。在某些实施方案中,施用的化合物选自包含式I、式II、式III、式IV或式V的化合物或组合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在某些实施方案中,施用的化合物选自表1中含有的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在一个实施方案中,CDK4/6复制依赖性细胞是造血干细胞和/或祖细胞(HSPC)。
在某些方面,提供用作化学治疗剂的化合物、方法和组合物,其限制受试者中CDK4/6抑制对CDK4/6复制依赖性健康细胞的有害作用,所述方法包括向患有Rb阳性癌症的受试者施用有效量的本文中描述的化合物,其中相当大部分的CDK4/6复制依赖性健康细胞在化合物的CDK4/6抑制作用消散后不到约24、30、36或40小时内同步再进入细胞周期。在一个实施方案中,施用化合物的IC50CDK4抑制浓度比其对CDK2的IC50抑制浓度小超过500倍。在某些实施方案中,相当大部分的CDK4/6复制依赖性健康细胞在离受试者血液中的化合物的浓度水平降到治疗有效浓度以下的时刻不到约24、30、36或40小时内同步再进入细胞周期。在某些实施方案中,施用的化合物选自包含式I、式II、式III、式IV或式V的化合物或组合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在某些实施方案中,施用的化合物选自表1中含有的化合物,或其药学上可接受的组合物、盐、同位素类似物或前药。在一个实施方案中,CDK4/6复制依赖性细胞是造血干细胞和/或祖细胞(HSPC)。在一个实施方案中,CDK4/6复制依赖性健康细胞是肾上皮细胞。
在某些实施方案中,施用的化合物选自包含式I、式II、式III、式IV或式V的化合物或组合物或其药学上可接受的组合物、盐、同位素类似物或前药,或表1中含有的化合物或其药学上可接受的组合物、盐、同位素类似物或前药,其中化合物的作用实际上是短暂且瞬时的,允许显著部分的CDK4/6复制依赖性健康细胞快速地同步再进入细胞周期,例如离化合物上次施用不到约24、30、36或40小时内。
用于所描述的方法的化合物是高度选择性的有效CDK4/6抑制剂,具有最低的CDK2抑制活性。在一个实施方案中,用于本文中描述的方法中的化合物的CDK4/CycD1IC50抑制浓度值比其对CDK2/CycE抑制相应的IC50浓度值低>1500倍、>1800倍、>2000倍、>2200倍、>2500倍、>2700倍、>3000倍、>3200倍或更大。在一个实施方案中,用于本文中描述的方法的化合物对CDK4/CycD1抑制的IC50浓度值为约<1.50nM、<1.25nM、<1.0nM、<0.90nM、<0.85nM、<0.80nM、<0.75nM、<0.70nM、<0.65nM、<0.60nM、<0.55nM或更少。在一个实施方案中,用于本文中描述的方法的CDK4/6抑制剂对CDK2/CycE抑制的IC50浓度值为约>1.0μM、>1.25μM、>1.50μM、>1.75μM、>2.0μM、>2.25μM、>2.50μM、>2.75μM、>3.0μM、>3.25μM、>3.5μM或更大。在一个实施方案中,用于本文中描述的方法的化合物对CDK2/CycA IC50的IC50浓度值为>0.80μM、>0.85μM、>0.90μM、>0.95μM、>1.0μM、>1.25μM、>1.50μM、>1.75μM、>2.0μM、>2.25μM、>2.50μM、>2.75uM、>3.0μM或更大。
在某些实施方案中,适用于所述方法的化合物可以提供CDK4/6复制依赖性健康细胞的瞬时和快速可逆的G1停滞,同时提供CDK4/6复制依赖性癌症的生长抑制。通过具有有限时间的瞬时效应,此类化合物用作化学治疗剂允许与例如长效CDK4/6抑制剂(例如PD0332991)相比,在治疗停止后CDK4/6复制依赖性健康细胞更快地再进入细胞周期。对CDK4/6复制依赖性健康细胞的G1停滞作用的更快消散使得此类化合物在以下情况下优于长效CDK4/6抑制剂:1)受试者将暴露于密集的治疗,其中使用长效CDK4/6抑制剂将阻止CDK4/6复制依赖性健康细胞在暴露之间循环;2)连续或长期的治疗方案,其中CDK4/6复制依赖性健康细胞的长期G1停滞是靶向癌症生长抑制的一种副作用,并且受试者将得益于CDK4/6复制依赖性健康细胞在治疗方案停止后、连续方案中抑制剂的给予之间或治疗中断之间快速再进入细胞周期,从而限制复制延迟,因此在治疗停止后减少、限制或改善进一步健康细胞损坏,例如骨髓抑制。根据本发明,使用本文中描述的选择性化合物的化学治疗方案可以通过大量不同的给药时程,包括循环/非循环方案和连续治疗方案实现。
在一个实施方案中,本文中描述的化合物用于其中受试者暴露于针对Rb阳性癌症的常规重复的化学治疗剂治疗的CDK4/6复制依赖性健康细胞循环策略中。此类循环允许CDK4/6复制依赖性细胞在常规重复治疗之间再生破坏的血细胞谱系,且降低与长期CDK4/6抑制有关的风险。在G1停滞状态与复制状态之间的此循环在使用更长效的CDK4/6抑制剂(例如PD0332991)的时间间隔有限的重复剂暴露中是不可能的,因为化合物的延缓G1停滞作用禁止在下一次暴露于CDK4/6抑制剂前CDK4/6复制依赖性细胞显著并有意义地再进入细胞周期或在治疗停止后延迟健康细胞进入细胞周期和复原破坏的组织或细胞。
在一个实施方案中,使用本文中描述的化合物提供CDK4/6复制依赖性健康细胞,例如HSPC快速再进入细胞周期,使得细胞在不到约40小时、36小时、30小时、28小时、24小时或更少时间内恢复至治疗前基线细胞周期活性。在一个实施方案中,使用本文中描述的化合物提供CDK4/6复制依赖性健康细胞,例如HSPC快速再进入细胞周期,使得细胞在不到约40小时、36小时、30小时、28小时、24小时、18小时、16小时、14小时、12小时或更少时间接近治疗前基线细胞周期活性。在一个实施方案中,使用本文中描述的化合物提供CDK4/6复制依赖性健康细胞,例如HSPC快速再进入细胞周期,使得细胞在离本文中描述的化合物的上次施用不到约40小时、36小时、30小时、28小时、24小时、18小时、16小时、14小时、12小时或更少时间内恢复至治疗前基线细胞周期活性。在一个实施方案中,使用本文中描述的化合物提供CDK4/6复制依赖性健康细胞快速再进入细胞周期,使得细胞在离化合物的上次施用不到约40小时、36小时、30小时、28小时、24小时、18小时、16小时、14小时、12小时或更少时间内接近治疗前基线细胞周期活性。在一个实施方案中,使用本文中描述的化合物提供CDK4/6复制依赖性健康细胞快速再进入细胞周期,使得细胞在离受试者血液中的化合物的浓度水平降到治疗有效浓度以下的时刻不到约40小时、36小时、30小时、28小时、24小时、18小时、16小时、14小时、12小时或更少时间内接近治疗前基线细胞周期活性。在一个实施方案中,CDK4/6复制依赖性健康细胞是HSPC。在一个实施方案中,CDK4/6复制依赖性健康细胞是肾上皮细胞。在一个实施方案中,快速再进入细胞周期是同步的。
在一个实施方案中,使用本文中描述的化合物提供CDK4/6复制依赖性健康细胞,例如HSPC快速再进入细胞周期,使得一部分细胞在连续治疗方案,例如化合物施用长时间,例如连续5天、连续7天、连续10天、连续14天、连续18天、连续21天、连续24天、连续28天、连续35天或更多天的治疗方案期间显示一定水平细胞周期活性,或能够进入细胞周期并增殖。在一个实施方案中,适用于所述方法的化合物连续一段时间施用,例如21、28、35天或更多天,不需要非循环期或药物假期。在一个实施方案中,使用本文中描述的化合物消除了对非循环期、药物假期的需要,或减少治疗期间共同施用的抗赘生性化合物浓度。
根据本发明,本文中描述的化合物可以作为化学治疗剂依任何治疗时程和以符合规定疗程的任何剂量施用患有Rb阳性增殖病症的受试者。例如,化合物可以一天一次、一天两次或一天三次施用。化合物可以依交替天数施用,或每三天或每四天或每五天或每六天或每隔一周施用。化合物可以每隔一周或每月施用。
组合疗法
在本发明的一个方面,本文公开的化合物可以有利地与治疗方案组合施用以达成有益、累加或协同效应。
在一个实施方案中,本发明的化合物/方法与治疗Rb阳性癌症的另一疗法组合使用。第二疗法可以是免疫疗法。如以下更详细地论述,化合物可以缀合于抗体、放射性试剂或将化合物引导至患病或异常增殖细胞的其它靶向剂。在另一个实施方案中,化合物与另一药物或生物试剂(例如抗体)组合使用以用组合或协同方法增加治疗的功效。在一个实施方案中,化合物可以与T细胞免疫接种一起使用,T细胞免疫接种典型地涉及用失活的自体反应性T细胞免疫接种以消除如本文中描述的Rb阳性癌细胞群体。在另一个实施方案中,化合物与双特异性T细胞衔接蛋白(BiTE)组合使用,双特异性T细胞衔接蛋白是设计成能同时缀合于内源性T细胞和如本文中描述的Rb阳性癌细胞上的特定抗原的抗体,连接两种类型细胞。
在一个实施方案中,另一疗法是单克隆抗体(MAb)。一些MAb刺激破坏癌细胞的免疫反应。类似于B细胞天然产生的抗体,这些MAb“涂布”癌细胞表面,触发免疫系统对其的破坏。例如,贝伐单抗(bevacizumab)靶向血管内皮生长因子(VEGF),VEGF是肿瘤细胞和肿瘤微环境中的其它细胞分泌的促进肿瘤血管发展的一种蛋白质。当缀合于贝伐单抗时,VEGF无法与其细胞受体相互作用,阻止引起新的血管生长的信号传导。类似地,西妥昔单抗(cetuximab)和帕尼单抗(panitumumab)靶向表皮生长因子受体(EGFR),并且曲妥珠单抗(trastuzumab)靶向人类表皮生长因子受体2(HER-2)。缀合于细胞表面生长因子受体的MAb阻止靶向受体发送其正常的促进生长的信号。其也可以触发细胞凋亡并活化免疫系统以破坏肿瘤细胞。
另一组癌症治疗性MAb是免疫结合物。有时称为抗毒素或抗体-药物结合物的这些MAb由附接到杀死细胞物质(例如植物或细菌毒素、化学疗法药物或放射性分子)的抗体组成。抗体锁定到癌细胞表面上的其特异性抗原,且杀死细胞物质被细胞吸收。以此方式工作的FDA批准的结合MAb包括阿多-曲妥珠单抗美坦辛(ado-trastuzumab emtansine),其靶向HER-2分子以将抑制细胞增殖的药物DM1递送至表达HER-2的转移性乳癌细胞。
使T细胞经工程化以经由双特异性抗体(bsAb)或嵌合抗原受体(CAR)识别癌细胞的免疫疗法是能够将分裂与非分裂/缓慢分裂亚群癌细胞分离的方法。
通过同时识别免疫效应细胞表面上的标靶抗原和活化受体的双特异性抗体提供了重定向免疫效应细胞以杀死癌细胞的机会。另一种方法为通过细胞外抗体与细胞内信号传导结构域融合来产生嵌合抗原受体。嵌合抗原受体工程化的T细胞能够以MHC非依赖性方式特异性杀死肿瘤细胞。
在一些实施方案中,化合物可以与其它化学治疗剂组合施用至受试者。合宜时,本文中描述的化合物可以与另一化学治疗剂同时施用,以简化治疗方案。在一些实施方案中,化合物和另一化学治疗剂可以提供于单一制剂中。在一个实施方案中,本文中描述的化合物的使用在治疗方案中与其它剂组合。此类剂可以包括(但不限于)他莫昔芬(tamoxifen)、咪达唑仑(midazolam)、来曲唑(letrozole)、硼替佐米(bortezomib)、阿那曲唑(anastrozole)、戈舍瑞林(goserelin)、mTOR抑制剂、PI3激酶抑制剂、双重mTOR-PI3K抑制剂、MEK抑制剂、RAS抑制剂、ALK抑制剂、HSP抑制剂(例如HSP70和HSP 90抑制剂或其组合)、BCL-2抑制剂、细胞凋亡诱发化合物、AKT抑制剂(包括(但不限于)MK-2206、GSK690693、哌立福新、KRX-0401、GDC-0068、曲西立滨(Triciribine)、AZD5363、和厚朴酚(Honokiol)、PF-04691502和米替福新(Miltefosine))、PD-1抑制剂(包括(但不限于)纳武单抗(Nivolumab)、CT-011、MK-3475、BMS936558和AMP-514)或FLT-3抑制剂(包括(但不限于)P406、多韦替尼(Dovitinib)、奎扎替尼(Quizartinib)(AC220)、阿木替尼(Amuvatinib)(MP-470)、唐杜替尼(Tandutinib)(MLN518)、ENMD-2076和KW-2449或其组合。mTOR抑制剂的实例包括(但不限于)雷帕霉素和其类似物、依维莫司(Afinitor)、坦西莫司、雷达莫司、西罗莫司和德佛莫司。P13激酶抑制剂的实例包括(但不限于)渥曼青霉素、脱甲绿胶酶素、哌立福新、依德西伯、PX-866、IPI-145(Infinity)、BAY 80-6946、BEZ235、RP6503、TGR 1202(RP5264)、MLN1117(INK1117)、皮提西伯、布帕西伯、SAR245408(XL147)、SAR245409(XL765)、帕洛米德529、ZSTK474、PWT33597、RP6530、CUDC-907和AEZS-136。MEK抑制剂的实例包括(但不限于)曲美替尼、司美替尼、MEK162、GDC-0973(XL518)和PD0325901。RAS抑制剂的实例包括(但不限于)瑞赖新和siG12D LODER。ALK抑制剂的实例包括(但不限于)克唑替尼、AP26113和LDK378。HSP抑制剂包括(但不限于)格尔德霉素或17-N-烯丙基氨基-17-脱氧基格尔德霉素(17AAG)和根赤壳菌素。在一个具体的实施方案中,本文中描述的化合物与来曲唑和/或他莫昔芬组合施用。可以与本文中描述的化合物组合使用的其它化学治疗剂包括(但不限于)抗赘生性作用不需要细胞周期活性的化学治疗剂。
在一个实施方案中,本文中描述的CDK4/6抑制剂可以与选自(但不限于)以下的化学治疗剂组合:甲磺酸伊马替尼(Imatinib mesylate)达沙替尼(Dasatinib)尼洛替尼(Nilotinib)博舒替尼(Bosutinib)曲妥珠单抗帕妥珠单抗(Pertuzumab)(PerjetaTM)、拉帕替尼(Lapatinib)吉非替尼(Gefitinib)厄罗替尼(Erlotinib)西妥昔单抗(Cetuximab)帕尼单抗(Panitumumab)凡德他尼(Vandetanib)威罗菲尼(Vemurafenib)伏立诺他(Vorinostat)罗米地辛(Romidepsin)蓓萨罗丁(Bexarotene)阿利维A酸(Alitretinoin)维甲酸(Tretinoin)卡非佐米(Carfilizomib)(KyprolisTM)、普拉曲沙(Pralatrexate)贝伐单抗(Bevacizumab)(Ziv-aflibercept)索拉非尼(Sorafenib)舒尼替尼(Sunitinib)帕唑帕尼(Pazopanib)瑞格菲尼(Regorafenib)和卡博替尼(Cabozantinib)(CometriqTM)。
在某些方面,其它治疗剂是抗炎剂、化学治疗剂、放射治疗、其它治疗剂或免疫抑制剂。
适合的化学治疗剂包括(但不限于)放射性分子、毒素(也称为细胞毒素或细胞毒性剂,其包括对细胞活力有害的任何剂)、剂和含有化学治疗化合物的脂质体或其它囊泡。一般抗癌药物包括:长春新碱(Vincristine)或脂质体长春新碱道诺霉素(Daunorubicin)(道诺霉素(daunomycin)或)或多柔比星)、阿糖胞苷(Cytarabine)(阿糖胞苷(cytosine arabinoside)、ara-C或)、L-天冬酰胺酶(L-asparaginase)或PEG-L-天冬酰胺酶(pegaspargase或)、依托泊苷(Etoposide)(VP-16)、替尼泊苷(Teniposide)6-巯基嘌呤(6-mercaptopurine)(6-MP或)、甲氨蝶呤(Methotrexate)、环磷酰胺(Cyclophosphamide)泼尼松(Prednisone)、地塞米松(Dexamethasone)(地卡特隆(Decadron))、伊马替尼(imatinib))、达沙替尼(dasatinib)尼洛替尼(nilotinib)博舒替尼和帕纳替尼(ponatinib)(IclusigTM)。其它适合的化学治疗剂的实例包括(但不限于)1-去氢睾固酮、5-氟尿嘧啶(5-fluorouracil)达卡巴嗪(decarbazine)、6-巯基嘌呤(6-mercaptopurine)、6-硫鸟嘌呤(6-thioguanine)、放线菌素D(actinomycin D)、阿霉素(adriamycin)、阿地白介素(aldesleukin)、烷基化剂、别嘌呤醇钠(allopurinol sodium)、六甲蜜胺(altretamine)、氨磷汀(amifostine)、阿那曲唑(anastrozole)、氨茴霉素(anthramycin,AMC)、抗有丝分裂剂、顺式-二氯二胺铂(II)(DDP)(顺铂)、二氨基二氯铂(diamino dichloro platinum)、蒽环霉素(anthracyclines)、抗生素(antibiotics)、抗代谢物(antimetabolite)、天冬酰胺酶(asparaginase)、活BCG(膀胱内)、倍他米松磷酸钠(betamethasone sodium phosphate)和乙酸倍他米松(betamethasone acetate)、比卡鲁胺(bicalutamide)、硫酸博来霉素(bleomycin sulfate)、白消安(busulfan)、甲酰四氢叶酸钙(calcium leucouorin)、加里刹霉素(calicheamicin)、卡培他滨(capecitabine)、卡铂(carboplatin)、洛莫司汀(lomustine)(CCNU)、卡莫司汀(carmustine)(BSNU)、苯丁酸氮芥(Chlorambucil)、顺铂、克拉屈滨(Cladribine)、秋水仙碱(Colchicin)、结合雌激素(conjugated estrogens)、环磷酰胺、环硫酰胺(Cyclothosphamide)、阿糖胞苷、阿糖胞苷、细胞分裂抑素B(cytochalasin B)、环磷酰胺(Cytoxan)、氮烯唑胺(Dacarbazine)、放线菌素D(Dactinomycin)、放线菌素D(以前为放线菌素)、盐酸道诺霉素(daunirubicin HCL)、柠檬酸道诺霉素(daunorucbicin citrate)、地尼白介素(denileukin diftitox)、得拉唑沙(Dexrazoxane)、二溴甘露醇(Dibromomannitol)、二羟基炭疽菌素二酮(dihydroxy anthracin dione)、多烯紫杉醇(Docetaxel)、甲磺酸多拉司琼(dolasetron mesylate)、盐酸多柔比星(doxorubicin HCL)、屈大麻酚(dronabinol)、大肠杆菌L-天冬酰胺酶(E.coli L-asparaginase)、吐根碱(emetine)、红细胞生成素-α(epoetin-α)、欧文氏菌属L-天冬酰胺酶(Erwinia L-asparaginase)、酯化雌激素(esterified estrogen)、雌二醇(estradiol)、雌莫司汀磷酸钠(estramustine phosphate sodium)、溴化乙锭(ethidium bromide)、乙炔雌二醇(ethinyl estradiol)、依替膦酸盐(etidronate)、依托泊苷嗜橙菌因子(etoposide citrororum factor)、磷酸依托泊苷(etoposide phosphate)、非格司亭(filgrastim)、氟尿核苷(floxuridine)、氟康唑(fluconazole)、磷酸氟达拉滨(fludarabine phosphate)、氟尿嘧啶(fluorouracil)、氟他胺(flutamide)、亚叶酸(folinic acid)、盐酸吉西他滨(gemcitabine HCL)、糖皮质激素(glucocorticoid)、盐酸戈舍瑞林(goserelin acetate)、短杆菌肽D(gramicidin D)、盐酸格兰西龙(granisetron HCL)、羟基脲(hydroxyurea)、盐酸艾达霉素(idarubicin HCL)、异环磷酰胺(ifosfamide)、干扰素α-2b(interferonα-2b)、盐酸伊立替康(irinotecan HCL)、来曲唑(letrozole)、甲酰四氢叶酸钙(leucovorin calcium)、乙酸亮脯利特(leuprolide acetate)、盐酸左旋四咪唑(levamisole HCL)、利多卡因(lidocaine)、洛莫司汀(lomustine)、类美登素(maytansinoid)、盐酸二氯甲二乙胺(mechlorethamine HCL)、醋酸甲羟孕酮(medroxyprogesterone acetate)、醋酸甲地孕酮(megestrol acetate)、盐酸美法仑(melphalan HCL)、巯基嘌呤(mercaptipurine)、美司钠(mesna)、甲氨蝶呤、甲基睾甾酮(methyltestosterone)、光神霉素(mithramycin)、丝裂霉素C(mitomycin C)、米托坦(mitotane)、米托蒽醌(mitoxantrone)、尼鲁米特(nilutamide)、乙酸奥曲肽(octreotide acetate)、盐酸奥坦西隆(ondansetron HCL)、紫杉醇(paclitaxel)、帕米膦酸二钠(pamidronate disodium)、喷司他汀(pentostatin)、盐酸匹鲁卡品(pilocarpine HCL)、皮利霉素(plimycin)、具有卡莫司汀植入物的聚苯丙生20(polifeprosan 20 with carmustine implant)、卟吩姆钠(porfimer sodium)、普鲁卡因(procaine)、盐酸丙卡巴肼(procarbazine HCL)、普萘洛尔(propranolol)、利妥昔单抗(rituximab)、沙莫司亭(sargramostim)、链脲佐菌素(streptozotocin)、他莫昔芬、紫杉酚(taxol)、替尼泊苷(teniposide)、替尼泊苷(tenoposide)、睾内酯(testolactone)、丁卡因(tetracaine)、噻依派(thioepa)苯丁酸氮芥(chlorambucil)、硫鸟嘌呤(thioguanine)、噻替派(thiotepa)、盐酸拓扑替康(topotecan HCL)、柠檬酸托瑞米芬(toremifene citrate)、曲妥珠单抗、维甲酸(tretinoin)、戊柔比星(valrubicin)、硫酸长春花碱(vinblastine sulfate)、硫酸长春新碱(vincristine sulfate)和酒石酸长春瑞滨(vinorelbine tartrate)。
可以与本文公开的化合物组合施用的其它治疗剂可以包括贝伐单抗(bevacizumab)、舒尼替尼(sutinib)、索拉非尼(sorafenib)、2-甲氧雌甾二醇(2-methoxyestradiol)或2ME2、非纳索特(finasunate)、瓦他拉尼(vatalanib)、凡德他尼(vandetanib)、阿非赛特(aflibercept)、伐洛昔单抗(volociximab)、依他珠单抗(etaracizumab)(MEDI-522)、斯莱替得(cilengitide)、厄罗替尼(erlotinib)、西妥昔单抗(cetuximab)、帕尼单抗(panitumumab)、吉非替尼(gefitinib)、曲妥珠单抗、多维替尼(dovitinib)、非格木单抗(figitumumab)、阿它赛特(atacicept)、利妥昔单抗(rituximab)、阿仑单抗(alemtuzumab)、阿地白介素(aldesleukine)、阿替珠单抗(atlizumab)、托珠单抗(tocilizumab)、坦西莫司(temsirolimus)、依维莫司(everolimus)、卢卡珠单抗(lucatumumab)、达昔珠单抗(dacetuzumab)、HLL1、huN901-DM1、阿替莫得(atiprimod)、那他珠单抗(natalizumab)、硼替佐米(bortezomib)、卡非米德(carfilzomib)、吗佐米德(marizomib)、他斯霉素(tanespimycin)、甲磺酸沙奎那韦(saquinavir mesylate)、利托那韦(ritonavir)、甲磺酸那非那韦(nelfinavir mesylate)、硫酸印地那韦(indinavir sulfate)、倍林司他(belinostat)、帕比司他(panobinostat)、吗帕珠单抗(mapatumumab)、莱沙珠单抗(lexatumumab)、杜拉能米(dulanermin)、ABT-737、奥利默森(oblimersen)、皮替德辛(plitidepsin)、塔吗皮莫(talmapimod)、P276-00、恩佐塔辛(enzastaurin)、替吡法尼(tipifarnib)、哌立福新、伊马替尼、达沙替尼、来那度胺(lenalidomide)、沙利窦迈(thalidomide)、辛伐他汀(simvastatin)和赛利考昔(celecoxib)。
在本发明的一方面,本文描述的化合物可以与至少一种免疫抑制剂组合。免疫抑制剂优选地选自钙调磷酸酶抑制剂,例如环孢菌素环孢菌素(cyclosporin)或子囊霉素(ascomycin),例如环孢菌素AFK506(塔可莫司(tacrolimus)、皮可莫司(pimecrolimus));mTOR抑制剂,例如雷帕霉素或其衍生物,例如西罗莫司依维莫司坦西莫司、佐它莫司(zotarolimus)、拜林莫司-7(biolimus-7)、拜林莫司-9(biolimus-9);雷帕霉素类似物(rapalog),例如雷达莫司(ridaforolimus)、咪唑硫嘌呤(azathioprine)、卡莫司1H(campath 1H);S1P受体调节剂,例如芬戈莫德(fingolimod)或其类似物、抗IL-8抗体、霉酚酸(mycophenolic acid)或其盐(例如钠盐)或其前药(例如麦考酚酸莫酯(Mycophenolate Mofetil))、OKT3(ORTHOCLONE)、泼尼松(Prednisone)、布喹那钠(Brequinar Sodium)、OKT4、T10B9.A-3A、33B3.1、15-去氧司加林(15-deoxyspergualin)、曲培莫司(tresperimus)、来氟米特(Leflunomide)CTLAI-Ig、抗CD25、抗IL2R、巴利昔单抗(Basiliximab)达珠单抗(Daclizumab)米佐滨(mizorbine)、甲氨蝶呤、地塞米松、ISAtx-247、SDZ ASM 981(吡美莫司(pimecrolimus),))、CTLA4lg(阿巴赛特(Abatacept))、倍拉赛特(belatacept)、LFA3lg、依那西普(etanercept)(Immunex以出售)、阿达木单抗(adalimumab)英利西单抗(infliximab)抗LFA-1抗体、那他珠单抗(natalizumab)恩莫单抗(Enlimomab)、加维利单抗(gavilimomab)、抗胸腺细胞免疫球蛋白(antithymocyte immunoglobulin)、希普利珠单抗(siplizumab)、阿法珠单抗(Alefacept efalizumab)、颇得斯安(pentasa)、美沙拉嗪(mesalazine)、亚沙可(asacol)、磷酸可待因(codeine phosphate)、扑炎痛(benorylate)、联苯丁酮酸(fenbufen)、那颇辛(naprosyn)、双氯芬酸(diclofenac)、依托度酸(etodolac)和吲哚美辛(indomethacin)、阿斯匹林(aspirin)和布洛芬(ibuprofen)。
在某些实施方案中,本文中描述的化合物在用另一化学治疗剂治疗前,在用另一化学治疗剂治疗期间,在施用另一化学治疗剂后或其组合施用至受试者。
在一些实施方案中,选择性化合物可以施用至受试者,使得其它化学治疗剂可以在更高剂量下(增加化学治疗剂剂量强度)或更频繁(增加化学治疗剂剂量密度)施用。剂量密集化学疗法是一种化学疗法治疗计划,其中药物在治疗之间的给予时间少于标准化学疗法治疗计划。化学疗法剂量强度表示每单位时间施用化学疗法之单位剂量。剂量强度可以通过改变施用的剂量、施用时间间隔或两者增加或减少。
在本发明的一个实施方案中,本文中描述的化合物可以在与另一剂的配合方案中施用,所述另一剂为例如非DNA破坏的靶向抗赘生剂或造血生长因子剂。近来已经报导造血生长因子不合时宜的施用可能具有严重的副作用。例如,EPO家族生长因子的使用已经与高动脉压、大脑惊厥、高血压性脑病、血栓栓塞、缺铁症、类流感综合症和静脉血栓形成相关联。生长因子的G-CSF家族已经与脾扩大和破裂、呼吸窘迫综合症、变态反应和镰刀形红细胞并发症有关。通过将本文中描述的短期选择性化合物和本发明的方法的施用与造血生长因子的及时施用组合,例如当患病细胞不再处于生长停滞的时候,保健医师可降低生长因子的量以使不需要的副作用减到最少,同时实现想要的治疗益处。在一个实施方案中,生长因子在化合物对CDK4/6复制依赖性健康细胞,例如HSPC的作用停止后施用。因此,在此实施方案中,在抗赘生性治疗方案中使用本文中描述的选择性化合物允许受试者接收减少量的生长因子,因为靶向的造血细胞将比例如PD0332991等其它化合物时更快地再进入细胞周期。另外,使用本文中描述的化合物使得在G1停滞后快速再进入细胞循环提供了定时造血生长因子的施用以帮助再造造血细胞系,从而最大化生长因子作用的能力,那就是说,此时生长因子将最有效。因而,在一个实施方案中,本文中描述的化合物和方法的使用与包括(但不限于)以下的造血生长因子的使用组合:粒细胞群落刺激因子(G-CSF,例如以Neupogen(非格司亭(filgrastin)、Neulasta(聚乙二醇化非格斯亭(peg-filgrastin)或来格司亭(lenograstin)出售)、粒细胞-巨噬细胞群落刺激因子(GM-CSF,例如以莫拉司亭(molgramostim)和沙莫司亭(sargramostim)(Leukine)出售)、M-CSF(巨噬细胞群落刺激因子)、促血小板生成素(巨核细胞生长发育因子(MGDF),例如以罗米司亭(Romiplostim)和艾曲波帕(Eltrombopag)出售)、介白素(IL)-12、介白素-3、介白素-11(脂肪形成抑制因子或奥普瑞白介素(oprelvekin))、SCF(干细胞因子、青灰因子、试剂盒配体或KL)和促红细胞生成素(EPO)以及其衍生物(例如红细胞生成素-α以Darbopoetin、Epocept、Nanokine、Epofit、Epogin、Eprex和Procrit出售;红细胞生成素-β以例如NeoRecormon、Recormon和Micera出售)、红细胞生成素-δ(以例如Dynepo出售)、红细胞生成素-ω(以例如Epomax出售)、红细胞生成素-ζ(以例如Silapo和Reacrit出售)以及例如Epocept、EPOTrust、Erypro Safe、Repoeitin、Vintor、Epofit、Erykine、Wepox、Espogen、Relipoeitin、Shanpoietin、Zyrop和EPIAO)。在一个实施方案中,CDK4/6抑制剂在施用造血生长因子之前施用。在一个实施方案中,造血生长因子施用时间经安排,使得化合物对HSPC的作用已经消散。在一个实施方案中,生长因子在本文中描述的化合物施用后至少20小时施用。
必要时,多剂的本文中描述的化合物可以施用至受试者。或者,可以给予受试者单剂的本文中描述的化合物。例如,可以施用化合物,以便CDK4/6复制依赖性健康细胞停滞在G1,其中由于化合物的G1停滞作用快速消散,所以很多健康细胞在暴露后不久,例如在约24-48小时或更少时间内再进入细胞循环且能够复制,并继续复制,直到随后施用化合物。在一个实施方案中,施用化合物以允许CDK4/6复制依赖性健康细胞在G1停滞与再进入细胞循环之间循环,从而适应重复给药的治疗方案,例如长期重复给药的治疗方案。
在一些实施方案中,CDK4/6复制依赖性健康细胞可以通过多次限制时间的本文中描述的化合物的分开施用,停滞更长时期,例如经数小时、数天、数周和/或数月。由于例如HSPC等CDK4/6复制依赖性健康细胞在化合物抑制性细胞内作用消散后快速并同步再进入细胞周期,所以细胞能够比例如PD0332991等具有更长G1停滞型态的化合物抑制剂更快地复原细胞谱系。
通过本文中描述的化合物提供的副作用、尤其是骨髓抑制的减少可以允许剂量加强(例如可以在固定时间段内给予更多疗法),其将转化为更佳的功效。因此,本发明公开的方法可以使化学治疗方案毒性更少且更有效。适当时,小分子可以调配用于经口、局部、鼻内、吸入、静脉内或任何其它期望施用形式。
适用于本文中描述的方法的化合物是选择性地抑制CDK4和CDK6中至少一者或通过抑制Rb阳性癌症的细胞复制的选择性CDK4/6抑制剂。在一个实施方案中,如CDK4/CycD1 IC50磷酸化分析中测量的本文中描述的化合物对CDK4的IC50比如CDK2/CycE IC50磷酸化分析中测量的所述化合物对CDK2的IC50低至少1500倍或更多倍。在一个实施方案中,CDK4/6抑制剂比PD0332991有效至少约10倍或倍数大得多(即在CDK4/CycD1磷酸化分析中IC50低至少10倍或更多)。
如本文中描述的化合物的使用可以诱发CDK4/6依赖性细胞中选择性G1停滞(例如如基于细胞的体外分析中测量)。在一个实施方案中,CDK4/6抑制剂能够增加G1期CDK4/6依赖性细胞的百分比,同时降低G2/M期和S期CDK4/6依赖性细胞的百分比。在一个实施方案中,化合物在CDK4/6依赖性细胞中诱发基本上纯(即“彻底”)G1细胞周期停滞(例如其中化合物的治疗诱发细胞周期停滞,使得如标准方法(例如碘化丙锭(PI)染色或其它)所定义,大部分细胞停滞在G1,其中组合的G2/M和S期细胞的群体不到总细胞群体的约30%、约25%、约20%、约15%、约10%、约5%、约3%或更少。评估细胞群体的细胞分裂期的方法为本领域中已知(参见例如美国专利申请公布No.2002/0224522)并包括血细胞计数分析、微观分析、梯度离心、淘洗、荧光技术(包括免疫荧光)和其组合。血细胞计数技术包括将细胞暴露于标记试剂或染色剂,例如DNA结合染料,例如PI,并通过流动式细胞测量术分析细胞DNA含量。免疫荧光技术包括用荧光抗体检测特定细胞周期指示物,例如胸苷类似物(例如5-溴-2-脱氧尿苷(BrdU)或碘代脱氧尿嘧啶核苷)。
在一些实施方案中,使用本文中描述的化合物使得尤其与除CDK4和或CDK6以外的激酶(例如CDK2)抑制相关的脱靶效应降低或基本上没有,因为本文中描述的化合物是CDK2的不良抑制剂(例如>1μM IC50)。此外,由于CDK4/6的高选择性,使用本文中描述的化合物不应诱发CDK4/6非依赖性细胞的细胞周期停滞。另外,由于G1停滞作用的短暂瞬时性质,故CDK4/6复制依赖性健康细胞比使用PD0332991相对更快速地再进入细胞周期,使得在一个实施方案中,在长期治疗方案期间由于HSPC能够在化学治疗剂治疗之间复制而降低血液不足的风险。
在本发明的一个方面,本文公开的化合物可以有利地与需要放射疗法、化学疗法或其它治疗剂的任何治疗方案组合施用。在其它实施方案中,本文公开的化合物可以有利地与靶向自体免疫病症的治疗剂组合施用。
药物缀合物
在一个实施方案中,达成本文中描述的目的的活性化合物的活性可以通过缀合于靶向患病或异常增殖细胞或以其它方式增强活性、递送、药物动力学或其它有益特性的剂来加强。
例如,化合物可以呈抗体-药物缀合物(ADC)施用。在某些实施方案中,本文中描述的所选化合物可以与抗体或抗体片段缀合或组合施用。抗体的片段可以通过化学或遗传机制产生。抗体片段可以是抗原结合片段。例如,抗原结合片段可以选自Fab、Fab'、(Fab')2或Fv。抗体片段可以是Fab。单价F(ab)片段具有一个抗原结合位点。抗体可以是二价(Fab’)2片段,其具有由二硫键连接的两个抗原结合区。在一个实施方案中,抗原片段是(Fab’)。F(ab')2片段的还原产生两个单价Fab'片段,其具有适用于缀合于其它分子的游离硫氢基。
本文中描述的所选化合物可以与Fv片段缀合或组合施用。Fv片段是由IgG和IgM类别抗体的酶裂解制成的最小片段。Fv片段具有由VH和VC区制成的抗原结合位点,但其缺乏CH1和CL区。VH和VL链通过非共价相互作用结合在Fv片段中。
在一个实施方案中,如本文中描述的所选化合物可以与选自ScFv、域抗体、双功能抗体、三功能抗体、四功能抗体、双scFv、微型抗体、Fab2或Fab3抗体片段的抗体片段组合。在一个实施方案中,抗体片段是ScFv。基因工程方法允许产生单链可变片段(ScFv),其是包括用柔性肽连接的VH和VL结构域的Fv类型片段。当连接子是至少12个残基长时,ScFv片段主要是单体。V-结构域的取向和连接子长度的操纵产生3-11个残基长的各种Fv分子连接子,产生不会折叠到功能性Fv结构域中的scFv分子。这些分子可以与第二scFv分子缔合,产生二价双功能抗体。在一个实施方案中,与本文中描述的所选化合物组合施用的抗体片段是二价双功能抗体。如果连接子长度小于三个残基,那么scFv分子缔合成三功能抗体或四功能抗体。在一个实施方案中,抗体片段是三功能抗体。在一个实施方案中,抗体片段是四功能抗体。通过再多缀合于两个标靶抗原,减少抗体片段的解离速率,多价scFv对其标靶抗原的功能性结合亲和力比其单价对应物大。在一个实施方案中,抗体片段为微型抗体。微型抗体是装配成二价二聚体的scFv-CH3融合蛋白。在一个实施方案中,抗体片段是双scFv片段。双scFv片段是双特异性的。可以产生具有两个不同的可变域,从而允许这些双scFv分子同时结合于两个不同的抗原决定基的小型化ScFv片段。
在一个实施方案中,本文中描述的所选化合物与双特异性二聚体(Fab2)或三特异性二聚体(Fab3)缀合或组合施用。基因方法也用于产生双特异性Fab二聚体(Fab2)和三特异性Fab三聚体(Fab3)。这些抗体片段能够同时结合2个(Fab2)或3个(Fab3)不同抗原。
在一个实施方案中,本文中描述的所选化合物可以与rIgG抗体片段缀合或组合施用。rIgG抗体片段是指还原的IgG(75,000道尔顿)或一半IgG。其是仅仅选择性地还原铰链区二硫键的产物。虽然若干二硫键存在于IgG,但铰链区中那些二硫键最易接近和容易还原,尤其是用弱还原剂,如2-巯基乙胺(2-MEA)。一半IgG经常是为了靶向可以靶向以用于缀合、抗体固定或酶标记的暴露的铰链区硫氢基而制备。
在其它实施方案中,本文中描述的所选活性化合物可以使用本领域中众所周知的方法连接于放射性同位素以增加功效。适用于Rb阳性癌细胞的任何放射性同位素可以掺入缀合物中,例如(但不限于)131I、123I、192Ir、32P、90Sr、198Au、226Ra、90Y、241Am、252Cf、60Co或137Cs。
值得注意地,连接子化学可能对药物缀合物的功效和可耐受性来说是重要。硫醚连接的T-DM1相对于二硫化物连接子形式增加血清稳定性,且似乎进行内涵体降解,引起细胞毒性剂的细胞内释放,由此改善功效和可耐受性。参见Barginear,M.F.和Budman,D.R.,Trastuzumab-DM1:A review of the novel immune-conjugate for HER2-overexpressing breast cancer,The Open Breast Cancer Journal,1:25-30,2009。
可以用于本发明的用于产品研发的早期和近来的抗体-药物缀合物、论述药物、连接子化学和标靶类别的实例可以见于以下评述中:Casi,G.和Neri,D.,Antibody-drug conjugates:basic concepts,examples and future perspectives,J.Control Release 161(2):422-428,2012;Chari,R.V.,Targeted cancer therapy:conferring specificity to cytotoxic drugs,Acc.Chem.Rev.,41(1):98-107,2008;Sapra,P.和Shor,B.,Monoclonal antibody-based therapies in cancer:advances and challenges,Pharmacol.Ther.,138(3):452-69,2013;Schliemann,C.和Neri,D.,Antibody-based targeting of the tumor vasculature,Biochim.Biophys.Acta.,1776(2):175-92,2007;Sun,Y.,Yu,F.和Sun,B.W.,Antibody-drug conjugates as targeted cancer therapeutics,Yao Xue Xue Bao,44(9):943-52,2009;Teicher,B.A.和Chari,R.V.,Antibody conjugate therapeutics:challenges and potential,Clin.Cancer Res.,17(20):6389-97,2011;Firer,M.A.和Gellerman,G.J.,Targeted drug delivery for cancer therapy:the other side of antibodies,J.Hematol.Oncol.,5:70,2012;Vlachakis,D.和Kossida,S.,Antibody Drug Conjugate bioinformatics:drug delivery through the letterbox,Comput.Math.Methods Med.,2013;2013:282398,Epub 2013年6月19日;Lambert,J.M.,Drug-conjugated antibodies for the treatment of cancer,Br.J.Clin.Pharmacol.,76(2):248-62,2013;Concalves,A.,Tredan,O.,Villanueva,C.和Dumontet,C.,Antibody-drug conjugates in oncology:from the concept to trastuzumab emtansine(T-DM1),Bull.Cancer,99(12):1183-1191,2012;Newland,A.M.,Brentuximab vedotin:a CD-30-directed antibody-cytotoxic drug conjugate,Pharmacotherapy,33(1):93-104,2013;Lopus,M.,Antibody-DM1 conjugates as cancer therapeutics,Cancer Lett.,307(2):113-118,2011;Chu,Y.W.和Poison,A.,Antibody-drug conjugates for the treatment of B-cell non-Hodgkin’s lymphoma and leukemia,Future Oncol.,9(3):355-368,2013;Bertholjotti,I.,Antibody-drug conjugate--a new age for personalized cancer treatment,Chimia,65(9):746-748,2011;Vincent,K.J.和Zurini,M.,Current strategies in antibody engineering:Fc engineering and pH–dependent antigen binding,bispecific antibodies and antibody drug conjugates,Biotechnol.J.,7(12):1444-1450,2012;Haeuw,J.F.,Caussanel,V.和Beck,A.,Immunoconjugates,drug-armed antibodies to fight against cancer,Med.Sci.,25(12):1046-1052,2009;以及Govindan,S.V.和Goldenberg,D.M.,Designing immunoconjugates for cancer therapy,Expert Opin.Biol.Ther.,12(7):873-890,2012。
药物组合物和剂型
如本文所述的活性化合物或其盐、同位素类似物或前药可以使用实现所需治疗结果的任何适合的方法以有效量施用于主体。施用的活性化合物的量和时间安排当然将取决于治疗的主体、监督专科医师的指导、暴露时程、施用方式、具体活性化合物的药物动力学特性和主治医师的判断。因此,由于主体与主体之间的变化性,以下给出的剂量是指导且医师可以调整化合物的剂量以实现医师认为适合于主体的治疗。在考虑所需治疗程度时,医师可以平衡多种因素,例如主体的年龄和重量、先前存在的疾病的存在以及其它疾病的存在。药物制剂可以制备用于任何所需的施用途径,包括(但不限于)经口、静脉内或气溶胶施用,如以下更详细地论述。
本文中描述的任何活性化合物的治疗有效剂量将由保健执业医师,依赖于患者的状况、体型和年龄以及递送途径确定。在一个非限制性实施方案中,约0.1至约200mg/kg的剂量具有治疗功效,其中所有重量都是基于活性化合物的重量计算,包括采用盐的情况。在一些实施方案中,剂量可以是提供至多约1与5、10、20、30或40μM之间的活性化合物的血清浓度所需的化合物量。在一些实施方案中,约10mg/kg至约50mg/kg的剂量可以用于经口施用。典型地,约0.5mg/kg至5mg/kg的剂量可以用于肌肉内注射。在一些实施方案中,剂量可以为约1μmol/kg至约50μmol/kg,或任选地,约22μmol/kg与约33μmol/kg之间的化合物,用于静脉内或经口施用。口服剂型可以包括任何适当量的活性物质,每个片剂或其它固体剂型包括5mg至50、100、200或500mg。
根据本发明公开的方法,如本文中描述的药学活性化合物可以呈固体形式或呈液体形式经口施用,或可以呈溶液、混悬液或乳液形式肌肉内、静脉内或通过吸入施用。在一些实施方案中,化合物或盐也也可以通过呈脂质体混悬液形式吸入、静脉内或肌肉内施用。当通过吸入施用时,活性化合物或盐可以呈具有任何所需粒度,且例如约0.01、0.1或0.5至约5、10、20或更多微米且任选地约1至约2微米的多个固体颗粒或小滴形式。如本发明公开的化合物已经证明例如当通过经口或静脉内途径施用时优良的药物动力学和药效学特性。
药物制剂可以包含本文中描述的活性化合物或其药学上可接受的盐于任何药学上可接受的载体中。如果想要溶液,那么水可以是精选用于水溶性化合物或盐的载体。关于水溶性化合物或盐,例如甘油、丙二醇、聚乙二醇或其混合物等有机媒介物可以是适合的。在后一情况下,有机媒介物可以含有相当大量的水。接着任一情况下的溶液可以用本领域的技术人员已知的适合方式杀菌,且为了说明,通过0.22微孔过滤器过滤杀菌。在杀菌之后,溶液可以分配至适当容器,例如去除热原的玻璃小瓶。分配任选地通过无菌方法进行。接着可以将杀菌盖板放于小瓶上且必要时可以冻干小瓶内容物。
除活性化合物或其盐之外,药物制剂可以含有其它添加剂,例如pH值调整添加剂。具体地说,适用pH值调整试剂包括例如盐酸等酸、碱或缓冲剂,例如乳酸钠、乙酸钠、磷酸钠、柠檬酸钠、硼酸钠或葡糖酸钠。此外,制剂可以含有抗微生物防腐剂。适用抗微生物防腐剂包括对羟基苯甲酸甲酯、对羟基苯甲酸丙酯和苯甲醇。当制剂放于打算多剂量使用的小瓶时典型地采用抗微生物防腐剂。本文中描述的药物制剂可以使用本领域中众所周知的技术冻干。
为经口施用,药物组合物可以采取溶液、混悬液、片剂、丸剂、胶囊、粉末等形式。可以采用含有例如柠檬酸钠、碳酸钙和磷酸钙等各种赋形剂以及例如淀粉(例如马铃薯或木薯淀粉)和某些复合硅酸盐等各种崩解剂以及例如聚乙烯吡咯烷酮、蔗糖、明胶和阿拉伯胶等粘合剂的片剂。另外,例如硬脂酸镁、十二烷基硫酸钠和滑石等润滑剂常常非常适用于达成压片目的。可以采用类似类型的固体组合物作为软和硬填充明胶胶囊中的填料。在这方面的物质还包括乳糖或乳糖以及高分子量聚乙二醇。当希望水性混悬液和/或酏剂经口施用时,本发明公开的主题的化合物可以与各种甜味剂、调味剂、着色剂、乳化剂和/或悬浮剂以及例如水、乙醇、丙二醇、甘油等稀释剂和其各种类似组合进行组合。
在本文中描述的主题的又一个实施方案中,提供一种可注射的稳定无菌制剂,其包含如本文中描述的活性化合物或其盐,该制剂呈单位剂型于密封容器中。化合物或盐以冻干物形式提供,其能够用适合的药学上可接受的载体复原以形成适于其注射至主体的液体制剂。当化合物或盐基本上不溶于水时,可以采用足以使化合物或盐在水性载体中乳化的量的足够量的在生理学上可接受的乳化剂。尤其适用的乳化剂包括磷脂酰胆碱和卵磷脂。
本文中提供的其它实施方案包括本文公开的活性化合物的脂质体制剂。用于形成脂质体混悬液的技术为本领域中众所周知。当化合物是水溶性盐时使用常规的脂质体技术,可以将其并入脂质小泡中。在此类情况下,由于活性化合物的水溶性,活性化合物可以基本上夹在脂质体的亲水性中心或核心内。所用脂质层可以具有任何常规的组合物且可以含有胆固醇或者可以不含胆固醇。当相关活性化合物不溶于水时,再次采用常规的脂质体形成技术,盐可以基本上夹在形成脂质体结构的疏水性双层脂质内。在任一情况下,如通过使用标准超声处理和均化技术,可以减小产生的脂质体的尺寸。包含本文公开的活性化合物的脂质体制剂可以冻干以产生冻干物,其可以用例如水等药学上可接受的载体复原,再生脂质体混悬液。
还提供适合于呈气溶胶形式通过吸入施用的药物制剂。这些制剂包含本文中描述的希望化合物或其盐的溶液或混悬液或化合物或盐的多个固体颗粒。希望制剂可以放于小的腔室中并雾化。雾化可以通过压缩空气或通过超声波能实现以形成包含化合物或盐的多个液滴或固体颗粒。液滴或固体颗粒可以例如具有在约0.5至约10微米且任选约0.5至约5微米范围内的粒度。在一个实施方案中,固体颗粒通过使用可降解的聚合物提供控制释放。固体颗粒可以通过用本领域中已知的任何适当方式,例如通过微粉化加工固体化合物或其盐获得。任选地,固体颗粒或小滴的尺寸可以为约1到约2微米。在这方面,商业喷雾器可以用来实现此目的。化合物可以经由可呼吸粒子的气溶胶混悬液用美国专利No.5,628,984中阐述的方式施用,其公开内容以引用的方式整体并入本文中。
还提供药物制剂,其提供本文所述的化合物的控制释放,包括通过使用如本领域中所知的可降解的聚合物。
当适于呈气溶胶形式施用的药物制剂呈液体形式时,制剂可以包含水溶性活性化合物于包含水的载体中。可以存在表面活性剂,其降低制剂的表面张力,当经受雾化时足以形成希望尺寸范围内的小滴。
如本文所用,术语“药学上可接受的盐”是指在合理医学判断范围内,适用于与主体(例如人类主体)接触,无过度毒性、刺激、过敏反应等,与合理益处/风险比相称,且有效用于其预期用途的盐,以及可能时本发明公开的主题的化合物的两性离子形式。
因此,术语“盐”是指本发明公开的化合物的相对无毒的无机和有机酸加成盐。这些盐可以在化合物最终分离和纯化期间原位制备,或通过分开使呈游离碱形式的纯化化合物与适合的有机或无机酸反应且分离由此形成的盐来制备。碱性化合物能够与各种无机和有机酸形成各种不同的盐。碱性化合物的酸加成盐通过以常规的方式使游离碱形式与足够量的所需酸接触以产生盐来制备。游离碱形式可以通过使盐形式与碱接触并以常规的方式分离游离碱来再生。游离碱形式在例如极性溶剂中的溶解性等某些物理特性方面可以不同于其相应的盐形式。药学上可接受的碱加成盐可以用金属或胺,例如碱金属和碱土金属氢氧化物或有机胺形成。用作阳离子的金属的实例包括(但不限于)钠、钾、镁、钙等等。适合胺的实例包括(但不限于)N,N'-二苯甲基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、N-甲基葡糖胺和普鲁卡因。酸性化合物的碱加成盐通过以常规的方式使游离酸形式与足够量的所需碱接触以产生盐来制备。游离酸形式可以通过使盐形式与酸接触并以常规的方式分离游离酸来再生。游离酸形式在例如极性溶剂中的溶解性等某些物理特性方面略微不同于其相应的盐形式。
盐可以由无机酸硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、硝酸盐、磷酸盐、磷酸一氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物(例如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、磷酸等等)。代表性盐包括氢溴化物、盐酸盐、硫酸盐、硫酸氢盐、硝酸盐、乙酸盐、草酸盐、戊酸盐、油酸盐、棕榈酸盐、硬脂酸酯、月桂酸盐、硼酸盐、苯甲酸盐、乳酸盐、磷酸盐、甲苯磺酸盐、柠檬酸盐、顺丁烯二酸盐、反丁烯二酸盐、丁二酸盐、酒石酸盐、萘甲酸盐、甲磺酸盐、葡糖庚酸盐、乳糖酸盐、十二烷基硫酸盐和羟乙基磺酸盐等等。盐还可以从有机酸制备,例如脂肪族单羧酸和二羧酸、经苯基取代的链烷酸、羟基链烷酸、链烷二羧酸、芳香族酸、脂肪族和芳香族磺酸等等。代表性盐包括乙酸盐、丙酸盐、辛酸盐、异丁酸盐、草酸盐、丙二酸盐、丁二酸盐、辛二酸盐、癸二酸盐、反丁烯二酸盐、顺丁烯二酸盐、扁桃酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、邻苯二甲酸盐、苯磺酸盐、甲苯磺酸盐、苯乙酸盐、柠檬酸盐、乳酸盐、顺丁烯二酸盐、酒石酸盐、甲烷磺酸盐等等。药学上可接受的盐可以包括基于碱金属和碱土金属的阳离子,例如钠、锂、钙、镁等等,以及无毒铵、季铵和胺阳离子,包括(但不限于)铵、四甲铵、四乙铵、甲胺、二甲胺、三甲胺、三乙胺、乙胺等等。还涵盖氨基酸的盐,例如精氨酸盐、葡糖酸盐、半乳糖醛酸酯等等。参见例如Berge等人,J.Pharm.Sci.,1977,66,1-19,以引用的方式并入本文中。
制备活性化合物
合成
公开的化合物可以通过以下一般方案制造:
方案1
在方案1中,Ref-1为WO 2010/020675 A1;Ref-2为White,J.D.等人J.Org.Chem.1995,60,3600;并且Ref-3为Presser,A.和Hufner,A.Monatshefte für Chemie 2004,135,1015。
方案2
在方案2中,Ref-1为WO 2010/020675 A1;Ref-4为WO 2005/040166 A1;且Ref-5为Schoenauer,K和Zbiral,E.Tetrahedron Letters 1983,24,573。
方案3
在方案3中,Ref-1为WO 2010/020675 A1。
方案4
方案5
方案6
方案7
方案8
在方案8中,Ref-1为WO 2010/020675 A1;Ref-2为WO 2005/040166 A1;且Ref-3为Schoenauer,K和Zbiral,E.Tetrahedron Letters 1983,24,573。
或者,内酰胺可以通过羧酸与保护胺在强酸和脱水剂存在下反应产生,强酸和脱水剂可以一起在一个部分中作为强酸酸酐。强酸酸酐的实例包括(但不限于)三氟乙酸酐、三溴乙酸酐、三氯乙酸酐或混合酸酐。脱水剂可以是基于碳化二亚胺的化合物,例如(但不限于)DCC(N,N-二环己基碳化二亚胺)、EDC(1-乙基-3-(3-二甲基氨基丙基)碳化二亚胺或DIC(N,N-二异丙基碳化二亚胺)。可能需要额外的步骤来去掉N-保护基且方法为本领域的技术人员已知。
或者,键结至嘧啶环的卤素部分可以经可以被伯胺置换的任何离去基团取代,例如产生最终产物的中间体,例如Br、I、F、SMe、SO2Me、SO烷基、SO2烷基。参见例如Tavares的PCT/US2013/037878。
其它胺中间体和最终胺化合物可以通过本领域的技术人员合成。应了解在本发明的时候所述化学可以采用包含可以保护和脱保护的反应官能团并为本领域的技术人员已知的试剂。参见例如Greene,T.W.和Wuts,P.G.M.,Greene’s Protective Groups in Organic Synthesis,第4版,John Wiley and Sons。
方案9
本发明的CDK4/6抑制剂可以根据通用方案9合成。经取代的2-氨基嘧啶的特定合成和表征可见于例如WO2012/061156。
化合物T、Q、GG和U如上制备并通过质谱分析和NMR表征,如下所示:
化合物T
1H NMR(600MHz,DMSO-d6)δppm 1.47(br.s.,6H)1.72(br.s.,2H)1.92(br.s.,2H)2.77(br.s.,3H)3.18(br.s.,2H)3.46(br.s.,2H)3.63(br.s.,2H)3.66(d,J=6.15Hz,2H)3.80(br.s.,2H)7.25(s,1H)7.63(br.s.,2H)7.94(br.s.,1H)8.10(br.s.,1H)8.39(br.s.,1H)9.08(br.s.,1H)11.59(br.s.,1H)。LCMS ESI(M+H)447。
化合物Q
1H NMR(600MHz,DMSO-d6)δppm 0.82(d,J=7.32Hz,2H)1.08-1.37(m,3H)1.38-1.64(m,2H)1.71(br.s.,1H)1.91(br.s.,1H)2.80(br.s.,1H)3.12(s,1H)3.41(br.s.,4H)3.65(br.s.,4H)4.09(br.s.,1H)7.26(s,1H)7.52-7.74(m,2H)7.94(br.s.,1H)8.13(br.s.,1H)8.40(br.s.,1H)9.09(br.s.,1H)9.62(br.s.,1H)11.71(br.s.,1H)。LCMS ESI(M+H)433。
化合物GG
1H NMR(600MHz,DMSO-d6)δppm 0.85(br.s.,1H)1.17-1.39(m,7H)1.42-1.58(m,2H)1.67-1.84(m,3H)1.88-2.02(m,1H)2.76-2.93(m,1H)3.07-3.22(m,1H)3.29-3.39(m,1H)3.41-3.61(m,4H)3.62-3.76(m,4H)3.78-3.88(m,1H)4.12(br.s.,1H)7.28(s,1H)7.60-7.76(m,2H)7.98(s,1H)8.13(br.s.,1H)8.41(s,1H)9.10(br.s.,1H)11.21(br.s.,1H)11.54(s,1H)。LCMS ESI(M+H)475。
化合物U
1H NMR(600MHz,DMSO-d6)δppm 0.84(t,J=7.61Hz,2H)1.13-1.39(m,4H)1.46(d,J=14.05Hz,2H)1.64-1.99(m,6H)2.21(br.s.,1H)2.66-2.89(m,2H)3.06(br.s.,1H)3.24-3.36(m,1H)3.37-3.50(m,2H)3.56-3.72(m,2H)3.77-4.00(m,4H)4.02-4.19(m,2H)7.25(s,1H)7.50-7.75(m,2H)7.89(d,J=2.93Hz,1H)8.14(d,J=7.32Hz,1H)8.38(br.s.,1H)9.06(s,1H)11.53(br.s.,1H)。LCMS ESI(M+H)517。
实施例
中间体B、E、K、L、1A、1F和1CA根据Tavares,F.X.和Strum,J.C.的标题为CDK Inhibitors的US 8,598,186合成。
Tavares,F.X.的标题为Lactam Kinase Inhibitors的专利WO 2013/148748、Tavares,F.X.的标题为Synthesis of Lactams的WO 2013/163239和Tavares,F.X.和Strum,J.C.的标题为CDK Inhibitors的US 8,598,186以引用的方式整体并入本文中。
实施例1
合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯,化合物1
向5-溴-2,4-二氯嘧啶(3.2g,0.0135mol)于乙醇(80mL)中的溶液中添加亨尼格碱(Hunig’s base)(3.0mL),接着添加N-(叔丁氧羰基)-1,2-二氨基乙烷(2.5g,0.0156摩尔)于乙醇(20mL)中的溶液。将内容物搅拌过夜,历时20小时。在真空下蒸发溶剂。添加乙酸乙酯(200mL)和水(100mL)且分离各层。有机层经硫酸镁干燥并接着真空浓缩。使用己烷/乙酸乙酯(0-60%)的硅胶柱色谱法得到N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯。1HNMR(d6-DMSO)δppm 8.21(s,1H),7.62(brs,1H),7.27(brs,1H),3.39(m,2H),3.12(m,2H),1.34(s,9H)。LCMS(ESI)351(M+H)。
实施例2
合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯,化合物2
向含N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯(1.265g,3.6mmol)的THF(10mL)中添加缩醛(0.778mL,5.43mmol)、Pd(dppf)CH2Cl2(148mg)和三乙胺(0.757mL,5.43mmol)。将内容物脱气并接着用氮气净化。接着向其中添加CuI(29mg)。将反应混合物在回流下加热48小时。冷却后,内容物经CELITETM过滤并浓缩。使用己烷/乙酸乙酯(0-30%),所得残余物进行柱色谱法,得到N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯。1HNMR(d6-DMSO)δppm 8.18(s,1H),7.63(brs,1H),7.40(brs,1H),5.55(s,1H),3.70(m,2H),3.60(m,2H),3.42(m,2H),3.15(m,2H),1.19-1.16(m,15H)。LCMS(ESI)399(M+H)。
实施例3
合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯,化合物3
向偶合产物(2.1g,0.00526摩尔)于THF(30mL)中的溶液中添加TBAF固体(7.0g)。将内容物加热至并保持在65℃,历时2小时。浓缩,接着使用乙酸乙酯/己烷(0-50%)进行柱色谱法,得到呈浅棕色液体状的N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(1.1g)。1HNMR(d6-DMSO)δppm 8.88(s,1H),6.95(brs,1H),6.69(s,1H),5.79(s,1H),4.29(m,2H),3.59(m,4H),3.34(m,1H),3.18(m,1H),1.19(m,9H),1.17(m,6H)。LCMS(ESI)399(M+H)。
实施例4
合成N-[2-(2-氯-6-甲酰基-吡咯并[2,3-d]嘧啶-7-基)乙基]氨基甲酸叔丁酯,化合物4
向来自前一步骤的缩醛(900mg)中添加AcOH(8.0mL)和水(1.0mL)。将反应物在室温下搅拌16小时。浓缩并使用乙酸乙酯/己烷(0-60%),进行硅胶柱色谱法,得到呈泡沫状的N-[2-(2-氯-6-甲酰基-吡咯并[2,3-d]嘧啶-7-基)乙基]氨基甲酸叔丁酯(0.510g)。1HNMR(d6-DMSO)δppm 9.98(s,1H),9.18(s,1H),7.66(s,1H),6.80(brs,1H),4.52(m,2H),4.36(m,2H),1.14(s,9H)。LCMS(ESI)325(M+H)。
实施例5
合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸,化合物5
向含来自前一步骤的醛(0.940g)的DMF(4mL中)添加过硫酸氢钾(1.95g,1.1eq)。将内容物在室温下搅拌7小时。使用己烷/乙酸乙酯(0-100%)进行硅胶柱色谱法,得到7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸(0.545g)。1HNMR(d6-DMSO)δppm 9.11(s,1H),7.39(s,1H),4.38(m,2H),4.15(m,2H),1.48(m,9H)。LCMS(ESI)341(M+H)。
实施例6
合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸甲酯,化合物6
向来自前一步骤的2-氯-7-丙基-吡咯并[2,3-d]嘧啶-6-甲酸(0.545g,0.00156摩尔)于甲苯(3.5mL)和MeOH(1mL)中的溶液中添加TMS-重氮甲烷(1.2mL)。在室温下搅拌过夜后,将过量TMS-重氮甲烷用乙酸(3mL)淬灭且真空浓缩反应物。残余物通过硅胶柱色谱法,使用己烷/乙酸乙酯(0-70%)来纯化,得到呈灰白色固体状的7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸甲酯(0.52g)。1HNMR(d6-DMSO)δppm 9.10(s,1H),7.45(s,1H),6.81(brs,1H)4.60(m,2H),3.91(s,3H),3.29(m,2H),1.18(m,9H)LCMS(ESI)355(M+H)。
实施例7
合成氯三环状酰胺,化合物7
向含来自前一步骤的7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸甲酯(0.50g,0.0014摩尔)的二氯甲烷(2.0mL)中添加TFA(0.830mL)。将内容物在室温下搅拌1小时。真空浓缩得到粗氨基酯,其悬浮于甲苯(5mL)和亨尼格碱(0.5mL)中。将内容物在回流下加热2小时。浓缩,接着使用己烷/乙酸乙酯(0-50%)进行硅胶柱色谱法,得到所需氯三环状酰胺(0.260g)。1HNMR(d6-DMSO)δppm 9.08(s,1H),8.48(brs,1H),7.21(s,1H)4.33(m,2H),3.64(m,2H)。LCMS(ESI)223(M+H)。
实施例8
合成氯-N-甲基三环状酰胺,化合物8
向氯三环状内酰胺化合物7(185mg,0.00083摩尔)于DMF(2.0mL)中的溶液中添加氢化钠(55%于油中的悬浮液,52mg)。搅拌15分钟后,碘甲烷(62μL,1.2eq)。将内容物在室温下搅拌30分钟。添加甲醇(5mL)后,添加饱和NaHCO3,接着添加乙酸乙酯。分离有机层,接着经硫酸镁干燥并真空浓缩,得到N-甲基化酰胺,产率定量。1HNMR(d6-DMSO)δppm 9.05(s,1H),7.17(s,1H)4.38(m,2H),3.80(m,2H),3.05(s,3H)。LCMS(ESI)237(M+H)。
实施例9
合成1-甲基-4-(6-硝基-3-吡啶基)哌嗪,化合物9
向含5-溴-2-硝基吡啶(4.93g,24.3毫摩尔)的DMF(20mL)中添加N-甲基哌嗪(2.96g,1.1eq),接着添加DIPEA(4.65mL,26.7毫摩尔)。将内容物在90℃下加热24小时。添加乙酸乙酯(200mL)后,添加水(100mL)且分离各层。干燥,接着浓缩得到粗产物,通过硅胶柱色谱法,使用(0-10%)DCM/甲醇来纯化。1HNMR(d6-DMSO)δppm 8.26(s,1H),8.15(1H,d,J=9.3Hz),7.49(1H,d,J=9.4Hz),3.50(m,4H),2.49(m,4H),2.22(s,3H)。
实施例10
合成5-(4-甲基哌嗪-1-基)吡啶-2-胺,化合物10
向含1-甲基-4-(6-硝基-3-吡啶基)哌嗪(3.4g)的乙酸乙酯(100mL)和乙醇(100mL)中添加10%Pd/C(400mg)并接着将反应物在氢气(10psi)下搅拌过夜。经CELITETM过滤后,蒸发溶剂且粗产物通过硅胶柱色谱法,使用DCM/7N氨的MeOH溶液(0-5%)来纯化,得到5-(4-甲基哌嗪-1-基)吡啶-2-胺(2.2g)。1HNMR(d6-DMSO)δppm 7.56(1H,d,J=3Hz),7.13(1H,m),6.36(1H,d,J=8.8Hz),5.33(brs,2H),2.88(m,4H),2.47(m,4H),2.16(s,3H)。
实施例11
合成4-(6-氨基-3-吡啶基)哌嗪-1-甲酸叔丁酯,化合物11
此化合物如WO 2010/020675 A1中所述来制备。
实施例12
合成N-[2-(苯甲氧基羰基氨基)-3-甲基-丁基]氨基甲酸叔丁酯,化合物12
向冷却至0℃的含N-[1-(羟基甲基)-2-甲基-丙基]氨基甲酸苯甲酯(11.0g,0.0464摩尔)的二噁烷(100mL)中添加叠氮磷酸二苯酯(10.99mL,1.1eq),接着添加DBU(8.32mL,1.2eq)。使内容物升温至室温并搅拌16小时。添加乙酸乙酯(300mL)和水(100mL)后,将有机层分离并用饱和NaHCO3(100mL)洗涤。接着干燥有机层(硫酸镁)并真空浓缩。向含此中间体的DMSO(100mL)中添加叠氮化钠(7.54g)且接着将内容物加热至90℃,保持2小时。添加乙酸乙酯和水后分离各层。有机层经硫酸镁干燥,接着真空浓缩,得到油状物,通过硅胶柱色谱法,使用己烷/乙酸乙酯(0-70%)来纯化,得到6.9g呈无色油状的N-[1-(叠氮基甲基)-2-甲基-丙基]氨基甲酸苯甲酯。
向含N-[1-(叠氮基甲基)-2-甲基-丙基]氨基甲酸苯甲酯(6.9g,0.0263摩尔)的THF(100mL)中添加三苯基膦(7.59g,1.1eq)。将内容物搅拌20小时。添加水(10mL)并再搅拌6小时后,添加乙酸乙酯且分离各层。经硫酸镁干燥并真空浓缩后,粗产物通过硅胶柱色谱法,使用DCM/MeOH(0-10%)来纯化,得到呈黄色油状的N-[1-(氨基甲基)-2-甲基-丙基]氨基甲酸苯甲酯。
向含N-[1-(氨基甲基)-2-甲基-丙基]氨基甲酸苯甲酯(4.65g,0.019摩尔)的THF(70mL)中添加2N NaOH(20mL),接着添加二碳酸二叔丁酯(5.15g,1.2eq)。搅拌16小时后,添加乙酸乙酯且分离各层。经硫酸镁干燥并真空浓缩后,粗产物使用己烷/乙酸乙酯(0-40%),经硅胶柱来纯化,得到中间体A,N-[2-(苯甲氧基羰基氨基)-3-甲基-丁基]氨基甲酸叔丁酯(6.1g)。1HNMR(600MHz,氯仿-d)δppm 0.89(d,J=6.73Hz,3H)0.92(d,J=6.73Hz,3H)1.38(s,9H)1.70-1.81(m,1H)3.18(d,J=5.56Hz,2H)3.47-3.60(m,1H)4.76(s,1H)4.89(d,J=7.90Hz,1H)5.07(s,2H)7.25-7.36(m,5H)。LCMS(ESI)337(M+H)。
实施例13
合成N-[2-(苯甲氧基羰基氨基)-4-甲基-戊基]氨基甲酸叔丁酯,化合物13
在0℃下向N-[1-(羟基甲基)-3-甲基-丁基]氨基甲酸苯甲酯(6.3g,0.025摩尔)于DCM(100mL)中的溶液中添加二异丙基乙胺(5.25mL,1.2eq),接着添加甲烷磺酰氯(2.13mL,1.1eq)。搅拌3小时后,添加水(100mL)且分离有机层。经硫酸镁干燥并真空浓缩后,得到粗[2-(苯甲氧基羰基氨基)-4-甲基-戊基]甲烷磺酸酯,其直接用于下一步。
向含来自以上反应的粗[2-(苯甲氧基羰基氨基)-4-甲基-戊基]甲烷磺酸酯的DMF(50mL)中添加2.43g叠氮化钠。接着将反应混合物加热至85℃,保持3小时。冷却后,添加乙酸乙酯(300mL)和水。将有机层分离,经硫酸镁干燥并接着真空浓缩,得到粗N-[1-(叠氮基甲基)-3-甲基-丁基]氨基甲酸苯甲酯。向此粗中间体中添加THF(100mL),接着添加7.21g三苯基膦并在氮气下搅拌16小时。添加水(10mL)并再搅拌6小时后,添加乙酸乙酯且分离各层。经硫酸镁干燥并真空浓缩后,粗产物使用DCM/MeOH(0-10%)进行柱分离,得到N-[1-(氨基甲基)-3-甲基-丁基]氨基甲酸苯甲酯(4.5g)。
向含N-[1-(氨基甲基)-3-甲基-丁基]氨基甲酸苯甲酯(4.5g,0.018摩尔)的THF(60mL)中添加2N NaOH(18mL),接着添加二碳酸二叔丁酯(4.19g,1.07eq)。搅拌16小时后,添加乙酸乙酯且分离各层。经硫酸镁干燥并真空浓缩后,粗产物用于下一步。1HNMR(600MHz,氯仿-d)δppm 0.89(d,J=6.73Hz,6H)1.25-1.34(m,1H)1.39(s,9H)1.57-1.71(m,2H)3.04-3.26(m,2H)3.68-3.80(m,1H)4.72-4.89(m,2H)5.06(s,2H)7.25-7.38(m,5H)。LCMS(ESI)351(M+H)。
实施例14
合成N-[(2R)-2-(苯甲氧基羰基氨基)-3-甲基-丁基]氨基甲酸叔丁酯,化合物14
化合物14由N-[(1R)-1-(羟基甲基)-2-甲基-丙基]氨基甲酸苯甲酯,使用与针对化合物13所述类似的合成步骤合成。分析数据(NMR和质谱)与化合物12一致。
实施例15
合成N-[(2S)-2-(苯甲氧基羰基氨基)-3-甲基-丁基]氨基甲酸叔丁酯,化合物15
化合物15由N-[(1S)-1-(羟基甲基)-2-甲基-丙基]氨基甲酸苯甲酯,使用与针对化合物13所述类似的合成步骤合成。分析数据(NMR和质谱)与化合物12一致。
实施例16
合成N-[(1S)-1-(氨基甲基)-2-甲基-丙基]氨基甲酸叔丁酯,化合物16
向N-[(1S)-1-(羟基甲基)-2-甲基-丙基]氨基甲酸酯氨基甲酸叔丁酯(6.3g,0.025摩尔)于THF(100mL)中的溶液中添加二异丙基乙胺(5.25mL,1.2eq),接着在0℃下添加甲烷磺酰氯(2.13mL,1.1eq)。搅拌3小时后,添加水(100mL)且分离有机层。经硫酸镁干燥并真空浓缩后,粗[(2S)-2-(叔丁氧羰基氨基)-3-甲基-丁基]甲烷磺酸酯直接用于下一步。
向含来自以上反应的粗[(2S)-2-(叔丁氧羰基氨基)-3-甲基-丁基]甲烷磺酸酯的DMSO(50mL)中添加叠氮化钠(2.43g)。接着将反应混合物加热至85℃,保持3小时。冷却后,添加乙酸乙酯(300mL)和水。将有机层分离,经硫酸镁干燥并接着真空浓缩,得到粗N-[1-(叠氮基甲基)-3-甲基-丁基]氨基甲酸苯甲酯。向此粗中间体中添加THF(100mL),接着添加三苯基膦(7.21g)并将反应物在氮气下搅拌16小时。添加水(10mL)并再搅拌6小时后,添加乙酸乙酯且分离各层。经硫酸镁干燥并真空浓缩后,粗产物通过硅胶柱色谱法,使用DCM/MeOH(0-10%)来纯化,得到N-[1-(氨基甲基)-3-甲基-丁基]氨基甲酸苯甲酯(4.5g)。LCMS(ESI)203(M+H)。
实施例17
合成N-[(1R)-1-(氨基甲基)-2-甲基-丙基]氨基甲酸叔丁酯,化合物17
化合物17由N-[(1R)-1-(羟基甲基)-2-甲基-丙基]氨基甲酸叔丁酯,使用与针对化合物16所述类似的合成顺序合成。分析数据(NMR和质谱)与化合物16一致。
实施例18
合成N-[(2S)-2-(苯甲氧基羰基氨基)-4-甲基-戊基]氨基甲酸叔丁酯,化合物18
化合物18由N-[(1S)-1-(羟基甲基)-3-甲基-丁基]氨基甲酸苯甲酯,使用与针对化合物13所述类似的合成顺序合成。分析数据(NMR和质谱)与化合物13一致。
实施例19
合成N-[(2S)-2-(苯甲氧基羰基氨基)-2-苯基-乙基]氨基甲酸叔丁酯,化合物19
化合物19由N-[(1S)-2-羟基-1-苯基-乙基]氨基甲酸苯甲酯,使用与针对化合物13所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.20-1.33(m,9H)3.11(t,J=6.29Hz,2H)4.59-4.68(m,1H)4.88-5.01(m,2H)6.81(t,J=5.42Hz,1H)7.14-7.35(m,10H)7.69(d,J=8.49Hz,1H)。LCMS(ESI)371(M+H)。
实施例20
合成N-[(2S)-2-(苯甲氧基羰基氨基)-3-甲基-戊基]氨基甲酸叔丁酯,化合物20
化合物20由N-[(1S)-1-(羟基甲基)-2-甲基-丁基]氨基甲酸苯甲酯,使用与针对化合物13所述类似的合成顺序合成。1HNMR(600MHz,氯仿-d)δppm 0.85-0.92(m,6H)1.05-1.15(m,1H)1.35-1.41(m,9H)1.45-1.56(m,2H)3.14-3.24(m,2H)3.54-3.64(m,1H)4.78(s,1H)4.96(d,J=7.91Hz,1H)5.06(s,2H)7.27-7.37(m,5H)。LCMS(ESI)351(M+H)。
实施例21
合成N-[(2S)-2-(苯甲氧基羰基氨基)-3,3-二甲基-丁基]氨基甲酸叔丁酯,化合物21
化合物21由N-[(1S)-1-(羟基甲基)-2,2-二甲基-丙基]氨基甲酸苯甲酯,使用与针对化合物13所述类似的合成顺序合成。LCMS(ESI)351。
实施例22
合成N-[[1-(苯甲氧基羰基氨基)环己基]甲基]氨基甲酸叔丁酯,化合物22
向N-[1-(氨基甲基)环己基]氨基甲酸苯甲酯(10.0g,0.0381摩尔)于THF(150mL)中的溶液中添加二碳酸二叔丁酯(9.15g,1.1eq)且将内容物在室温下搅拌16小时。接着添加乙酸乙酯和水。将有机层分离,经硫酸镁干燥并接着真空浓缩,得到N-[[1-(苯甲氧基羰基氨基)环己基]甲基]氨基甲酸叔丁酯(13.1g)。1HNMR(600MHz,DMSO-d6)δppm 0.92-1.54(m,17H)1.76-2.06(m,2H)3.09(d,J=6.15Hz,2H)4.92(s,2H)6.63(d,J=17.27Hz,1H)7.16-7.49(m,6H)。LCMS(ESI)363(M+H)。
实施例23
合成N-[[1-(苯甲氧基羰基氨基)环戊基]甲基]氨基甲酸叔丁酯,化合物23
N-[[1-(苯甲氧基羰基氨基)环戊基]甲基]氨基甲酸叔丁酯以与N-[[1-(苯甲氧基羰基氨基)环己基]甲基]氨基甲酸叔丁酯类似的方式合成。LCMS(ESI)349(M+H)。
实施例24
合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶,化合物24
向含5-溴-2-硝基吡啶(1.2g,5.9mmol)的DMSO(4mL)中添加1-(4-哌啶基)哌啶(1.0g,5.9毫摩尔)和三乙胺(0.99mL,7.1毫摩尔)。将内容物在CEM Discovery微波系统中加热至120℃,保持3小时。接着粗反应物通过硅胶柱色谱法,使用DCM/甲醇(0-20%)来纯化,得到呈油状的2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶(457mg)。1HNMR(600MHz,DMSO-d6)δppm 1.26-1.36(m,2H)1.43(m,6H)1.76(m,2H)2.37(m,5H)2.94(t,J=12.74Hz,2H)4.06(d,J=13.47Hz,2H)7.41(dd,J=9.37,2.64Hz,1H)8.08(d,J=9.37Hz,1H)8.20(d,J=2.64Hz,1H)。
实施例25
合成5-[4-(1-哌啶基)-1-哌啶基]吡啶-2-胺,化合物25
5-[4-(1-哌啶基)-1-哌啶基]吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 1.13-1.37(m,6H)1.40-1.63(m,6H)1.71(m,2H),2.24(m,1H)2.43(m,2H)3.33(d,J=12.30Hz,2H)5.31(s,2H)6.33(d,J=8.78Hz,1H)7.10(dd,J=8.78,2.93Hz,1H)7.55(d,J=2.64Hz,1H)。LCMS(ESI)261(M+H)。
实施例26
合成4-[1-(6-硝基-3-吡啶基)-4-哌啶基]吗啉,化合物26
4-[1-(6-硝基-3-吡啶基)-4-哌啶基]吗啉以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 1.41(m,2H)1.82(m,2H)2.42(m,5H)2.98(t,J=12.44Hz,2H)3.52(s,4H)4.04(d,J=12.88Hz,2H)7.42(d,J=9.37Hz,1H)8.08(d,J=9.08Hz,1H)8.21(s,1H)。
实施例27
合成5-(4-吗啉代-1-哌啶基)吡啶-2-胺,化合物27
5-(4-吗啉代-1-哌啶基)吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 1.34-1.52(m,2H)1.78(m,2H)2.14(m,1H)2.43(m,4H)3.32(d,J=12.30Hz,4H)3.47-3.59(m,4H)5.32(s,2H)6.34(d,J=8.78Hz,1H)7.11(dd,J=8.93,2.78Hz,1H)7.47-7.62(m,1H)。LCMS(ESI)263(M+H)。
实施例28
合成4-[1-(6-硝基-3-吡啶基)-4-哌啶基]硫吗啉,化合物28
4-[1-(6-硝基-3-吡啶基)-4-哌啶基]硫吗啉以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 1.40-1.52(m,2H)1.71(m,2H)2.49-2.55(m,4H)2.56-2.63(m,1H)2.68-2.75(m,4H)2.88-2.98(m,2H)4.09(d,J=13.18Hz,2H)7.42(dd,J=9.22,3.07Hz,1H)8.08(d,J=9.37Hz,1H)8.20(d,J=3.22Hz,1H)。
实施例29
合成5-(4-硫吗啉基-1-哌啶基)吡啶-2-胺,化合物29
5-(4-硫吗啉基-1-哌啶基)吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 1.47-1.59(m,2H)1.65(m,2H)2.22-2.38(m,1H)2.50-2.59(m,6H)2.68-2.82(m,4H)3.33(d,J=12.00Hz,2H)5.31(s,2H)6.33(d,J=9.08Hz,1H)7.10(dd,J=8.78,2.93Hz,1H)7.55(d,J=2.64Hz,1H)。LCMS(ESI)279(M+H)。
实施例30
合成2-硝基-5-(1-哌啶基)吡啶,化合物30
2-硝基-5-(1-哌啶基)吡啶以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 1.56(m,6H)3.49(d,J=4.39Hz,4H)7.30-7.47(m,1H)8.02-8.12(m,1H)8.15-8.26(m,1H)。
实施例31
合成5-(1-哌啶基)吡啶-2-胺,化合物31
5-(1-哌啶基)吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 1.39-1.46(m,2H)1.51-1.62(m,4H)2.75-2.92(m,4H)5.30(s,2H)6.34(d,J=8.78Hz,1H)7.09(dd,J=8.78,2.93Hz,1H)7.54(d,J=2.93Hz,1H)。LCMS(ESI)178(M+H)。
实施例32
合成4-(6-硝基-3-吡啶基)硫吗啉,化合物32
4-(6-硝基-3-吡啶基)硫吗啉以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 2.56-2.69(m,4H)3.79-3.92(m,4H)7.43(dd,J=9.22,3.07Hz,1H)8.10(d,J=9.37Hz,1H)8.20(d,J=2.93Hz,1H)。
实施例33
合成5-硫吗啉基吡啶-2-胺,化合物33
5-硫吗啉基吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 2.59-2.73(m,4H)3.04-3.20(m,4H)5.41(s,2H)6.35(d,J=8.78Hz,1H)7.10(dd,J=8.78,2.93Hz,1H)7.57(d,J=2.64Hz,1H)。LCMS(ESI)196(M+H)。
实施例34
合成(4R)-5-(6-硝基-3-吡啶基)-2,5-二氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯,化合物34
(4R)-5-(6-硝基-3-吡啶基)-2,5-二氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 1.33(d,J=32.21Hz,11H)1.91(m,2H)3.15(d,J=10.25Hz,1H)3.58(m,1H)4.46(m,1H)4.83(s,1H)7.16(s,1H)7.94(s,1H)8.05-8.16(m,1H)。
实施例35
合成(4R)-5-(6-氨基-3-吡啶基)-2,5-二氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯,化合物35
(4R)-5-(6-氨基-3-吡啶基)-2,5-二氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 1.31(d,J=31.91Hz,11H)1.83(m,2H)2.71-2.82(m,1H)3.44(m,1H)4.30(d,2H)5.08(s,2H)6.35(d,J=8.78Hz,1H)6.77-6.91(m,1H)7.33(s,1H)。LCMS(ESI)291(M+H)。
实施例36
合成N,N-二甲基-1-(6-硝基-3-吡啶基)哌啶-4-胺,化合物36
N,N-二甲基-1-(6-硝基-3-吡啶基)哌啶-4-胺以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 1.30-1.45(m,2H)1.79(m,2H)2.14(s,6H)2.33(m,1H)2.92-3.04(m,2H)4.03(d,J=13.76Hz,2H)7.42(dd,J=9.22,3.07Hz,1H)8.04-8.11(m,1H)8.21(d,J=2.93Hz,1H)。
实施例37
合成5-[4-(二甲基氨基)-1-哌啶基]吡啶-2-胺,化合物37
5-[4-(二甲基氨基)-1-哌啶基]吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,DMSO-d6)δppm 1.35-1.50(m,2H)1.69-1.81(m,2H)2.00-2.10(m,1H)2.11-2.22(s,6H)3.17-3.36(m,4H)5.19-5.38(s,2H)6.34(d,J=8.78Hz,1H)7.10(dd,J=8.78,2.93Hz,1H)7.55(d,J=2.63Hz,1H)。LCMS(ESI)221(M+H)。
实施例38
合成4-(6-硝基-3-吡啶基)吗啉,化合物38
4-(6-硝基-3-吡啶基)吗啉以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成。
实施例39
合成5-吗啉代吡啶-2-胺,化合物39
5-吗啉代吡啶-2-胺以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式制备。1HNMR(600MHz,氯仿-d)δppm 2.91-3.00(m,4H)3.76-3.84(m,4H)4.19(br.s.,2H)6.45(d,J=8.78Hz,1H)7.12(dd,J=8.78,2.93Hz,1H)7.72(d,J=2.93Hz,1H)。
实施例40
合成5-(4-异丁基哌嗪-1-基)吡啶-2-胺,化合物40
1-异丁基-4-(6-硝基-3-吡啶基)哌嗪以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中类似的方式合成,接着其以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式转化成5-(4-异丁基哌嗪-1-基)吡啶-2-胺。1HNMR(600MHz,氯仿-d)δppm 0.88(d,J=6.73Hz,6H)1.71-1.84(m,1H)2.10(d,J=7.32Hz,2H)2.46-2.58(m,4H)2.97-3.07(m,4H)4.12(s,2H)6.45(d,J=8.78Hz,1H)7.14(dd,J=8.78,2.93Hz,1H)7.75(d,J=2.93Hz,1H)。LCMS(ESI)235(M+H)。
实施例41
合成5-(4-异丙基哌嗪-1-基)吡啶-2-胺,化合物41
1-异丙基-4-(6-硝基-3-吡啶基)哌嗪以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中类似的方式合成,接着其以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式转化成5-(4-异丙基哌嗪-1-基)吡啶-2-胺。1HNMR(600MHz,氯仿-d)δppm 1.06(d,J=6.44Hz,6H)2.59-2.75(m,5H)2.97-3.10(m,4H)4.13(s,2H)6.45(d,J=8.78Hz,1H)7.15(dd,J=9.08,2.93Hz,1H)7.76(d,J=2.93Hz,1H)。LCMS(ESI)221(M+H)。
实施例42
合成5-[(2R,6S)-2,6-二甲基吗啉-4-基]吡啶-2-胺,化合物42
(2S,6R)-2,6-二甲基-4-(6-硝基-3-吡啶基)吗啉以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中所用类似的方式合成,接着其以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式转化成5-[(2R,6S)-2,6-二甲基吗啉-4-基]吡啶-2-胺。1HNMR(600MHz,氯仿-d)δppm 1.20(d,J=6.44Hz,6H)2.27-2.39(m,2H)3.11-3.21(m,2H)3.70-3.84(m,2H)4.15(s,2H)6.45(d,J=8.78Hz,1H)7.12(dd,J=8.78,2.93Hz,1H)7.72(d,J=2.63Hz,1H)。LCMS(ESI)208(M+H)。
实施例43
合成5-[(3R,5S)-3,5-二甲基哌嗪-1-基]吡啶-2-胺,化合物43
(3S,5R)-3,5-二甲基-1-(6-硝基-3-吡啶基)哌嗪以与合成2-硝基-5-[4-(1-哌啶基)-1-哌啶基]吡啶中类似的方式合成,接着其以与合成5-(4-甲基哌嗪-1-基)吡啶-2-胺中所用类似的方式转化成5-[(3R,5S)-3,5-二甲基哌嗪-1-基]吡啶-2-胺。1HNMR(600MHz,氯仿-d)δppm 1.09(d,J=6.44Hz,6H)2.20(t,J=10.83Hz,2H)2.95-3.08(m,2H)3.23(dd,J=11.71,2.05Hz,2H)4.13(s,2H)6.45(d,J=8.78Hz,1H)7.14(dd,J=8.78,2.93Hz,1H)7.73(d,J=2.63Hz,1H)。LCMS(ESI)207(M+H)。
实施例44
合成化合物44
N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯
中间体A于乙醇(100mL)中的溶液在压力计中在30psi氢气下使用10%Pd/C(0.7g)氢化7小时。反应混合物经CELITETM过滤后,有机层真空浓缩,得到N-(2-氨基-3-甲基-丁基)氨基甲酸叔丁酯(3.8g)。
向5-溴-2,4-二氯-嘧啶(7.11g,0.0312摩尔)于乙醇(100mL)中的溶液中添加二异丙基乙胺(5.45mL,1.0eq)和N-(2-氨基-3-甲基-丁基)氨基甲酸叔丁酯(6.31g,0.0312摩尔)。将反应混合物在室温下搅拌20小时。真空浓缩后,添加乙酸乙酯和水。将有机层分离,经硫酸镁干燥并接着真空浓缩。粗产物通过硅胶柱色谱法,使用己烷/乙酸乙酯(0-30%)来纯化,得到N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯。1HNMR(600MHz,DMSO-d6)δppm 0.77-0.85(d,J=6.5Hz,3H)0.87(d,J=6.73Hz,3H)1.31-1.39(m,9H)1.82-1.93(m,1H)2.94(d,J=5.56Hz,1H)3.08-3.22(m,2H)3.98(d,J=8.20Hz,1H)6.96(d,J=8.78Hz,1H)8.21(s,1H)。LCMS(ESI)393(M+H)。
N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]-3-甲基-丁基]氨基甲酸叔丁酯
N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]-3-甲基-丁基]氨基甲酸叔丁酯通过使N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯经受如针对N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯所述的Sonogoshira条件,接着随后如合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯中所述用TBAF处理来合成。1HNMR(600MHz,DMSO-d6)δppm 1.11(d,J=6.44Hz,3H)1.18(t,J=7.03Hz,6H)1.21-1.26(m,12H)2.88(br.s.,1H)3.43-3.78(m,6H)3.97-4.08(m,1H)5.61(s,1H)6.65(s,1H)6.71-6.78(m,1H)8.87(s,1H)。LCMS(ESI)441(M+H)。
7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
向N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯于THF中的溶液中添加TBAF且将内容物在回流下加热3小时。接着添加乙酸乙酯和水且分离有机层,经硫酸镁干燥并接着真空浓缩。向此粗反应物中添加乙酸/水(9:1)且将内容物在室温下搅拌12小时。真空浓缩后,添加饱和NaHCO3和乙酸乙酯。将有机层分离,干燥并接着真空浓缩。由此获得的粗反应产物溶解于DMF中,接着添加过硫酸氢钾且将内容物搅拌3小时。添加乙酸乙酯后,反应混合物经CELITETM过滤并真空浓缩。粗产物使用己烷/乙酸乙酯(0-100%)进行硅胶柱色谱法,得到7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸。1HNMR(600MHz,DMSO-d6)δppm 0.85(d,J=7.03Hz,3H)0.97(d,J=6.73Hz,3H)1.52(s,9H)1.99-2.23(m,1H)3.98(dd,J=14.05,3.51Hz,1H)4.47-4.71(m,2H)7.47(s,1H)9.17(s,1H)。LCMS(ESI)383(M+H)。
化合物44
向含7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸(0.050g,0.00013摩尔)的DCM(1.5mL)中添加DIC(32.7mg)和DMAP(10mg)。将内容物搅拌2小时。接着添加三氟乙酸(0.4mL)且再继续搅拌30分钟。添加饱和NaHCO3以中和过量酸后,添加乙酸乙酯且分离有机层,使用硫酸镁干燥并接着真空浓缩。粗产物通过硅胶柱色谱法,使用己烷/乙酸乙酯(0-100%)来纯化,得到产物。1HNMR(600MHz,DMSO-d6)δppm 0.72(d,J=6.73Hz,3H)0.97(d,J=6.73Hz,3H)2.09-2.22(m,1H)3.57(dd,J=13.18,4.98Hz,1H)3.72(dd,J=13.61,4.25Hz,1H)4.53(dd,J=8.05,3.95Hz,1H)7.20(s,1H)8.34(d,J=4.98Hz,1H)9.08(s,1H)。LCMS(ESI)265(M+H)。
实施例45
合成化合物45
化合物14用10%Pd/C氢化,得到中间体N-[(2R)-2-氨基-3-甲基-丁基]氨基甲酸叔丁酯,其接着,使用与针对化合物44所述类似的反应条件用5-溴-2,4-二氯-嘧啶处理,得到化合物45。分析数据与针对外消旋体(中间体1A)所报导一致。
实施例46
合成化合物46
化合物15用10%Pd/C氢化,得到中间体N-[(2S)-2-氨基-3-甲基-丁基]氨基甲酸叔丁酯,接着其使用与针对化合物44所述类似的反应条件,用5-溴-2,4-二氯-嘧啶处理,得到化合物46。分析数据(NMR和LCMS)与针对外消旋体化合物44所报导一致。
实施例47
合成化合物47
向化合物44(80mg,0.00030摩尔)于DMF(3mL)中的溶液中添加氢化钠于油中的60%分散体(40mg)。搅拌15分钟后,添加碘甲烷(37μL,2eq)。将内容物在室温下搅拌30分钟。接着添加饱和NaHCO3,接着乙酸乙酯。有机层经硫酸镁干燥并接着真空浓缩,得到产物。1HNMR(600MHz,DMSO-d6)δppm 0.74(d,J=6.73Hz,3H)0.91(d,J=6.73Hz,3H)2.04-2.20(m,1H)3.04(s,3H)3.69(dd,J=13.76,1.17Hz,1H)3.96(dd,J=13.76,4.68Hz,1H)4.58(dd,J=7.32,3.51Hz,1H)7.16(s,1H)9.05(s,1H)。LCMS(ESI)279(M+H)。
实施例48
合成化合物48
N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-4-甲基-戊基]氨基甲酸叔丁酯
化合物18在乙醇中在压力计中在50psi氢气罩下用10%Pd/C氢化,得到N-[(2S)-2-氨基-4-甲基-戊基]氨基甲酸叔丁酯,其接着使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件与5-溴-2,4-二氯-嘧啶反应,得到N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-4-甲基-戊基]氨基甲酸叔丁酯。1HNMR(600MHz,氯仿-d)δppm 0.91(d,J=6.44Hz,3H)0.94(d,J=6.44Hz,3H)1.32-1.51(m,11H)1.55-1.67(m,1H)3.28(t,J=5.86Hz,2H)4.21-4.42(m,1H)4.84(s,1H)5.84(d,J=7.32Hz,1H)8.07(s,1H)。LCMS(ESI)407(M+H)。
在氮气下向N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-4-甲基-戊基]氨基甲酸叔丁酯(5.0g,12.3毫摩尔)于甲苯(36mL)和三乙胺(7.2mL)中的溶液中添加3,3-二乙氧基丙-1-炔(2.8mL,19.7毫摩尔)、Pd2(dba)3(1.1g,1.23毫摩尔)和三苯基胂(3.8g,12.3毫摩尔)。将内容物加热至70℃,保持24小时。冷却到室温后,反应混合物经CELITETM过滤并接着真空浓缩。粗产物通过硅胶柱色谱法,使用己烷/乙酸乙酯(0-30%)来纯化,得到(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺。LCMS(ESI)455(M+H)。
7-[(1S)-1-[(叔丁氧羰基氨基)甲基]-3-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 0.88(d,J=6.44Hz,3H)0.97(d,J=6.44Hz,3H)1.47(s,9H)1.49-1.54(m,1H)1.56(t,J=7.17Hz,2H)3.98(dd,J=13.91,3.07Hz,1H)3.76(dd,J=13.31,4.13Hz,1H)4.38(d,J=14.05Hz,1H)4.90(t,J=7.17Hz,1H)7.41(s,1H)9.11(s,1H)。LCMS(M+H)397。
化合物48使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 0.82(d,J=6.73Hz,3H)0.97(d,J=6.44Hz,3H)1.34-1.46(m,1H)1.48-1.65(m,2H)3.40(dd,J=13.32,5.42Hz,1H)3.76(dd,J=13.47,4.10Hz,1H)4.76-4.92(m,1H)7.17(s,1H)8.34(d,J=5.27Hz,1H)9.04(s,1H)。LCMS(ESI)279(M+H)。
实施例49
合成化合物49
化合物49以与针对化合物47所述类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 0.82(d,J=6.44Hz,3H)0.97(d,J=6.44Hz,3H)1.37-1.68(m,3H)3.04(s,3H)3.56(d,J=13.47Hz,1H)4.00(dd,J=13.32,4.25Hz,1H)4.82-4.94(m,1H)7.16(s,1H)9.03(s,1H)。LCMS(ESI)293(M+H)。
实施例50
合成化合物50
N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-戊基]氨基甲酸叔丁酯
化合物20在压力容器中在50psi氢气下使用10%Pd/C氢化,得到N-[(2S)-2-氨基-3-甲基-戊基]氨基甲酸叔丁酯,其使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件与5-溴-2,4-二氯-嘧啶反应,得到N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-戊基]氨基甲酸叔丁酯。1HNMR(600MHz,氯仿-d)δppm 0.88-0.95(m,6H)1.11-1.20(m,1H)1.34(s,9H)1.44-1.54(m,1H)1.64-1.72(m,1H)3.17-3.27(m,1H)3.33-3.43(m,1H)4.11-4.21(m,1H)4.81(s,1H)5.92(d,J=8.20Hz,1H)8.05(s,1H)。LCMS(ESI)407。
N-[(2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-3-甲基-戊基]氨基甲酸叔丁酯
N-[(2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-3-甲基-戊基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。1HNMR(600MHz,DMSO-d6)δppm 0.76-0.89(m,6H)1.03(q,J=7.22Hz,3H)1.10-1.17(m,3H)1.25-1.42(m,11H)1.59-1.73(m,1H)3.35-3.47(m,4H)3.51-3.73(m,2H)3.99-4.11(m,1H)5.52-5.56(m,1H)6.76-7.03(m,2H)8.12-8.23(m,1H)。LCMS(ESI)455(M+H)。
7-[(1S)-1-[(叔丁氧羰基氨基)甲基]-2-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[(1S)-1-[(叔丁氧羰基氨基)甲基]-2-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 0.80(t,J=7.47Hz,3H)0.86(d,J=7.03Hz,3H)1.06-1.30(m,2H)1.48(s,9H)1.79-1.96(m,1H)3.95(dd,J=14.05,3.22Hz,1H)4.52(d,J=14.35Hz,1H)4.61-4.73(m,1H)7.43(s,1H)9.13(s,1H)。LCMS(ESI)397(M+H)。
化合物50使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 0.74(t,J=7.32Hz,3H)0.89(d,J=6.73Hz,3H)1.00-1.12(m,2H)1.82-1.94(m,1H)3.55(dd,J=13.91,4.83Hz,1H)3.70(dd,J=13.61,4.25Hz,1H)4.57(dd,J=7.91,4.10Hz,1H)7.17(s,1H)8.31(d,J=5.27Hz,1H)9.05(s,1H)。LCMS(ESI)279(M+H)。
实施例51
合成化合物51
化合物51以与化合物47类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 0.77(t,J=7.47Hz,3H)0.84(d,J=6.73Hz,3H)1.07-1.16(m,2H)1.82-1.95(m,1H)3.03(s,3H)3.68(d,J=13.76Hz,1H)3.96(dd,J=13.76,4.39Hz,1H)4.59-4.70(m,1H)7.16(s,1H)9.04(s,1H)。LCMS(ESI)293(M+H)。
实施例52
合成化合物52
N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3,3-二甲基-丁基]氨基甲酸叔丁酯
化合物21在压力容器中在50psi氢气下使用10%Pd/C氢化,得到N-[(2S)-2-氨基-3,3-二甲基-丁基]氨基甲酸叔丁酯,其接着使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件与5-溴-2,4-二氯-嘧啶反应,得到N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3,3-二甲基-丁基]氨基甲酸叔丁酯。LCMS(ESI)407(M+H)。
N-[(2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-3,3-二甲基-丁基]氨基甲酸叔丁酯
N-[(2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-3,3-二甲基-丁基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。LCMS(ESI)455(M+H)。
7-[(1S)-1-[(叔丁氧羰基氨基)甲基]-2,2-二甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[(1S)-1-[(叔丁氧羰基氨基)甲基]-2,2-二甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。LCMS(ESI)397(M+H)。
中间体1F使用与针对中间体1A所述类似的合成顺序合成。LCMS(ESI)279(M+H)。
实施例53
合成化合物53
化合物53以与针对中间体1CA所述类似的方式合成。LCMS(ESI)293(M+H)。
实施例54
合成化合物54
N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-苯基-乙基]氨基甲酸叔丁酯
化合物21在压力容器中在50psi氢气下使用10%Pd/C氢化,得到N-[(2S)-2-氨基-2-苯基-乙基]氨基甲酸叔丁酯,其接着使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件与5-溴-2,4-二氯-嘧啶反应,得到N-[(2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-苯基-乙基]氨基甲酸叔丁酯。1HNMR(600MHz,DMSO-d6)δppm 1.32(s,9H)3.29-3.50(m,2H)5.12-5.24(m,1H)7.10(t,J=5.27Hz,1H)7.21(t,J=6.88Hz,1H)7.26-7.34(m,4H)7.89(d,J=7.32Hz,1H)8.24(s,1H)。LCMS(ESI)427(M+H)。
N-[(2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-苯基-乙基]氨基甲酸叔丁酯
N-[(2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-苯基-乙基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。1HNMR(600MHz,DMSO-d6)δppm 1.14(t,J=7.03Hz,6H)1.32(s,9H)3.39(s,2H)3.52-3.61(m,2H)3.64-3.73(m,2H)5.17-5.26(m,1H)5.57(s,1H)7.07-7.14(m,1H)7.20-7.25(m,1H)7.26-7.33(m,4H)7.90(d,J=7.61Hz,1H)8.19(s,1H)。LCMS(ESI)475(M+H)。
7-[(1S)-2-(叔丁氧羰基氨基)-1-苯基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[(1S)-2-(叔丁氧羰基氨基)-1-苯基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。LCMS(ESI)417(M+H)。
化合物54
化合物54使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 3.58-3.69(m,1H)4.13(dd,J=13.47,4.39Hz,1H)6.07(d,J=3.81Hz,1H)6.85(d,J=7.32Hz,2H)7.19-7.31(m,3H)7.34(s,1H)8.27(d,J=5.27Hz,1H)9.13(s,1H)。LCMS(ESI)299(M+H)。
实施例55
合成化合物55
N-[(1S)-1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]-2-甲基-丙基]氨基甲酸叔丁酯
N-[(1S)-1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]-2-甲基-丙基]氨基甲酸叔丁酯使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件,使用5-溴-2,4-二氯-嘧啶和中间体E合成。1HNMR(600MHz,氯仿-d)δppm 0.95-1.02(m,6H)1.35-1.45(m,9H)1.75-1.90(m,1H)3.35-3.48(m,1H)3.52-3.61(m,1H)3.64-3.76(m,1H)4.56(d,J=8.49Hz,1H)6.47(s,1H)8.07(s,1H)。LCMS(ESI)393(M+H)。
N-[(1S)-1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]-2-甲基-丙基]氨基甲酸叔丁酯
N-[(1S)-1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]-2-甲基-丙基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。1HNMR(600MHz,氯仿-d)δppm 0.90-1.00(m,6H)1.18-1.25(m,6H)1.34-1.36(m,9H)1.69-1.90(m,1H)3.34-3.82(m,6H)4.53-4.77(m,1H)5.45-5.55(m,1H)6.37(dd,J=15.37,6.59Hz,1H)6.56(s,1H)8.05(s,1H)。LCMS(ESI)441(M+H)。
7-[(2S)-2-(叔丁氧羰基氨基)-3-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[(2S)-2-(叔丁氧羰基氨基)-3-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。1HNMR(600MHz,氯仿-d)δppm 0.90(d,J=6.73Hz,3H)0.96(d,J=7.03Hz,3H)1.55-1.66(m,10H)4.14(dd,J=13.61,3.95Hz,1H)4.52-4.63(m,1H)4.84(dd,J=13.61,1.32Hz,1H)7.37(s,1H)8.95(s,1H)。LCMS(ESI)383(M+H)。
化合物55
化合物55使用与针对化合物44所述类似的合成顺序合成。LCMS(ESI)265(M+H)。
实施例56
合成化合物56
化合物56使用5-溴-2,4-二氯-嘧啶和化合物17作为起始物质且根据与化合物55类似的合成步骤顺序合成。分析数据与针对其所述的对映体一致(化合物55)。1HNMR(600MHz,DMSO-d6)δppm 0.88(d,J=6.44Hz,6H)1.73-1.86(m,1H)3.67-3.76(m,2H)4.11-4.21(m,1H)7.13-7.19(m,1H)8.56(s,1H)9.05(s,1H)。LCMS(ESI)265(M+H)。
实施例57
合成化合物57
N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-甲基-丙基]氨基甲酸叔丁酯
N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-甲基-丙基]氨基甲酸叔丁酯使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件,使用5-溴-2,4-二氯-嘧啶和N-(2-氨基-2-甲基-丙基)氨基甲酸叔丁酯合成。LCMS(ESI)379(M+H)。
N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯
N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。1HNMR(600MHz,DMSO-d6)δppm 1.11-1.22(m,6H)1.31-1.45(m,15H)3.10-3.24(m,2H)3.51-3.76(m,4H)5.60(s,1H)6.94(s,1H)7.33(t,J=6.44Hz,1H)8.18(s,1H)。LCMS(ESI)427(M+H)。
7-[2-(叔丁氧羰基氨基)-1,1-二甲基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[2-(叔丁氧羰基氨基)-1,1-二甲基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.43(s,9H)1.73(s,6H)4.06(s,2H)7.46(s,1H)9.23(s,1H)。LCMS(ESI)369(M+H)。
化合物57
化合物57使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.73(s,6H)3.50(d,J=2.93Hz,2H)7.25(s,1H)8.46-8.55(m,1H)9.07(s,1H)。LCMS(ESI)251(M+H)。
实施例58
合成化合物58
N-[[1-[(5-溴-2-氯-嘧啶-4-基)氨基]环己基]甲基]氨基甲酸叔丁酯
N-[[1-[(5-溴-2-氯-嘧啶-4-基)氨基]环己基]甲基]氨基甲酸叔丁酯,使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件,使用5-溴-2,4-二氯-嘧啶和中间体K合成。1HNMR(600MHz,DMSO-d6)δppm 1.18-1.54(m,17H)2.23(d,J=14.35Hz,2H)3.36(d,J=6.44Hz,2H)5.82(s,1H)6.93(s,1H)8.22(s,1H)。LCMS(ESI)419(M+H)。
N-[[1-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环己基]甲基]氨基甲酸叔丁酯
N-[[1-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环己基]甲基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。1HNMR(600MHz,DMSO-d6)δppm 1.08-1.16(m,6H)1.17-1.54(m,17H)2.13(br.s.,2H)3.36(d,J=6.73Hz,2H)3.50-3.69(m,4H)5.72(s,1H)6.94(s,1H)5.72(br.s.,1H)8.17(s,1H)。LCMS(ESI)467(M+H)。
7-[1-[(叔丁氧羰基氨基)甲基]环己基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[1-[(叔丁氧羰基氨基)甲基]环己基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸,使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.37-1.54(m,13H)1.75(br.s.,4H)2.74(br.s.,2H)3.78-3.84(m,2H)7.44-7.51(m,1H)8.23(s,1H)9.11(s,1H)。LCMS(ESI)409(M+H)。
化合物58
化合物58使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.28(br.s.,2H)1.42(br.s.,2H)1.70(br.s.,4H)1.85-1.95(m,2H)2.69(m,2H)7.16-7.25(m,1H)8.41(br.s.,1H)9.04(s,1H)。LCMS 291(M+H)。
实施例59
合成化合物59
N-[[1-[(5-溴-2-氯-嘧啶-4-基)氨基]环戊基]甲基]氨基甲酸叔丁酯
N-[[1-[(5-溴-2-氯-嘧啶-4-基)氨基]环戊基]甲基]氨基甲酸叔丁酯使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件,使用5-溴-2,4-二氯-嘧啶和中间体L合成。1HNMR(600MHz,DMSO-d6)δppm 1.34(s,9H)1.50-1.58(m,2H)1.63-1.78(m,4H)1.96-2.06(m,2H)3.25(d,J=6.15Hz,2H)6.71(s,1H)7.18(t,J=6.29Hz,1H)8.20(s,1H)。LCMS(ESI)405(M+H)。
N-[[1-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环戊基]甲基]氨基甲酸叔丁酯
N-[[1-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环戊基]甲基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。LCMS(ESI)453(M+H)。
7-[1-[(叔丁氧羰基氨基)甲基]环戊基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[1-[(叔丁氧羰基氨基)甲基]环戊基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对所述7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.47(s,9H)1.74(br.s.,2H)1.88(br.s.,2H)2.04(br.s.,2H)2.41-2.45(m,2H)4.06(s,2H)7.45(s,1H)9.11(s,1H)。LCMS(ESI)395(M+H)。
化合物59
化合物59使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.72(br.s.,2H)1.86-1.93(m,2H)1.99(d,J=3.81Hz,2H)2.40(br.s.,2H)3.48(d,J=2.34Hz,2H)7.22(s,1H)8.53(br.s.,1H)9.05(s,1H)。LCMS(ESI)277(M+H)。
实施例60
合成化合物60
N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-4-甲基-戊基]氨基甲酸叔丁酯
N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-4-甲基-戊基]氨基甲酸叔丁酯使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件,使用5-溴-2,4-二氯-嘧啶和中间体B合成。分析数据与针对L-对映异构体所述一致。
N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-4-甲基-戊基]氨基甲酸叔丁酯
N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-4-甲基-戊基]氨基甲酸叔丁酯使用与合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯中所用类似的实验条件合成。1HNMR(600MHz,氯仿-d)δppm 1.21-1.31(m,12H)1.38-1.46(m,11H)1.70(m,1H)3.24(m,2H)3.65-3.82(m,4H)4.86(br s.,1H),5.65(s,1H)5.85(br s.,1H)6.94(s,1H)8.21(s,1H)。LCMS(ESI)455(M+H)。
7-[1-[(叔丁氧羰基氨基)甲基]-3-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[1-[(叔丁氧羰基氨基)甲基]-3-甲基-丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。分析数据与针对L-异构体所述一致。
化合物60
化合物60使用与针对化合物44所述类似的合成顺序合成。分析数据与针对L-异构体所述一致。
实施例61
合成化合物61
向化合物60(100mg,0.00024摩尔)于DMF(3.0mL)中的溶液中添加氢化钠(60%于油中的分散体)(27.6mg,3eq)。搅拌15分钟后,添加碘甲烷(30,2eq)。将内容物在室温下搅拌30分钟。添加饱和NaHCO3后,添加乙酸乙酯。分离有机层,接着经硫酸镁干燥并真空浓缩,得到产物。分析数据类似于所述化合物49。
实施例62
合成化合物62
N-[(1S,2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]环戊基]氨基甲酸叔丁酯
N-[(1S,2S)-2-[(5-溴-2-氯-嘧啶-4-基)氨基]环戊基]氨基甲酸叔丁酯通过使用与针对N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-3-甲基-丁基]氨基甲酸叔丁酯所述类似的反应条件,用5-溴-2,4-二氯-嘧啶处理N-[(1S,2S)-2-氨基环戊基]氨基甲酸叔丁酯合成。1HNMR(600MHz,DMSO-d6)δppm 1.27(s,9H)1.42-1.54(m,2H)1.56-1.65(m,2H)1.80-1.88(m,1H)1.96-2.01(m,1H)3.88-3.96(m,1H)4.03-4.09(m,1H)6.91(d,J=8.20Hz,1H)7.41(d,J=7.32Hz,1H)8.18(s,1H)。LCMS(ESI)391(M+H)。
N-[(1S,2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环戊基]氨基甲酸叔丁酯
N-[(1S,2S)-2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环戊基]氨基甲酸叔丁酯使用与合成(2S)-N2-[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]-4-甲基-戊-1,2-二胺中所用类似的实验条件合成。1HNMR(600MHz,DMSO-d6)δppm 1.13(t,6H)1.28(s,9H)1.42-1.52(m,2H)1.58-1.65(m,2H)1.81-1.90(m,1H)1.99-2.08(m,1H)3.49-3.60(m,2H)3.63-3.71(m,2H)3.84-3.93(m,1H)3.96-4.04(m,1H)5.53(s,1H)6.96(d,J=7.90Hz,1H)7.34(d,J=7.03Hz,1H)8.14(s,1H)。LCMS(ESI)439(M+H)。
7-[(1S,2S)-2-(叔丁氧羰基氨基)环戊基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸
7-[(1S,2S)-2-(叔丁氧羰基氨基)环戊基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对7-[1-[(叔丁氧羰基氨基)甲基]-2-甲基-丙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.41-1.52(m,9H)1.55-1.68(m,1H)1.88-2.00(m,2H)2.05-2.15(m,1H)2.26-2.35(m,1H)2.71-2.89(m,1H)4.01-4.16(m,1H)4.28-4.45(m,1H)7.41(s,1H)9.11(s,1H)。LCMS(ESI)381(M+H)。
化合物62
化合物62使用与针对化合物44所述类似的合成顺序合成。1HNMR(600MHz,DMSO-d6)δppm 1.48-1.60(m,1H)1.88-1.98(m,3H)1.99-2.08(m,1H)2.66-2.75(m,1H)3.63-3.74(m,1H)3.99-4.12(m,1H)7.21(s,1H)8.89(s,1H)9.04(s,1H)。LCMS(ESI)263(M+H)。
实施例63
合成化合物63
在氮气下向含氯三环状内酰胺(0.050g,0.225毫摩尔)的二噁烷(2.0mL)添加5-(4-甲基哌嗪-1-基)吡啶-2-胺(0.052g,1.2eq,0.270毫摩尔),接着添加Pd2(dba)3(18.5mg)、BINAP(25mg)和叔丁醇钠(31mg,0.324毫摩尔)。使烧瓶的内容物脱气10分钟并接着加热至100℃,保持12小时。粗反应负载在硅胶柱上并用DCM/MeOH(0-15%)洗脱,得到所需产物(26mg)。向溶解于DCM/MeOH(10%)中的此化合物中添加3N HCl的异丙醇溶液(2eq)并将反应物搅拌过夜。真空浓缩得到盐酸盐。1HNMR(d6-DMSO)δppm 11.13(brs,1H),9.07(s,1H),8.42(s,1H),8.03(br m 1H),7.99(s,1H),7.67(brm,1H),7.18(s,1H),4.33(m,2H),3.79(m,2H),3.64(m,2H),3.50(m,2H),3.16(m,4H),2.79(s,3H)。LCMS(ESI)379(M+H)。
实施例64
合成化合物64
在氮气下向含氯三环状内酰胺(0.075g,0.338毫摩尔)的二噁烷(3.5mL)中添加4-(6-氨基-3-吡啶基)哌嗪-1-甲酸叔丁酯(0.098g,1.05eq),接着添加Pd2(dba)3(27mg)、BINAP(36mg)和叔丁醇钠(45mg)。将内容物在回流下加热11小时。粗反应物负载至硅胶柱上并用DCM/MeOH(0-10%)洗脱,得到所需产物(32mg)。1HNMR(d6-DMSO)δppm 9.48(s,1H),8.84(s,1H),8.29(s,1H),8.18(s,1H),7.99(s,1H),7.42(m,1H),6.98(s,1H),4.23(m,2H),3.59(m,2H),3.45(m,4H),3.50(m,2H),3.05(m,4H)。LCMS(ESI)465(M+H)。
实施例65
合成化合物65
向化合物64(23mg)于10%DCM/MeOH中的溶液中添加10mL3M HCl的异丙醇溶液。将内容物搅拌16小时。浓缩反应混合物得到盐酸盐。1HNMR(d6-DMSO)δppm 9.01(s,1H),7.94(m,1H),7.86(m,1H),7.23(s,1H),4.30(m,2H),3.64(m,2H),3.36(m,4H),3.25(m,4H)。LCMS(ESI)465(M+H)。
实施例66
合成化合物66
在氮气下向含氯-N-甲基三环状酰胺(0.080g,0.338毫摩尔)的二噁烷(3.5mL)添加4-(6-氨基-3-吡啶基)哌嗪-1-甲酸叔丁酯(0.102g(1.1eq),接着添加Pd2(dba)3(27mg)、BINAP(36mg)和叔丁醇钠(45mg)。将内容物在回流下加热11小时。粗产物使用硅胶柱色谱法,使用洗脱剂二氯甲烷/甲醇(0-5%)来纯化,得到所需产物(44mg)。1HNMR(d6-DMSO)δppm 9.49(s,1H),8.85(s,1H),8.32(m,1H),8.02(s,1H),7.44(m,1H),7.00(s,1H),4.33(m,2H),3.80(m,2H),3.48(m,4H),3.07(m,4H),3.05(s,3H),1.42(s,9H)。LCMS(ESI)479(M+H)。
实施例67
合成化合物67
向化合物66(32mg)中添加3N HCL(10mL)的异丙醇溶液且将内容物搅拌在室温下过夜,历时16小时。浓缩得到盐酸盐。1HNMR(d6-DMSO)δppm 9.13(m,2H),8.11(m,1H),8.10(s,1H),7.62(m,1H),7.21(s,1H),4.43(m,2H),3.85(m,2H),3.41(m,4H),3.28(m,4H),3.08(s,3H)。LCMS(ESI)379(M+H)。
实施例68
合成化合物68
化合物68使用与针对化合物64所述类似的实验条件合成。1HNMR(600MHz,DMSO-d6)δppm 0.79(d,J=7.03Hz,3H)1.01(d,J=6.73Hz,3H)1.35-1.48(m,9H)2.16(dd,J=14.64,6.73Hz,1H)3.00-3.14(m,4H)3.40-3.51(m,4H)3.51-3.60(m,1H)3.63-3.74(m,1H)4.44(dd,J=7.90,3.81Hz,1H)6.99(s,1H)7.46(dd,J=8.93,2.78Hz,1H)7.94-8.09(m,2H)8.31(dd,J=9.08,1.46Hz,1H)8.85(s,1H)9.46(s,1H)。LCMS(ESI)507(M+H)。
实施例69
合成化合物69
化合物69使用与针对化合物63所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.77-0.86(m,3H)0.96(d,J=7.03Hz,3H)2.10-2.24(m,1H)3.07(s,3H)3.37-3.79(m,8H)4.00(dd,J=13.61,4.54Hz,2H)4.63-4.73(m,1H)7.20(s,1H)7.58-7.71(m,1H)7.99(d,J=2.34Hz,1H)8.12(d,J=9.37Hz,1H)9.11(s,1H)9.41(br.s.,2H)11.76(br.s.,1H)。LCMS(ESI)421(M+H)。
实施例70
合成化合物70
化合物70使用与针对化合物64和65所述类似的实验条件合成并呈盐酸盐形式回收。表征数据(NMR和LCMS)与针对化合物71所报导一致。
实施例71
合成化合物71
化合物71使用与针对化合物64和65所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.79(d,J=6.73Hz,3H)1.01(d,J=6.73Hz,3H)2.18(dd,J=14.49,7.17Hz,1H)3.18-3.84(m,10H)4.53-4.71(m,1H)7.24(s,1H)7.65(d,J=9.37Hz,1H)8.01(d,J=2.64Hz,1H)8.14(d,J=1.46Hz,1H)8.35(d,J=5.27Hz,1H)9.14(s,1H)9.46(s,2H)11.80(s,1H)LCMS(ESI)407(M+H)。
实施例72
合成化合物72(化合物UUU)
化合物72使用与针对化合物64和65所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.77(d,J=7.03Hz,3H)0.99(d,J=6.73Hz,3H)2.10-2.24(m,1H)3.18-3.81(m,10H)4.54-4.69(m,1H)7.22(s,1H)7.63(d,J=9.08Hz,1H)7.99(d,J=2.63Hz,1H)8.11(s,1H)8.33(d,J=5.27Hz,1H)9.12(s,1H)9.43(s,2H)11.77(s,1H)。LCMS(ESI)407(M+H)。
实施例73
合成化合物73
化合物73使用与针对化合物64和65所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.84(d,J=6.73Hz,3H)0.98(d,J=6.73Hz,3H)2.12-2.26(m,1H)3.09(s,3H)3.22-3.81(m,8H)4.01(dd,J=13.61,4.25Hz,2H)4.59-4.72(m,1H)7.19(s,1H)7.74(s,1H)7.96-8.10(m,2H)9.08(s,1H)9.22(s,2H)。LCMS(ESI)421(M+H)。
实施例74
合成化合物74
化合物74使用与针对化合物63所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.85(d,J=4.98Hz,3H)0.95(d,J=4.98Hz,3H)1.42-1.70(m,3H)2.77(d,J=2.93Hz,3H)3.07-4.14(m,10H)4.95(s,1H)7.20(s,1H)7.66(d,J=9.66Hz,1H)7.94(s,1H)8.08-8.16(m,1H)8.33(d,J=4.68Hz,1H)9.09(s,1H)11.38(s,1H)11.71(s,1H)。LCMS(ESI)435(M+H)。
实施例75
合成化合物75
化合物75使用与针对化合物64和65所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.87(d,J=6.15Hz,3H)0.94(d,J=6.15Hz,3H)1.57(d,J=84.61Hz,3H)3.05(s,3H)3.13-3.55(m,8H)3.69(d,J=78.17Hz,2H)4.90(s,1H)7.15(s,1H)7.63-7.85(m,1H)7.93(s,1H)8.26(s,1H)9.03(s,1H)9.20(s,2H)。LCMS(ESI)421(M+H)。
实施例76
合成化合物76
化合物76使用与针对化合物63所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.85(d,J=6.44Hz,3H)0.95(d,J=6.44Hz,3H)1.43-1.70(m,3H)2.78(d,J=2.93Hz,3H)3.05(s,3H)3.24-3.84(m,8H)4.01(d,J=9.66Hz,2H)4.89-5.01(m,1H)7.15(s,1H)7.77(s,1H)7.91-8.05(m,2H)9.03(s,1H)10.96-11.55(m,2H)。LCMS(ESI)449(M+H)。
实施例77
合成化合物77
化合物77使用与针对化合物64和65所述类似的实验条件合成并呈盐酸盐形式回收。1HNMR(600MHz,DMSO-d6)δppm 0.83-0.88(d,J=6.15Hz,3H)0.95(d,J=6.15Hz,3H)1.40-1.71(m,3H)3.28-3.83(m,8H)4.00(d,J=3.22Hz,2H)4.91-5.08(m,1H)7.17(s,1H)7.68(d,J=9.66Hz,1H)7.93(s,1H)8.07(s,1H)9.06(s,1H)9.40(s,2H)11.59(s,1H)。LCMS(ESI)435(M+H)。
实施例78
合成化合物78
向化合物500.060g(0.205毫摩尔)中添加5-(4-甲基哌嗪-1-基)吡啶-2-胺(35.42mg,0.9eq),接着添加1,4-二噁烷(3mL)。用氮气脱气后,添加Pd2dba3(12mg)、BINAP(16mg)和叔丁醇钠(24mg)。接着将内容物在CEM Discovery微波中在90℃下加热3小时。接着将反应物负载至硅胶柱并通过用DCM/MeOH(0-15%)洗脱来纯化。1HNMR(600MHz,DMSO-d6)δppm 0.75(t,J=7.47Hz,3H)0.91(d,J=6.73Hz,3H)1.04-1.20(m,2H)1.80-1.98(m,1H)2.77(d,J=3.81Hz,3H)2.94-3.90(m,10H)4.54-4.68(m,1H)7.06-7.23(m,2H)7.56-7.75(m,1H)7.90-8.12(m,2H)8.29(s,1H)9.07(s,1H)10.98-11.74(m,2H)。LCMS(ESI)435(M+H)。
实施例79
合成化合物79
化合物79以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 0.75(t,J=7.32Hz,3H)0.90(d,J=6.73Hz,3H)1.07-1.15(m,2H)1.85-1.94(m,1H)3.17-3.75(m,10H)4.58-4.67(m,1H)7.17(s,1H)7.71(s,1H)7.96(s,1H)7.98-8.05(m,1H)8.28(d,J=4.10Hz,1H)9.06(s,1H)9.39(s,2H)。LCMS(ESI)421(M+H)。
实施例80
合成化合物80
化合物80以与针对化合物78所述类似的方式合成。1HNMR(600MHz,DMSO-d6)δppm 0.78(t,J=7.32Hz,3H)0.86(d,J=6.73Hz,3H)1.13-1.21(m,2H)1.84-1.96(m,1H)2.77(d,J=4.39Hz,3H)3.04(s,3H)3.11-3.84(m,8H)3.98(dd,J=13.61,4.25Hz,2H)4.66-4.74(m,1H)7.17(s,1H)7.64(s,1H)7.96(d,J=2.34Hz,1H)8.03-8.13(m,1H)9.08(s,1H)11.26(s,1H)11.66(s,1H)。LCMS(ESI)449(M+H)。
实施例81
合成化合物81
所述化合物以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 0.78(t,J=7.32Hz,3H)0.85(d,J=6.73Hz,3H)1.10-1.27(m,2H)1.82-1.99(m,1H)3.04(s,3H)3.28-3.77(m,8H)3.97(dd,J=13.91,4.54Hz,2H)4.62-4.75(m,1H)7.07-7.24(m,1H)7.62-7.75(m,1H)7.94(d,J=2.34Hz,1H)7.97-8.08(m,1H)9.05(s,1H)9.29(s,2H)。LCMS(ESI)435(M+H)。
实施例82
合成化合物82
所述化合物以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 0.96(s,9H)3.15-3.87(m,10H)4.42-4.53(m,1H)6.99(s,1H)7.24(s,1H)8.06(s,1H)8.11-8.21(m,1H)8.79-8.98(m,2H)9.25(s,2H)9.88(s,1H)。LCMS(ESI)421(M+H)。
实施例83
合成化合物83
化合物83以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 0.95(s,9H)2.79(d,J=4.10Hz,3H)3.06-3.86(m,10H)4.56-4.67(m,1H)7.17(s,1H)7.70(s,1H)7.96(d,J=2.63Hz,1H)7.99-8.08(m,1H)8.26(s,1H)9.06(s,1H)10.80(s,1H)。LCMS(ESI)435(M+H)。
实施例84
合成化合物84
化合物84以与针对化合物78所述类似的方式合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 2.75-2.81(m,3H)3.12-3.16(m,2H)3.46-3.54(m,4H)3.60-3.69(m,2H)3.72-3.79(m,1H)4.07-4.18(m,2H)6.06-6.09(m,1H)6.90(d,J=7.61Hz,2H)7.20-7.31(m,3H)7.33(s,1H)7.49-7.55(m,1H)7.62-7.70(m,1H)7.92(d,J=2.93Hz,1H)8.22(s,1H)9.14(s,1H)。LCMS(ESI)455(M+H)。
实施例85
合成化合物85
化合物85以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 3.21(s,4H)3.35-3.67(m,5H)4.07-4.20(m,2H)6.13(s,1H)6.90(d,J=7.32Hz,2H)7.22-7.31(m,3H)7.36(s,1H)7.48(d,J=9.37Hz,1H)7.93(d,J=2.34Hz,1H)8.04-8.11(m,1H)8.25(d,J=4.98Hz,1H)9.17(s,1H)11.77(br,s.,1H)。LCMS(ESI)441(M+H)。
实施例86
合成化合物86
化合物86以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 0.90(d,J=6.15Hz,6H)1.72-1.89(m,1H)3.15-3.92(m,9H)4.10-4.46(m,2H)7.18(s,1H)7.59(d,J=8.78Hz,1H)8.00(s,1H)8.13(d,J=9.37Hz,1H)8.55(s,1H)9.09(s,1H)9.67(s,2H)11.91(s,1H)。LCMS(ESI)407(ESI)。
实施例87
合成化合物87
化合物87以与化合物86类似的方式合成并转化成盐酸盐。表征数据(NMR和LCMS)与针对对映体所获得类似化合物86。
实施例88
合成化合物88
化合物88以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.78(s,6H)3.40-3.53(m,6H)3.64-3.73(m,4H)7.27(s,1H)7.66(d,J=9.37Hz,1H)7.98(d,J=2.34Hz,1H)8.12(br.s.,1H)8.47(br.s.,1H)9.11(s,1H)9.45(br.s.,2H)11.62(br.s.,1H)。LCMS(ESI)393(M+H)。
实施例89
合成化合物89(又称为化合物T)
化合物89以与针对化合物78所述类似的方式合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.47(br.s.,6H)1.72(br.s.,2H)1.92(br.s.,2H)2.77(br.s.,3H)3.18(br.s.,2H)3.46(br.s.,2H)3.63(br.s.,2H)3.66(d,J=6.15Hz,2H)3.80(br.s.,2H)7.25(s,1H)7.63(br.s.,2H)7.94(br.s.,1H)8.10(br.s.,1H)8.39(br.s.,1H)9.08(br.s.,1H)11.59(br.s.,1H)。LCMS(ESI)447(M+H)。
实施例90
合成化合物90(又称为化合物Q)
化合物90以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.27-1.64(m,6H)1.71(br.s.,2H)1.91(br.s.,2H)2.80(br.s.,1H)3.17-3.24(m,2H)3.41(br.s.,4H)3.65(br.s.,4H)7.26(br.s.,1H)7.63(br.s.,1H)7.94(br.s.,1H)8.13(br.s.,1H)8.40(br.s.,1H)9.09(br.s.,1H)9.62(br.s.,1H)11.71(br.s.,1H)。LCMS(ESI)433(M+H)。
实施例91
合成化合物91(又称为化合物ZZ)
化合物91使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.64-1.75(m,2H)1.83-1.92(m,2H)1.96-2.06(m,2H)2.49-2.58(m,2H)2.79(d,J=3.81Hz,3H)3.06-3.18(m,4H)3.59-3.69(m,2H)3.73-3.83(m,2H)4.04-4.12(m,2H)7.17(br.s.,1H)7.60-7.70(m,2H)7.70-7.92(m,2H)7.96(br.s.,1H)8.41(br.s.,1H)8.98(br.s.,1H)10.77(br.s.,1H)。LCMS(ESI)433(M+H)。
实施例92
合成化合物92
化合物92以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.64-1.75(m,2H)1.84-1.92(m,2H)1.96-2.05(m,2H)2.48-2.56(m,2H)3.22(br.s.,4H)3.42-3.48(m,4H)3.60-3.69(m,2H)4.05-4.13(m,1H)7.18(s,1H)7.65(d,J=13.47Hz,1H)7.70-7.77(m,1H)7.94(d,J=1.76Hz,1H)8.42(br.s.,1H)9.00(s,1H)9.15(br.s.,2H)。LCMS(ESI)419(M+H)。
实施例93
合成化合物93
化合物93以与针对化合物78所述类似的方式,接着针对化合物65所述的解封闭步骤合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.76(br.s.,2H)1.89(br.s.,2H)2.03(br.s.,2H)2.47-2.58(m,2H)3.04(s,3H)3.22(br.s.,4H)3.39(br.s.,4H)3.66(s,2H)7.21(s,1H)7.67(d,J=9.37Hz,1H)7.93(br.s.,1H)7.98-8.09(m,1H)9.04(s,1H)9.34(br.s.,2H)11.31(br.s.,1H)。LCMS(ESI)433(M+H)。
实施例94
合成化合物94
化合物94使用与针对化合物78所述类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.66-1.77(m,2H)1.84-1.94(m,2H)1.96-2.08(m,2H)2.48-2.57(m,2H)3.36-3.52(m,4H)3.60-3.80(m,6H)7.21(s,1H)7.53-7.74(m,2H)7.86(s,1H)8.02(s,1H)8.45(s,1H)9.03(s,1H)11.19(br.s.,1H)。LCMS(ESI)420(M+H)。
实施例95
合成化合物95
化合物95使用与针对化合物78所述类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.65-1.79(m,2H)1.85-1.95(m,2H)1.97-2.08(m,2H)2.47-2.54(m,2H)3.40-3.58(m,5H)3.65(dd,J=21.67,5.56Hz,1H)3.69-3.78(m,4H)7.24(s,1H)7.97-8.17(m,2H)8.48(s,1H)9.08(s,1H)11.81(s,1H)。LCMS(ESI)421(M+H)。
实施例96
合成化合物96
化合物96使用与针对化合物78所述类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.55-1.74(m,2H)1.80-1.98(m,4H)2.48-2.60(m,2H)3.40-3.50(m,4H)3.57-3.72(m,2H)3.90-4.20(m,4H)7.08(s,1H)7.37-7.57(m,2H)7.70(m,2H)8.32(s,1H)8.88(s,1H)9.98(s,1H)。LCMS(ESI)419(M+H)。
实施例97
合成化合物97(又称为化合物III)
化合物97使用与针对化合物78所述类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.30(d,J=5.27Hz,6H)1.65-1.78(m,2H)1.83-1.95(m,2H)1.97-2.10(m,2H)2.45-2.55(m,2H)3.25-3.36(m,1H)3.39-3.48(m,4H)3.60-3.70(m,4H)3.75-4.15(m,2H)7.24(s,1H)7.54-7.75(m,2H)7.95(s,1H)8.10(s,1H)8.49(s,1H)9.07(s,1H)11.25(s,1H)11.48(s,1H)。LCMS(ESI)461(M+H)。
实施例98
合成化合物98
化合物98使用与针对化合物78所述类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 0.99(d,J=6.15Hz,6H)1.65-1.78(m,2H)1.90(m,2H)1.97-2.08(m,2H)2.08-2.17(m,1H)2.45-2.55(m,2H)2.88-3.02(m,2H)3.33-3.48(m,4H)3.50-3.90(m,6H)7.24(s,1H)7.67(s,2H)7.94(s,1H)8.12(s,1H)8.49(s,1H)9.07(s,1H)10.77(s,1H)11.51(s,1H)。LCMS(ESI)475(M+H)。
实施例99
合成化合物99
化合物99使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.13(d,J=5.86Hz,6H)1.66-1.77(m,2H)1.84-1.94(m,2H)1.97-2.09(m,2H)2.40-2.53(m,2H)3.37-3.49(m,2H)3.50-3.59(m,2H)3.59-3.73(m,4H)7.23(s,1H)7.64(m,3H)7.85(s,1H)8.11(s,1H)8.47(s,1H)9.05(s,1H).11.35(br s.,1H)。LCMS(ESI)448(M+H)。
实施例100
合成化合物100
化合物100使用与针对化合物78所述类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.50-1.57(m,2H)1.62-1.68(m,3H)1.68-1.75(m,2H)1.84-1.92(m,2H)1.97-2.08(m,2H)2.48-2.53(m,2H)3.14-3.23(m,4H)3.43-3.47(m,2H)3.58-3.70(m,2H)7.22(s,1H)7.58-7.70(m,2H)7.85-8.00(m,1H)8.16(d,1H)8.46(s,1H)9.04(s,1H)11.37(br s.,1H)。LCMS(ESI)418(M+H)。
实施例101
合成化合物101(又称为化合物WW)
化合物101使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.72(s,2H)1.90(s,4H)2.03(s,2H)2.21(s,2H)2.48-2.54(m,2H)2.73(s,2H)3.03(s,2H)3.25-3.35(m,1H)3.38-3.48(m,4H)3.65-3.99(m,5H)7.23(s,1H)7.63(d,J=9.66Hz,1H)7.90(s,1H)8.13(s,1H)8.47(s,1H)9.06(s,1H)10.50(br s.,1H)。LCMS(ESI)503(M+H)。
实施例102
合成化合物102(又称为化合物HHH)
化合物102使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.63-1.85(m,6H)1.87-1.92(m,2H)1.99-2.06(m,2H)2.15-2.23(m,2H)2.47-2.53(m,1H)2.69-2.79(m,2H)2.81-2.91(m,2H)2.98-3.08(m,2H)3.32-3.48(m,4H)3.57-3.72(m,4H)3.77-3.85(m,2H)7.22(s,1H)7.60-7.68(m,2H)7.90(s,1H)8.07(s,1H)8.46(s,1H)9.04(s,1H).11.41(br s.,1H)。LCMS(ESI)501(M+H)。
实施例103
合成化合物103
化合物103使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.64-1.76(m,2H)1.87-1.93(m,2H)2.00-2.07(m,2H)2.48-2.53(m,2H)2.67-2.72(m,4H)3.44-3.47(m,2H)3.50-3.55(m,4H)7.24(s,1H)7.61(d,J=9.37Hz,2H)7.86(d,J=2.63Hz,1H)8.09(d,J=12.88Hz,1H)8.48(s,1H)9.06(s,1H)11.41(br s.,1H)。LCMS(ESI)436(M+H)。
实施例104
合成化合物104
化合物104使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.29(d,J=6.73Hz,6H)1.66-1.79(m,2H)1.84-1.95(m,2H)1.98-2.09(m,2H)2.46-2.55(m,2H)3.29-3.39(m,2H)3.58-3.70(m,4H)3.77-3.86(m,4H)7.24(s,1H)7.66(d,J=9.37Hz,1H)7.96(d,J=2.93Hz,1H)8.08(s,1H)8.48(s,1H)9.06(s,1H)9.28(s,1H)9.67(s,1H)11.36(s,1H)。LCMS(ESI)447(M+H)。
实施例105
合成化合物105
化合物105使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.73(s,2H)1.76-1.85(m,2H)1.85-1.94(m,2H)1.98-2.07(m,2H)2.19-2.26(m,2H)2.48-2.52(m,1H)2.70-2.81(m,4H)3.13-3.20(m,1H)3.30-3.48(m,3H)3.58-3.71(m,4H)3.78-3.84(m,4H)7.24(s,1H)7.62(d,J=9.37Hz,2H)7.89(d,J=1.17Hz,1H)8.09-8.18(m,1H)8.48(s,1H)9.06(s,1H)11.46(br s.,1H)。LCMS(ESI)519(M+H)。
实施例106
合成化合物106
化合物106使用与针对化合物78所述条件类似的条件合成,接着针对化合物65所述的解封闭步骤并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.65-1.75(m,2H)1.85-1.93(m,2H)1.93-1.99(m,1H)2.00-2.06(m,2H)2.08-2.14(m,1H)2.47-2.55(m,2H)3.07-3.25(m,2H)3.25-3.69(m,5H)4.46(s,1H)4.67(s,1H)7.22(s,1H)7.58-7.69(m,2H)8.46(s,1H)9.02(s,1H)9.34(s,1H)9.65(s,1H)。LCMS(ESI)431(M+H)。
实施例107
合成化合物107(又称为化合物YY)
化合物107使用与针对化合物78所述条件类似的条件合成并转化成盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.65-1.82(m,3H)1.89(br.s.,2H)1.98-2.08(m,2H)2.13(br.s.,2H)2.47-2.55(m,2H)2.68(d,J=4.98Hz,6H)2.71-2.80(m,2H)3.29-3.71(m,10H)7.16-7.26(m,1H)7.67(d,J=9.66Hz,2H)7.91(d,J=2.05Hz,1H)8.14(br.s.,1H)8.48(br.s.,1H)9.05(s,1H)11.14(br.s.,1H)11.43(br.s.,1H)。LCMS(ESI)461(M+H)。
实施例108
合成化合物108
化合物108以与针对化合物64和65所述类似的方式合成并呈盐酸盐形式回收。分析数据与针对对映体化合物75所述一致。
实施例109
合成化合物109
化合物109以与针对化合物64和65所述类似的方式合成并呈盐酸盐形式回收。分析数据与针对对映体化合物75所述一致。
实施例110
合成化合物110
化合物110以与针对化合物78所述类似的方式合成并接着转化成其盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.50-1.65(m,1H)1.92-2.02(m,3H)2.06-2.15(m,1H)2.78(d,J=3.81Hz,4H)3.10-3.20(m,4H)3.47-3.51(m,2H)3.64-3.71(m,1H)3.76-3.83(m,2H)3.98-4.14(m,1H)7.20(s,2H)7.77(s,1H)7.97(s,2H)8.81(s,1H)9.03(s,1H)10.97(br s.,1H)。LCMS(ESI)419(M+H)。
实施例111
合成化合物111
化合物111以与针对化合物78所述类似的方式合成并接着转化成其盐酸盐。1HNMR(600MHz,DMSO-d6)δppm 1.54-1.59(m,1H)1.92-2.01(m,3H)2.06-2.15(m,1H)2.76-2.84(m,1H)3.17-3.24(m,6H)3.64-3.71(m,2H)4.02-4.11(m,2H)7.22(s,2H)7.64(s,1H)7.97(s,2H)8.75(s,1H)8.97(s,1H)9.21(s,1H)。LCMS(ESI)405(M+H)。
实施例112
合成化合物112
化合物112使用与针对化合物64所述类似的实验条件合成。
实施例113
合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯,化合物113
向5-溴-2,4-二氯嘧啶(12.80g,0.054摩尔)于乙醇(250mL)中的溶液中添加亨尼格碱(12.0mL),接着添加N-(叔丁氧羰基)-1,2-二氨基乙烷(10g,0.0624摩尔)于乙醇(80mL)中的溶液。将内容物搅拌过夜,历时20小时。在真空下蒸发溶剂。添加乙酸乙酯(800mL)和水(300mL)且分离各层。有机层经硫酸镁干燥并接着真空浓缩。使用己烷/乙酸乙酯(0-60%)的硅胶柱色谱法,得到N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯。LCMS(ESI)351(M+H)。
实施例114
合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯,化合物114
在氮气下向含N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯(5g,14.23毫摩尔)的甲苯(42mL)和三乙胺(8.33mL)添加三苯基胂(4.39g)、3,3-二乙氧基丙-1-炔(3.24mL)和Pddba(1.27g)。将内容物在70℃下加热24小时。经过滤后,粗反应物使用己烷/乙酸乙酯(0-20%)进行柱分离,得到所需产物3.9g。所得残余物使用己烷/乙酸乙酯(0-30%)进行柱色谱法,得到N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯。LCMS(ESI)399(M+H)。
实施例115
合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯,化合物115
向化合物114(3.9g,0.00976摩尔)于THF(60mL)中的溶液中添加TBAF(68.3mL,7eq)。内容物加热至45℃,保持2小时。浓缩,接着使用乙酸乙酯/己烷(0-50%)进行柱色谱法,得到呈浅棕色液体状的N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(1.1g)。1HNMR(d6-DMSO)δppm 8.88(s,1H),6.95(brs,1H),6.69(s,1H),5.79(s,1H),4.29(m,2H),3.59(m,4H),3.34(m,1H),3.18(m,1H),1.19(m,9H),1.17(m,6H)。LCMS(ESI)399(M+H)。
实施例116
合成N-[2-[2-氯-6-(二乙氧基甲基)-5-碘-吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯,化合物116
向含N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(0.1g,0.00025mol)的乙腈(2mL)中添加1,3-二碘-5,5-二甲基乙内酰脲(95mg,1eq)和固体NaHCO3(63mg,3eq)。将反应物在室温下搅拌16小时。将反应物过滤并真空浓缩。产物通过硅胶柱色谱法,使用己烷/乙酸乙酯(0-50%)来纯化,得到呈浅黄色固体状的N-[2-[2-氯-6-(二乙氧基甲基)-5-碘-吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(0.03g)。LCMS(ESI)525(M+H)。
实施例117
合成N-[2-[2-氯-6-(二乙氧基甲基)-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯,化合物117
向含N-[2-[2-氯-6-(二乙氧基甲基)-5-碘-吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(0.1g,0.19毫摩尔)的二噁烷(3mL)中添加含2-甲基苯基硼酸(28mg)、四(三苯基膦)钯(25mg)和磷酸钾(250mg)的水(0.3mL)。将反应物在CEM Discovery微波中90℃下加热3小时。粗反应负载到硅胶上并使用己烷/乙酸乙酯(0-30%)进行柱分离,得到N-[2-[2-氯-6-(二乙氧基甲基)-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(0.06g)。LCMS(ESI)489(M+H)。
实施例118
合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸,化合物118
向含N-[2-[2-氯-6-(二乙氧基甲基)-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯(0.85g,1.74毫摩尔)的AcOH(10mL)中添加水(1.5mL)。将反应物在室温下搅拌16小时。接着真空浓缩粗反应物。添加乙酸乙酯(50mL)后,有机层用饱和NaHCO3洗涤。有机层经硫酸镁干燥并接着真空浓缩,得到粗中间体,N-[2-[2-氯-6-甲酰基-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯。向含此粗中间体的DMF(5mL)中添加过硫酸氢钾(1.3g)。搅拌2.5小时后,添加水(20mL)和乙酸乙酯(100mL)。将有机层分离,干燥并接着真空浓缩,得到粗产物,其使用己烷/乙酸乙酯(0-50%)进行硅胶柱分离,得到7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸(0.112g)。LCMS(ESI)431(M+H)。
实施例119
合成化合物119
向含7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸(0.1g,0.261mmol)的DCM(4.1mL)中添加DMAP(20mg),接着添加N,N’-二异丙基碳化二亚胺(0.081mL,2eq)。搅拌3小时后,添加TFA(0.723mL)。接着再继续搅拌30分钟。反应混合物用饱和NaHCO3中和。接着添加DCM(20mL)且分离有机层,经硫酸镁干燥并接着真空浓缩,得到粗产物,其使用己烷/乙酸乙酯(0-100%)进行柱分离,得到氯三环状酰胺化合物119(0.65g)。LCMS(ESI)313(M+H)。
实施例120
合成化合物120
在氮气下向含氯三环状酰胺(0.040g,0.128毫摩尔)(化合物119)的二噁烷(2.5mL)中添加Pd2(dba)3(12mg)、叔丁醇钠(16mg)、BINAP(16mg)和4-吗啉代苯胺(22.7mg,1eq)。将反应混合物在CEM Discovery微波中在90℃下加热3.0小时。粗反应物负载至硅胶柱上且内容物用DCM/MeOH(0-6%)洗脱,得到产物(10mg)。LCMS(ESI)455(M+H)。1HNMR(600MHz,DMSO-d6)δppm 2.14(s,3H)3.23-3.50(m,2H)3.57-3.73(m,2H),3.81-3.92(m,8H),7.11-7.31(m,4H)7.31-7.48(m,1H)7.58-7.73(m,1H)7.77-7.95(m,2H)8.05-8.21(m,1H)8.44(s,1H)9.85-10.01(m,1H)。
实施例121
合成化合物121
向含氯三环状酰胺(0.024g)(化合物119)的N-甲基-2-吡咯烷酮(NMP)(1.5mL)添加反式-4-氨基环己醇(0.0768mmol,26.54mg,3eq)和亨尼格碱(0.4mL)。反应在CEM Discovery微波容器中在150℃下加热1.2小时。粗反应物负载至硅胶柱上且内容物用DCM/MeOH(0-10%)洗脱,得到产物(21mg)。LCMS(ESI)392(M+H)。1HNMR(600MHz,DMSO-d6)δppm 1.23(d,J=8.78Hz,4H)1.84(br.s.,4H)2.11(s,3H)3.34-3.43(m,1H)3.55(br.s.,2H)3.72(br.s.,1H)4.13(br.s.,2H)4.50(br.s.,1H)7.03(br.s.,1H)7.12-7.28(m,4H)7.96(br.s.,1H)8.18(br.s.,1H)。
实施例122
合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸,化合物122
7-[2-(叔丁氧羰基氨基)乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸所述类似的实验程序合成。LCMS(ESI)341(M+H)。
实施例123
合成化合物123
氯三环状酰胺化合物123使用与针对合成氯三环状酰胺(化合物119)所述类似的实验程序合成。LCMS(ESI)223(M+H)。
实施例124
合成化合物124
向含氯三环状酰胺,化合物123(0.035g,0.00157摩尔)的NMP(1.5mL)中添加亨尼格碱(0.3mL),接着添加反式-4-氨基环己醇(54.2mg)。反应混合物在150℃下加热1.5小时。粗反应物负载至硅胶柱上且柱用DCM/MeOH(0-10%)洗脱,得到产物(5mg)。LCMS(ESI)302(M+H)。
实施例125
合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-甲基-丙基]氨基甲酸叔丁酯,化合物125
N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-甲基-丙基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用N-(2-氨基-2-甲基-丙基)氨基甲酸叔丁酯处理5-溴-2,4-二氯嘧啶来合成。LCMS(ESI)(M+H)379。
实施例126
合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯,化合物126
N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯使用与针对合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,通过在例如Pddba等催化剂存在下用3,3-二乙氧基丙-1-炔处理N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-甲基-丙基]氨基甲酸叔丁酯来合成。
LCMS(ESI)(M+H)427。
实施例127
合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]-2-甲基-丙基]氨基甲酸叔丁酯,化合物127
N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]-2-甲基-丙基]氨基甲酸叔丁酯使用与针对合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]乙基]氨基甲酸叔丁酯所述类似的实验条件,通过用TBAF处理N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯来合成。LCMS(ESI)(M+H)427。
实施例128
合成7-[2-(叔丁氧羰基氨基)-1,1-二甲基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸,化合物128
7-[2-(叔丁氧羰基氨基)-1,1-二甲基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸所述类似的实验程序合成。LCMS(ESI)369(M+H)。
实施例129
合成化合物129
氯三环状酰胺化合物129使用与针对合成氯三环状酰胺化合物119所述类似的程序合成。LCMS(ESI)251(M+H)。
实施例130
合成化合物130
化合物130通过使用与化合物124类似的实验条件,用反式-4-氨基环己醇处理氯三环状胺化合物129来合成。LCMS(ESI)330(M+H)。1HNMR(600MHz,DMSO-d6)δppm 1.07-1.34(m,4H)1.47-2.05(m,10H)3.09(m,1H)3.51(d,J=2.91Hz,2H)3.57(m,1H)4.50(br.s.,1H)6.89(s,1H)6.94-7.05(m,1H)8.04(br.s.,1H)8.60(s,1H)9.00(br.s.,1H)。
实施例131
合成N-[1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]丙基]氨基甲酸苯甲酯,化合物131
N-[1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]丙基]氨基甲酸苯甲酯通过使用与针对合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用N-[1-(氨基甲基)丙基]氨基甲酸苯甲酯处理5-溴-2,4-二氯嘧啶基来合成。LCMS(ESI)(M+H)413。
实施例132
合成N-[1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]丙基]氨基甲酸苯甲酯,化合物132
N-[1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]丙基]氨基甲酸苯甲酯通过使用与针对合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,在例如Pddba等催化剂存在下用3,3-二乙氧基丙-1-炔处理N-[1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]丙基]-氨基甲酸苯甲酯来制备。LCMS(ESI)(M+H)461。
实施例133
合成N-[1-[[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]甲基]丙基]氨基甲酸苯甲酯,化合物133
N-[1-[[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]甲基]丙基]氨基甲酸苯甲酯通过使用与针对合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3d]嘧啶-7-基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用TBAF处理N-[1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]丙基]氨基甲酸苯甲酯来合成。LCMS(ESI)(M+H)461。
实施例134
合成7-[2-(苯甲氧基羰基氨基)丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸,化合物134
7-[2-(苯甲氧基羰基氨基)丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸所述类似的实验程序合成。LCMS(ESI)403(M+H)。
实施例135
合成化合物135
向7-[2-(苯甲氧基羰基氨基)丁基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸于二氯甲烷中的溶液中添加HBr,将反应物在45℃下搅拌3小时。浓缩后,添加2N NaOH以碱化(pH=8.0)反应物,接着添加THF(20mL)。接着添加Boc2O(1.2eq)并将反应物搅拌16小时。接着向粗反应混合物中添加乙酸乙酯(100mL)和水(50mL)并分离有机相,干燥(硫酸镁)并接着真空浓缩。向粗产物中添加二氯甲烷(30mL),接着添加DIC和DMAP。搅拌2小时后,添加TFA且内容物搅拌一小时。将溶剂真空蒸发并用饱和NaHCO3碱化残余物。接着添加乙酸乙酯且分离有机层,干燥(硫酸镁)并接着真空浓缩。用己烷/乙酸乙酯(0-100%)进行柱色谱法,得到所需氯三环状核心,化合物135。LCMS(ESI)251(M+H)。
实施例136
合成化合物136
化合物136通过使用与化合物124类似的实验条件,用反式-4-氨基环己醇处理氯三环状胺化合物135合成。LCMS(ESI)330(M+H)。1HNMR(600MHz,DMSO-d6)δppm 0.80-0.95(m,3H)1.35-1.92(m,10H)3.66(br.m.,3H)4.17(br.s.,2H)4.47(br.s.,1H)6.85(s,1H)6.96(br.s.,1H)8.15(br.s.,1H)8.62(br.s.,1H)。
实施例137
合成N-[1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]环戊基]氨基甲酸叔丁酯,化合物137
N-[1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]环戊基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用N-[1-(氨基甲基)环戊基]氨基甲酸叔丁酯处理5-溴-2,4-二氯嘧啶来合成。LCMS(ESI)405(M+H)。
实施例138
合成N-[1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]环戊基]氨基甲酸叔丁酯,化合物138
N-[1-[[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]甲基]环戊基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,在例如Pddba等催化剂存在下,用3,3-二乙氧基丙-1-炔处理N-[1-[[(5-溴-2-氯-嘧啶-4-基)氨基]甲基]环戊基]氨基甲酸叔丁酯来合成。LCMS(ESI)453(M+H)。
实施例139
合成N-[1-[[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]甲基]环戊基]氨基甲酸叔丁酯,化合物139
N-[1-[[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]甲基]环戊基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3d]嘧啶-7-基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用TBAF处理N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯来合成。LCMS(ESI)453(M+H)。
实施例140
合成7-[[1-(叔丁氧羰基氨基)环戊基]甲基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸,化合物140
7-[[1-(叔丁氧羰基氨基)环戊基]甲基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸所述类似的实验程序合成。LCMS(ESI)395(M+H)。
实施例141
合成化合物141
氯三环状核心化合物141使用与针对合成氯三环状酰胺化合物119所述类似的实验程序合成。LCMS(ESI)277(M+H)。
实施例142
合成化合物142
化合物142通过使用与化合物124类似的实验条件,用反式-4-氨基环己醇处理氯三环状胺化合物141合成。LCMS(ESI)356(M+H)。1HNMR(600MHz,DMSO-d6)δppm 1.08-1.32(m,8H)1.60-2.09(m,8H)3.03-3.17(m,1H)3.35(s,2H)3.54-3.62(m,1H)4.51(d,J=4.39Hz,1H)6.88(s,1H)6.96(br.s.,1H)8.07(br.s.,1H)8.58(s,1H)。
实施例143
合成N-[[1-[(5-溴-2-氯-嘧啶-4-基)氨基]环戊基]甲基]氨基甲酸叔丁酯,化合物143
N-[[1-[(5-溴-2-氯-嘧啶-4-基)氨基]环戊基]甲基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用N-[(1-氨基环戊基)甲基]氨基甲酸叔丁酯处理5-溴-2,4-二氯嘧啶来合成。LCMS(ESI)405(M+H)。
实施例144
合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯,化合物144
N-[[1-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]环戊基]甲基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]乙基]氨基甲酸叔丁酯所述类似的实验条件,在例如Pddba等催化剂存在下用3,3-二乙氧基丙-1-炔处理N-[2-[(5-溴-2-氯-嘧啶-4-基)氨基]-2-甲基-丙基]氨基甲酸叔丁酯来合成。
LCMS(ESI)453(M+H)。
实施例145
合成N-[[1-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]环戊基]甲基]氨基甲酸叔丁酯,化合物145
N-[[1-[2-氯-6-(二乙氧基甲基)吡咯并[2,3-d]嘧啶-7-基]环戊基]甲基]氨基甲酸叔丁酯通过使用与针对合成N-[2-[2-氯-6-(二乙氧基甲基)吡咯并[2,3d]嘧啶-7-基]乙基]氨基甲酸叔丁酯所述类似的实验条件,用TBAF处理N-[2-[[2-氯-5-(3,3-二乙氧基丙-1-炔基)嘧啶-4-基]氨基]-2-甲基-丙基]氨基甲酸叔丁酯来合成。LCMS(ESI)4534(M+H)。
实施例146
合成7-[2-(叔丁氧羰基氨基)-1,1-二甲基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6甲酸,化合物146
7-[2-(叔丁氧基羰基氨基)-1,1-二甲基-乙基]-2-氯-吡咯并[2,3-d]嘧啶-6-甲酸使用与针对合成7-[2-(叔丁氧羰基氨基)乙基]-2-氯-5-(邻甲苯基)吡咯并[2,3-d]嘧啶-6-甲酸所述类似的实验程序合成。LCMS(ESI)395(M+H)。
实施例147
合成化合物147
氯三环状酰胺化合物147使用与针对氯三环状酰胺化合物119所述类似的实验程序合成。LCMS(ESI)277(M+H)。
实施例148
合成化合物148
化合物148通过使用与化合物124类似的实验条件,用反式-4-氨基环己醇处理氯三环状胺化合物147来合成。LCMS(ESI)356(M+H)。1HNMR(600MHz,DMSO-d6)δppm 1.06-1.35(m,8H)1.45-1.95(m,8H)3.10(m,1H)3.58(br.s.,2H)3.95(br.s.,1H)4.49(br.s.,1H)6.84(s,1H)6.85-6.93(m,1H)8.29(s,1H)8.61(br.s.,1H)。
实施例149
合成化合物149
步骤1:根据A.Sarkar等人(JOC,2011,76,7132-7140)的方法,将化合物59进行Boc保护。
步骤2:经Boc保护的化合物59用5mol%NiCl2(Ph3)2、0.1eq三苯基膦、3eq Mn、0.1eq碘化四乙基铵在DMI中在CO2(1atm)下在25℃下处理20小时,以将芳基卤化物衍生物转化成甲酸。
步骤3:使用标准条件将来自步骤2的甲酸转化成相应酸氯化物。
步骤4:来自步骤3的酸氯化物与N-甲基哌嗪反应,产生相应酰胺。
步骤5:使用含三氟乙酸的亚甲基氯将来自步骤4的酰胺脱保护,产生目标化合物。化合物149通过硅胶柱色谱法,用二氯甲烷-甲醇梯度洗脱来纯化,得到化合物149。
化合物119至147每一者和具有各种R8、R1和Z定义的相应化合物可以与氢化钠和烷基卤化物或其它卤化物反应,以在与胺反应前插入所需R取代,例如以上针对合成化合物120所述,产生所需的式I、II、III、IV或V的产物。
实施例150
CDK4/6抑制体外分析
通过Nanosyn(Santa Clara,CA),在CDK4/细胞周期蛋白D1、CDK2/CycA和CDK2/细胞周期蛋白E激酶分析中测试本文公开的所选化合物以确定其对这些CDK的抑制作用。使用微流体激酶检测技术(Caliper Assay Platform)进行分析。以12点剂量-反应格式,在Km下,针对ATP单一测试化合物。对于所有分析,使用的磷酸化受体底物肽浓度都是1μM,且对于所有分析,星孢菌素用作参考化合物。每一分析的细节如下所述:
CDK2/细胞周期蛋白A:酶浓度:0.2nM;ATP浓度:50μM;孵育时间:3小时。
CDK2/细胞周期蛋白E:酶浓度:0.28nM;ATP浓度:100μM;孵育时间:1小时。
CDK4/细胞周期蛋白D1:酶浓度:1nM;ATP浓度:200μM;孵育时间:10小时。
表1中化合物对CDK4/CycD1、CDK2/CycE、CDK2/CycA的抑制IC50值以及选择性倍数呈现于表2。
表2:CDK4的选择性抑制
为进一步表征其激酶活性,使用DiscoveRx’s KINOMEscanTM谱图分析服务,针对456种(395种非突变)激酶筛选化合物T。使用1000nM的单一浓度(是对CDK4的IC50的>1000倍)筛选化合物。来自此筛选的结果证实针对CDK4的高效力和相对于CDK2的高选择性。另外,激酶组谱图分析展示与其它测试的激酶相比,化合物T对CDK4和CDK6具有相对选择性。确切地说,当使用65%、90%或99%的抑制阈值时,化合物T对应地抑制395种非突变激酶中的92(23.3%)、31种(7.8%)或6种(1.5%)。
除CDK4激酶活性之外,还针对CDK6激酶活性测试若干化合物。针对PD0332991(参考)和化合物T、Q、GG和U,CDK6/CycD3激酶分析以及CDK4/细胞周期蛋白D1、CDK2/CycA和CDK2/细胞周期蛋白E激酶分析的结果展示在表3中。对CDK4/细胞周期蛋白D1 10nM和对CDK12/细胞周期蛋白E 10uM的IC50与先前针对PD0332991的公布报导非常一致(Fry等人Molecular Cancer Therapeutics(2004)3(11)1427-1437;Toogood等人Journal of Medicinal Chemistry(2005)48,2388-2406)。相对于参考化合物(PD0332991),化合物T、Q、GG和U更有效(IC50更低),并证明相对于参考化合物选择性倍数更高(CDK2/CycE IC50除以CDK4/CycD1IC50)。
表3:化合物T、Q、GG和U对CDK激酶的抑制
实施例151
G1停滞(细胞G1和S期)分析
为测定在各种处理后处于细胞周期的各种阶段的细胞分率,将HS68细胞(人类皮肤成纤维细胞系(Rb阳性))用碘化丙锭染色液染色并在Dako Cyan流动式细胞测量仪上操作。使用FlowJo 7.2.2分析测定G0-G1 DNA细胞周期中细胞的分率对比S期DNA细胞周期中的分率。
测试表1中列出的化合物使HS68细胞停滞在细胞周期的G1期的能力。从细胞G1停滞分析的结果,HS68细胞的G1停滞所需的抑制EC50值的范围为22nM至1500nM(参见表4中标题为“细胞G1停滞EC50”的列)。
实施例152
细胞增殖的抑制
使用以下癌细胞系进行细胞增殖分析:MCF7(乳腺癌-Rb阳性)、ZR-75-1(乳管癌瘤-Rb阳性)、H69(人类小细胞肺癌-Rb阴性)细胞或A2058(人类转移性黑色素瘤细胞-Rb阴性)。这些细胞在Costar(Tewksbury,Massachusetts)309396孔经组织培养物处理的白壁/透明底板中接种。将细胞用表1的化合物,以10uM到1nM的九点剂量反应连续稀释液处理。将细胞暴露于化合物,并接着在四天(H69)或六天(MCF7、ZR75-1、A2058)后,如所指示,使用发光细胞活力分析(CTG;Promega,Madison,Wisconsin,United States of America),按照制造商建议,测定细胞活力。在BioTek(Winooski,Vermont)Syngergy2多模盘式读数器上读取盘。将相对光单位(RLU)作为可变摩尔浓度的结果绘图,且使用Graphpad(LaJolla,Californaia)Prism 5统计软件分析数据以确定每一化合物的EC50。
两个Rb阳性乳癌细胞系(MCF7和ZR75-1)的细胞抑制分析的结果展示于表4中。抑制MCF7乳癌细胞增殖所需的抑制EC50值的范围是28nM至257nM。抑制ZR75-1乳癌细胞增殖所需的抑制EC50值的范围是24nM至581nM。
高效针对MCF7乳腺癌细胞增殖的代表性化合物的实施例展示于图21-24中。图21-24中测试的化合物(化合物T、Q、GG、U、H、MM、OO和PD-332991)都展示对MCF-7细胞的细胞增殖的显著抑制。如图21中可以看到,化合物T展示针对MCF-7细胞的活性比PD0332991更有效。
针对ZR75-1(乳房导管癌瘤(Rb阳性))细胞增殖高效的代表性化合物的实施例展示于图25-28中。图25-28中测试的化合物(化合物T、Q、GG、U、H、MM、OO和PD-332991)都展示对ZR75-1细胞的细胞增殖的显著抑制。如图25中可以看到,化合物T展示针对ZR75-1细胞的活性比PD0332991更有效。
除乳癌细胞系外,还针对小细胞肺癌细胞系(H69)和人类转移性黑色素瘤细胞系(A2058)(两种Rb缺乏的(Rb阴性)细胞系)评估大量本文公开的所选化合物。这些细胞抑制分析的结果展示于表4中。抑制H69小细胞肺癌细胞所需的抑制EC50值的范围是2040nM至>3000nM。抑制A2058恶性黑色素瘤细胞增殖所需的抑制EC50值的范围是1313nM至>3000nM。与在两种Rb阳性乳癌细胞系上看到的显著抑制相比,发现测试的化合物不显著有效地抑制小细胞肺癌或黑色素瘤细胞的增殖。
表4:对癌细胞增殖的抑制
实施例153
HSPC生长抑制研究
先前已经证明PD0332991对HSPC的作用。图2展示在通过口腔管饲法的单一剂量150mg/kg PD0332991后小鼠HSPC和脊髓祖细胞的EdU掺入以评估瞬时CDK4/6抑制对骨髓停滞的暂时作用,如Roberts等人Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy.JCNI 2012;104(6):476-487中报导(图2A)。如图2中可见,单一口服剂量的PD0332991引起HSPC(LKS+)和脊髓祖细胞(LKS-)持久减少,超过36小时。直到口服后48小时,HSPC和脊髓祖细胞才恢复基线细胞分裂。
实施例154
抗赘生性化合物的药物动力学和药效学特性
本发明的化合物证明优良的药物动力学和药效学特性。化合物T、Q、GG和U以30mg/kg通过口腔管饲法或10mg/kg通过静脉内注射给予。在给药后0、0.25、0.5、1.0、2.0、4.0和8.0小时取血液样品并通过HPLC测定化合物T、Q、GG或U的血浆浓度。如表5中所示,化合物T、GG和U证明具有极好的口腔药物动力学和药效学特性。此包括52%至80%的极高口腔生物利用率(F(%))和经口施用后3至5小时的血浆半衰期。当通过静脉内施用递送时,化合物T、Q、GG和U证明具有极好的口腔药物动力学和药效学特性。所有四种化合物的代表性IV和口腔PK曲线展示于图3中。
表5:抗赘生性化合物的药物动力学和药效学特性
实施例155
细胞洗脱实验
在第1天将HS68细胞以40,000个细胞/孔在60mm皿中在含有10%胎牛血清、100U/ml青霉素(penicillin)/链霉素(streptomycin)和1x Glutamax(Invitrogen)的DMEM中结晶析出,如所述(Brookes等人EMBO J,21(12)2936-2945(2002)和Ruas等人Mol Cell Biol,27(12)4273-4282(2007))。接种24小时后,将细胞用化合物T、化合物Q、化合物GG、化合物U、PD0332991或单独DMSO媒介物在300nM最终浓度的测试化合物下处理。在第3天,一式三份地收获一组处理的细胞样品(0小时样品)。剩余细胞在PBS-CMF中洗涤两次,并回到缺乏测试化合物的培养基。在24、40和48小时一式三份地收获成组样品。
或者,使用从美国典型菌种保藏中心(ATCC,Manassas,VA)中获得的正常肾近端小管上皮细胞(Rb阳性)进行相同的实验。细胞在孵育箱中在37℃下在5%CO2潮湿气氛中37℃潮湿孵育箱中补充有肾上皮细胞生长试剂盒(ATCC)的肾上皮细胞基础培养基(ATCC)中生长。
收获细胞后,将样品用碘化丙锭染色液染色且样品在Dako Cyan流动式细胞测量仪上操作。使用FlowJo 7.2.2分析测定G0-G1 DNA细胞周期中细胞的分率对比S期DNA细胞周期中的分率。
图4展示细胞洗脱实验,其证明本发明的抑制剂化合物在不同的细胞类型中具有短暂、瞬时的G1停滞作用。在人类成纤维细胞(Rb阳性)(图4A和4B)或者人类肾近端小管上皮细胞(Rb阳性)(图4C和4D)中化合物T、Q、GG和U与PD0332991相比,且在24、36、40和48小时测定化合物洗脱后对细胞周期的作用。
如图4中所示且类似于如图2中所示的体内结果,PD0332991需要细胞在洗脱后超过48小时恢复至正常基线细胞分裂。此在图4A和图4B中可见,因为获得的值对应地等于细胞分裂的G0-G1分率或者S期的DMSO对照的值。相比之下,在少至24小时或40小时内,用本发明的化合物处理的HS68细胞恢复至正常基线细胞分裂,不同于PD0332991在这些相同时间点。使用人类肾近端小管上皮细胞的结果(图4C和4D)也展示与用化合物T、Q、GG或U处理的细胞相比,经PD0332991处理的细胞花费显著更长的时间来恢复至基线水平。
实施例156
如使用EdU掺入和流动式细胞测量术分析评估的骨髓增殖
对于HSPC增殖实验,将青年雌性FVB/N小鼠用如指示的单一剂量的化合物T、化合物Q、化合物GG或PD0332991通过口腔管饲法处理。接着在指示的时间(化合物施用后0、12、24、36或48小时)处死小鼠,并如先前描述收获骨髓(每个时间点n=3只小鼠)(Johnson等人J.Clin.Invest.(2010)120(7),2528-2536)。在收获骨髓前四个小时,将小鼠用100μg EdU通过腹膜内注射(Invitrogen)处理。使用先前描述的方法,收获骨髓单核细胞并进行免疫表型分析,且接着确定EdU阳性细胞百分比(Johnson等人J.Clin.Invest.(2010)120(7),2528-2536)。简单地说,通过谱系标记物(Lin-)、Sca1(S+)和c-Kit(K+)的表达鉴别HSPC。
小鼠中的分析确定化合物T、化合物Q和化合物GG证明骨髓干细胞(HSPC)的剂量依赖性、瞬时且可逆的G1停滞(图5)。通过口腔管饲法,以150mg/kg化合物T、化合物Q、化合物GG或仅仅媒介物,给予每组六只小鼠。在处死动物并收获骨髓前四小时,将小鼠用100μg EdU通过腹膜内注射处理。每组三只小鼠在12小时处死且在24小时处死每组剩余三只动物。结果展示于图5A中,呈与对照相比,在12或24小时时间点,处理动物的EdU阳性细胞的比率。化合物T和GG证明在12小时EdU掺入减少,其在24小时开始恢复正常。化合物Q也证明在12小时少许减少,且在24小时开始恢复至基线,不过化合物Q的口服生物利用率低。
将其它实验用检验剂量反应的化合物T完成且化合物处理时期更长。通过口腔管饲法,给予50、100或150mg/kg化合物T,并如上所述,在12和24小时,测定骨髓中的EdU掺入。或者通过口腔管饲法,给予150mg/kg化合物T,并在12、24、36和48小时,测定骨髓中的EdU掺入。如图5B和5C中可见并类似于细胞洗脱实验,如通过EdU掺入确定,在口腔管饲多剂后24小时中骨髓细胞和尤其HSPC恢复至正常细胞分裂。图5C中150mg/kg口服剂量的化合物T可以直接与图2中展示的相同剂量的PD0332991的结果比较,其中在24和36小时细胞仍然未分裂(如通过低EdU掺入确定),仅仅在48小时恢复至正常值。
实施例157
比较化合物T和PD0332991的HSPC生长抑制研究
图6是用PD0332991(三角形)或化合物T(倒三角形)处理的小鼠的EdU阳性HSPC细胞百分比对比施用化合物后的时间(小时)的图。通过口腔管饲法,施用150mg/kg的两种化合物。在收获骨髓之前一小时,EdU经腹膜内注射以标记循环细胞。在化合物处理后12、24、36和48小时收获骨髓,且在每个时间点,测定EdU阳性HSPC细胞的百分比。
如图6中所见,单一口服剂量的PD0332991引起HSPC持久减少,超过36小时。相比之下,单一口服剂量的化合物T引起HSPC增殖在12小时初期减少,但到给予化合物T后24小时,HSPC增殖重新开始。
实施例158
代谢稳定性
与PD0332991相比,化合物T的代谢稳定性在人类、犬、大鼠、猴和小鼠肝微粒体中测定。人类、小鼠和犬肝微粒体从Xenotech购买,且通过Absorption Systems制备史泊格-多利大鼠(Sprague-Dawleyrat)肝微粒体。制备包含0.5mg/mL肝微粒体、100mM磷酸钾pH 7.4、5mM氯化镁和1uM测试化合物的反应混合物。将测试化合物以1uM最终浓度添加至反应混合物中。反应混合物(没有辅因子)的等分试样在振荡水浴中在37℃下孵育3分钟。对照化合物睾固酮与测试化合物同时在分开的反应中操作。通过添加辅因子(NADPH)引发反应,且接着在振荡水浴中在37℃下孵育混合物。对于测试化合物,在0、10、20、30和60分钟取等分试样(100μL),且对于睾固酮,在0、10、30和60分钟取等分试样(100μL)。测试化合物样品立刻与100μL含有内标的冰冷乙腈组合以终止反应。睾固酮样品立刻与800μL含有0.1%甲酸和内标的冰冷50/50乙腈/dH2O组合以终止反应。使用有效的LC-MS/MS方法分析样品。使用Orbitrap高分辨率质谱仪分析测试化合物样品以定量母体测试化合物的消失并检测代谢物的出现。与内标的峰面积响应比(PARR)与0时PARR相比,以确定保持在时间点的测试化合物或阳性对照的百分比。使用与单相指数式衰减方程式拟合的GraphPad软件计算半衰期。
基于t1/2=0.693k计算半衰期,其中k是基于剩余自然对数百分比对比孵育时间的曲线斜率的消除速度常数。当计算的半衰期比实验的持续时间长时,半衰期表达为>最长孵育时间。计算的半衰期也在括号中列出。如果计算的半衰期>实验的持续时间2x时,那么不报导半衰期。细胞增殖的及时重新开始为组织修复所必需,因此在例如HSPC等健康细胞中过度长期的停滞是不希望有的。决定其停滞持续时间的CDK4/6抑制剂的特征是其药物动力学(PK)和酶半衰期。一旦引发,体内G1停滞将维持,只要循环化合物保持在抑制水平下且只要化合物占用酶。PD032991例如具有整体长久的PK半衰期和相当缓慢的酶解离速率。人类中,PD0332991显示27小时的PK半衰期(参见Schwartz,GK等人(2011)BJC,104:1862-1868)。人类中,PD0332991的单一施用产生HSPC持续大概一周的细胞周期停滞。此反映了6天清除化合物(5个半衰期×27小时半衰期)以及酶CDK4/6功能的另外1.5至2天的抑制。此计算表明正常骨髓功能恢复花费总共7+天,在此期间新血液的产生减少。这些观测可以说明在临床中在PD0332991下见到的严重粒性白细胞减少症。
其它实验用化合物T和PD0332991完成以比较人类、犬、大鼠、猴和小鼠肝微粒体中的代谢稳定性(半衰期)。如图7中所示,当分析化合物在肝微粒体中跨越物种的稳定性时,在每一物种中化合物T的可测定半衰期比针对PD0332991所报导的半衰期短。此外,如先前和图4所述,似乎PD0332991也具有延长的酶半衰期,如人类细胞中持续超过四十小时的显著细胞周期停滞产生所证明,甚至在化合物从细胞培养基(即在体外洗脱实验中)去除后。如图4中进一步展示,本文中描述的化合物从培养基的去除使得增殖迅速重新开始,与快速的酶解离速率一致。酶解离速率的这些差异转变成药效学(PD)作用的显著区别,如图2、5C和6中所示。如所示,单一口服剂量的PD0332991使得鼠类骨髓中造血干细胞和祖细胞(HSPC)36+小时生长停滞,此超过PD0332991在小鼠中6小时PK半衰期所说明。相比之下,化合物T的作用短得多,允许快速再进入细胞周期,提供了HSPC增殖良好的体内控制。
实施例159
CDK4/6抑制剂化合物T在HER2驱动的乳房肿瘤中的功效
表达由MMTV启动子驱动的c-neu(人类HER2的小鼠直系同源物)的HER2驱动乳癌模型(Rb阳性)(Muller WJ,Sinn E,Pattengale PK,Wallace R,Leder P.Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene.Cell 1988;54:105-15)用于以下实施例。选择此模型是因为在鼠类(Yu Q,Geng Y,Sicinski P.Specific protection against breast cancers by cyclin D1ablation.Nature 2001;411:1017-21;Landis MW,Pawlyk BS,Li T,Sicinski P,Hinds PW.Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis.Cancer Cell 2006;9:13-22;Reddy HK,Mettus RV,Rane SG,Grana X,Litvin J,Reddy EP.Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis.Cancer Res 2005;65:10174-8;Yu Q,Sicinska E,Geng Y,Ahnstrom M,Zagozdzon A,Kong Y等人,Requirement for CDK4 kinase function in breast cancer.Cancer Cell 2006;9:23-32)和人类HER2阳性乳癌(An HX,Beckmann MW,Reifenberger G,Bender HG,Niederacher D.Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation.Am J Pathol 1999;154:113-8;Samady L,Dennis J,Budhram-Mahadeo V,Latchman DS.Activation of CDK4 gene expression in human breast cancer cells by the Brn-3b POU family transcription factor.Cancer Biol Ther 2004;3:317-23;Takano Y,Takenaka H,Kato Y,Masuda M,Mikami T,Saegusa M等人,Cyclin D1 overexpression in invasive breast cancers:correlation with cyclin-dependent kinase 4 and oestrogen receptor overexpression,and lack of correlation with mitotic activity.J Cancer Res Clin Oncol 1999;125:505-12)中的先前研究表明这些肿瘤需要CDK4/6和CCND1来进展和维持。
产生MMTV-neu小鼠并在哺乳后观测,其中肿瘤以大概25周的中位等待时间观测。当肿瘤到达允许容易连续评估的标准尺寸(50-60mm3)时小鼠进入疗法研究。负载肿瘤的小鼠连续用添加到其食物中的化合物T(100mg/kg/d或150mg/kg/d)处理。MMTV-c-neu(对照,n=9;化合物T 100mg/kg,n=7;化合物T 150mg/kg,n=6)小鼠每周检验以通过触诊评估肿瘤发展。肿瘤体积是通过下式计算:体积=[(宽度)2×长度]/2。负载肿瘤的小鼠因预定发病、肿瘤溃疡或肿瘤尺寸超过1.5cm直径而在指示时间处死。
如图8和9中所示,在28天疗法过程期间用化合物T连续处理(100mg/kg/d或150mg/kg/d)显著减小肿瘤体积。若干肿瘤显示完全的肿瘤消退并在28天治疗过程期间注意到对化合物T无抗性。这些数据展示此HER2驱动小鼠模型‘依赖于’CDK4/6活性来增殖且化合物T是CDK4/6依赖性的Rb阳性肿瘤中的有效剂。
实施例160
HER2驱动的乳房肿瘤中CDK4/6抑制剂(化合物T、化合物GG和化合物U)的功效
化合物T、化合物GG和化合物U的临床前表征指示其分别以0.7-1.0nM和5-6nM的IC50抑制CDK4和CDK6。在具有功能性Rb蛋白质的肿瘤细胞中,这些化合物有效地抑制Rb磷酸化,引起G1停滞。在乳腺腺腔乳癌的基因工程小鼠模型中测试CDK4/6抑制剂化合物T、化合物GG和化合物U的体内功效。每周使用测径规测量法来连续评估肿瘤。一旦肿瘤到达40-64mm3,就开始治疗介入。使用式((宽度2)×长度)/2计算肿瘤体积。所有三种化合物经由加入药物的饮食(100mg/kg/d)经口施用。加入药物的饮食连续施用28天,接着停止。RECIST准则用以评估客观反应率。客观反应率基于肿瘤体积变化百分比,使用以下类别分类:CR(完全反应)=100%反应;PR(部分反应)=至少30%降低;SD(稳定疾病)=无变化(无PR和无PD);和PD(进行性疾病)=20%增加。
如图10中所示,在用化合物T、化合物GG和化合物U处理的小鼠中注意到客观反应。处理组耐受良好,没有毒性的临床征象,没有重量减轻,且没有与毒性相关的死亡。线性回归t检验用以确定随着时间推移的肿瘤体积生长的统计显著性。所有三组与非处理组相比都是统计上显著的;化合物T,p<0.0001;化合物GG,p=0.0001;化合物U,p<0.0001。如图10中所示,确定每一客观类别中的动物数目。发现化合物T具有100%的客观反应率(n=7),发现化合物GG具有85%的客观反应率(n=7),且发现化合物U具有100%的客观反应率(n=8)。图11中,展示来自用化合物T、化合物GG和化合物U处理的MMTV-Neu小鼠的肿瘤体积,其中每七天测量肿瘤体积。图12中,展示每一个别肿瘤的最佳反应(14天或更迟)的数据。总的来说,这些数据展示,在28天疗法过程期间用化合物T(100mg/kg)、化合物GG(100mg/kg)或化合物U(100mg/kg)连续处理引起肿瘤体积显著下降。
实施例161
CDK4/6依赖性细胞中化合物T使细胞周期停滞
为测试CDK4/6抑制剂诱发彻底G1停滞的能力,使用基于细胞的筛选法,其由两种CDK4/6依赖性细胞系(tHS68和WM2664;Rb阳性)和一种CDK4/6非依赖性(A2058;Rb阴性)细胞系组成。涂铺后二十四小时,将每一细胞系用化合物T以剂量依赖性方式处理24小时。在实验结束时,收获细胞,固定并用碘化丙锭(DNA插入剂)染色,当通过488nm光激发时发出强烈红色荧光(发射最大值637nm)。样品在Dako Cyan流动式细胞测量仪上操作并针对每一样品,收集>10,000个事件。使用TreeStar,Inc.研发的FlowJo 2.2软件分析数据。
图29A中,结果展示化合物T在人类肾近端小管细胞中诱发稳健的G1细胞周期停滞,因为在用增加量的化合物T处理后发现几乎所有的细胞都在G0-G1期。图29A中,结果展示在CDK4/6依赖性细胞系中,化合物T以80nM的EC50诱发tHS68细胞中稳健的G1细胞周期停滞,其中展示S期对应减少在基线下28%至最高浓度下6%的范围内。在用化合物T(300nM)处理后,两种CDK4/6依赖性细胞系(tHS68(比较图29B和29E)和WM2664(比较图29C和29F))中S期群体类似减少和G1停滞细胞增加,但CDK4/6非依赖性(A2058;比较图29D和29G)细胞系中没有。CDK4/6非依赖性细胞系展示在抑制剂存在下无效。
实施例162
化合物T抑制RB的磷酸化
CDK4/6-细胞周期蛋白D复合物对DNA细胞周期从G1进展至S期来说是必不可少的。此复合物使成视网膜细胞瘤肿瘤抑制蛋白(Rb)磷酸化。为证明CDK4/6抑制对Rb磷酸化(pRb)的影响,化合物T暴露于三个细胞系,两个CDK4/6依赖性(tHS68、WM2664;Rb阳性)和一个CDK4/6非依赖性(A2058;Rb阴性)。接种后二十四小时,将细胞用300nM最终浓度的化合物T处理4、8、16和24小时。将样品溶解并通过蛋白质印迹分析进行分析。使用物种特异性的抗体,在CDK4/6细胞周期蛋白D复合物靶向的两个位点Ser780和Ser807/811,测量Rb磷酸化。结果证明至暴露后16小时,化合物T在Rb依赖性细胞系中阻断Rb磷酸化,同时对Rb非依赖性细胞没有影响(图30)。
已经参考本发明的实施方案描述本说明书。已经参考以随附实施例说明的分类实施方案描述本发明。然而,本发明可以用不同的形式具体化,且不应该被视作局限于本文中阐述的实施方案。在本文中的传授内容下,本领域技术人员将能够修改本发明以达到想要的目的且此类改变视为在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!