包含1-茚满硫酰胺衍生物的疼痛治疗剂和/或预防剂的制作方法
本发明涉及疼痛治疗剂和/或预防剂,其包含1-茚满硫酰胺衍生物、其盐或其前药。
背景技术:
根据国际疼痛研究协会(International Association for the Study of Pain,IASP)的分类所述,“疼痛”解释作“与实际或潜在的组织损伤相关的不愉快的感觉和情绪体验,或者依据这种损伤描述的不愉快的感觉和情绪体验”(非专利文献1)。
疼痛通常分为急性的或慢性的。急性疼痛是存在不超过三个月的疼痛。在大多数情况下,急性疼痛突然开始并且在特性方面是急剧的。急性疼痛可能由许多事件或情况所引起,如外科手术、断骨、牙科治疗、烧伤或割伤等。慢性疼痛是持续不少于三个月的疼痛。常见的慢性疼痛包括头痛、腰痛、癌性疼痛、关节痛、神经性疼痛、精神性疼痛(在无身体疼痛原因(如过去的疾病或损伤)的情况下发生的疼痛)。神经性疼痛,也翻译为神经源性疼痛,是由周围或中枢躯体感觉神经系统的损伤或疾病引起的顽固性疼痛,包括糖尿病神经病变、三叉神经痛、以及带状疱疹后遗神经痛(非专利文献2和3)。
疼痛是一项常见的医学问题,而缓解疼痛是一项重要的治疗目标。用镇痛药来治疗疼痛最为常见。镇痛药大致分为三个种类:(1)阿片类镇痛药;(2)非阿片类镇痛药,如抗炎类固醇、扑热息痛、以及安乃近;以及(3)“辅助镇痛药”(一组不同的药物,其被称为“不具有作为主要药理作用的镇痛作用,但当与镇痛药结合使用时可增强镇痛效果并且在选定的情况下显示出镇痛效果的药物”)。
尽管阿片类镇痛药通过作用于中枢神经系统中的阿片受体来提供强的镇痛效果;由于其严重的药品不良反应与依赖性,其用途受到限制。虽然非阿片类镇痛药具有镇痛效果,但效果微弱而且可引起多种药品不良反应。此外,尚未发现对慢性疼痛(如与糖尿病神经病变、三叉神经病变以及带状疱疹相关的神经性疼痛)有效的治疗药物,而且所期望的是开发一种对宽范围的疼痛(包括急性疼痛和慢性疼痛)有效的药物。
尽管目前多种镇痛药可供用于治疗疼痛,但在疼痛治疗方面仍存在巨大的、未满足的医疗需要。最近的报道估计四位经历外科治疗的患者中只有一位实现了对急性疼痛有充分的镇痛效果(非专利文献4和5)。此外,急性疼痛的不完全治疗可导致多种症状,包括焦虑、抑郁、失眠、疲劳、食欲下降、恶心和呕吐。另外,未被减轻的急性疼痛可进展成慢性疼痛。
从另一方面来说,关于慢性疼痛,WHO估计世界人口的20%患有某种程度的慢性疼痛。慢性疼痛对直接医疗成本与相关的间接成本(例如,伤残补偿、生产率损失)都有显著影响。由于大约40%患有慢性疼痛的患者无法实现充分的减轻,现在慢性疼痛被认为是一项重要的公共健康问题(非专利文献6)。
急性疼痛和慢性疼痛的有效治疗对于很多患者来说仍然是未满足的医疗需要。因此,强烈期望开发一种对急性疼痛和慢性疼痛有效的治疗剂。
作为急性疼痛的动物模型,用于评估瞬时疼痛的模型,如甩尾试验、退缩/顶锻试验(flinch/jump test)、热板试验、挤捏试验是已知的。作为用于急性持久性疼痛的模型,使用福尔马林测试。(非专利文献7)
从另一方面来说,作为慢性疼痛的模型,大鼠慢性压迫性损伤模型(CCI模型)等是已知的。利用疼痛原因进行分类,CCI模型被认为是与神经性疼痛对应的疾病模型。(非专利文献8)
因为硫酰胺衍生物具有镇痛效果,专利文献1和2中披露的低分子化合物是已知的。然而,具有镇痛效果的1-茚满硫酰胺衍生物是尚未知的。
引用清单
专利文献
专利文献1:WO 2007/095615
专利文献2:WO 2007/075752
非专利文献
非专利文献1:International Association for the Study of Pain(国际疼痛研究协会)(1979),“Pain Definitions(疼痛定义)”,Pain(疼痛)6(3):247-248,
非专利文献2:“Japanese translation of Neuropathic Pain-A report of Terminology Committee,the Japan Society of Pain Clinicians”(神经性疼痛的日语翻译-术语委员会报告书,日本疼痛医师协会),Journal of Japan Society of Pain Clinicians(日本疼痛医师协会杂志)(2009),16(4)509-514,
非专利文献3:Treede(特里德),R.D.,Jensen(詹森),T.S.,Campbell(坎贝尔),J.N.等人,(2008),“Neuropathic pain:Redefinition and a grading system for clinical and research purposes(神经性疼痛:临床和研究目的的重新定义与分级系统)”,Neurology(神经学)70:1630-1635,
非专利文献4:Phillip(菲利普),D.M.(2000),“JCAHO:Pain management standards are unveiled(JCAHO:揭露疼痛管理标准)”,JAMA 284(4):428-429
非专利文献5:Wu(吴),C.L.与Raja(拉贾),S.N.(2011),“Treatment of acute postoperative pain(术后剧烈疼痛的治疗)”,Lancet(柳叶刀)377(9784):2215-2225
非专利文献6:Breivik(布雷维克),H.,Collett(科利特),B.,Ventafridda(范塔弗里达),V.,Cohen(科恩),R.与Gallacher(加拉赫),D.(2006),“Survey of chronic pain in Europe:Prevalence,impact on daily life,and treatment(欧洲慢性疼痛调查:流行率、日常生活的影响、以及治疗)”,European Journal of Pain(欧洲疼痛杂志)10(4):287-333
非专利文献7:Dubuisson(迪比松),D.与Stephen(斯蒂芬),G.(1977),“The formalin test:A quantitative study of the analgesic effects of morphine,meperidine,and brain stem stimulation in rats and cats(福尔马林测试:吗啡、哌替啶、与脑干刺激对大鼠与猫的镇痛效果的定量研究)”,Pain(疼痛)4(2):161-174
非专利文献8:Wang(王),L.X.与(王),Z.(2003),“Animal and cellular models of chronic pain(慢性疼痛的动物与细胞模型)”Advanced Drug Delivery Reviews(先进药物递送评论)55(8):949-965
发明概述
技术问题
本发明的一个目的在于提供疼痛治疗剂和/或预防剂,其在不同动物模型中展示出镇痛效果并且可以适用于不同疼痛类型。
问题的解决方案
本发明诸位发明人进行了如下研究,分别使用小鼠热板试验以证实对急性疼痛的镇痛效果和使用大鼠慢性压迫性损伤(CCI)模型以证实对慢性疼痛的镇痛效果。
作为使用小鼠热板试验的研究结果,诸位发明人发现1-茚满硫酰胺衍生物对由疼痛性刺激诱发的急性疼痛具有抑制作用。此外,作为使用大鼠慢性压迫性损伤(CCI)模型的研究结果,诸位发明人发现1-茚满硫酰胺衍生物对由神经结扎导致的慢性疼痛具有抑制作用。诸位发明人因此完成了本发明。
具体地说,本发明涉及:
[1]一种疼痛治疗剂和/或预防剂,其包含选自下组的化合物:
(1)N-[(1S)-2,2,5,7-四氟-2,3-二氢-1H-茚-1-基]硫酰胺、
(2)(-)-N-(7-氯-2,2,5-三氟-2,3-二氢-1H-茚-1-基)硫酰胺、
(3)N-[(1S)-2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺、以及
(4)N-[(1S)-2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺、或其药学上可接受的盐;
[2]根据[1]所述的治疗剂和/或预防剂,其中该疼痛是急性疼痛或慢性疼痛;
[3]根据[1]所述的疼痛治疗剂和/或预防剂,其中该疼痛是神经性疼痛;
[4]根据[1]所述的疼痛治疗剂和/或预防剂,其中该疼痛是糖尿病神经病变、三叉神经病变或带状疱疹后遗神经痛;
[5]根据[1]至[4]中任一项所述的疼痛治疗剂和/或预防剂,其中该剂经口服地、舌下地、鼻内地、直肠地、齿龈内地、静脉内地、肌内地、关节内地、皮下地、吸入地、经皮地或硬膜外地给予;以及
[6]根据[1]至[4]中任一项所述的疼痛治疗剂和/或预防剂,其中该剂经口服地、舌下地、静脉内地、肌内地、关节内地、皮下地、经皮地或硬膜外地给予。
本发明的有利效果
根据本发明所述的剂在小鼠热板试验中对由疼痛性刺激诱发的急性疼痛以及在大鼠CCI模型中对由神经结扎导致的慢性疼痛具有抑制作用。此外,根据本发明所述的剂在小鼠福尔马林测试中展示出镇痛效果,该福尔马林测试是急性持久性疼痛的动物模型。本发明所述的剂因此可以用作急性疼痛和慢性疼痛的治疗剂和/或预防剂。
附图简要说明
图1是示出其中给予测试化合物1、2、3和4的测试实例1的结果的图。
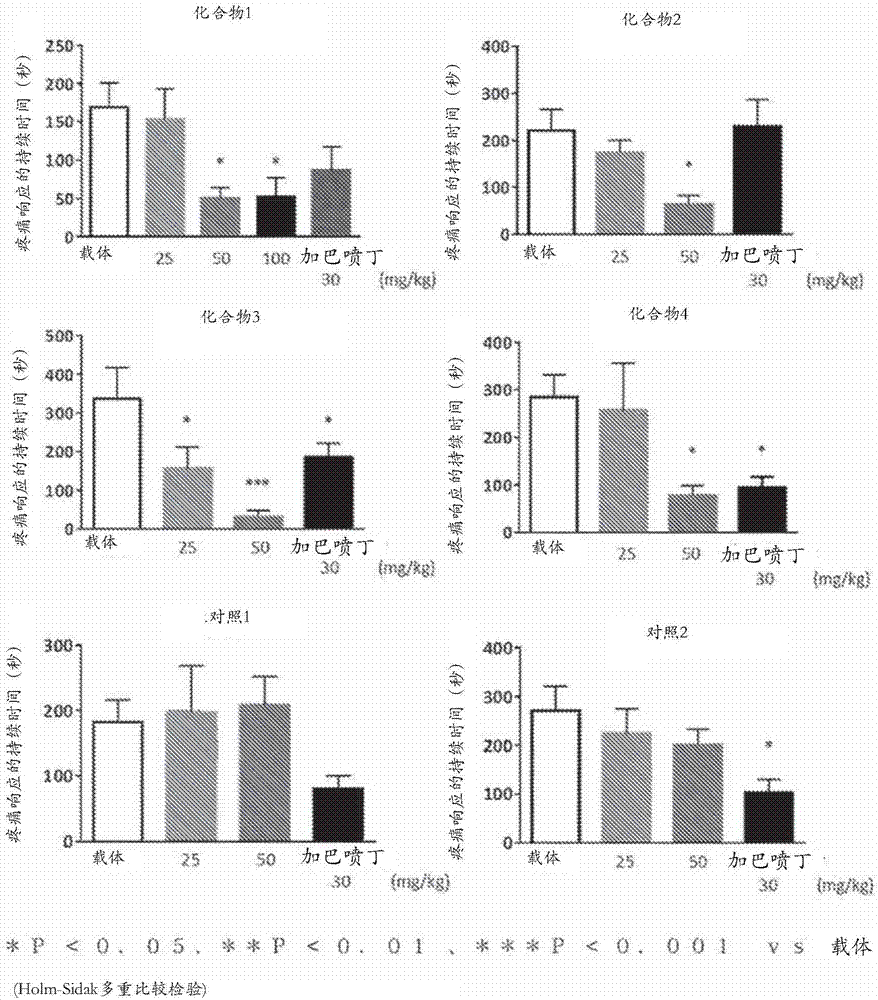
图2是示出其中给予测试化合物1、2、3和4的测试实例3的结果的图。
实施例的描述
下面将详细说明本发明。
尽管用于本发明中的1-茚满硫酰胺化合物可具有晶体多晶型物,但是该化合物并不被局限于这些多晶型物中的任一种并且可以作为单晶形式或单晶形式的混合物而存在。并且还包括非晶形式。
此外,该化合物可形成药学上可接受的盐以及不同的溶剂化物。
在下文中,将解释在本说明书中描述的术语、符号等的含义。
在本说明书中的“药学上可接受的盐”并不具体受限,只要它与化合物形成一种盐并且是药学上可接受的即可。
溶剂化物意指在反应或结晶中使用的溶剂被掺入在晶体中而不与化合物的分子或离子形成共价键的状态。溶剂化物的实例是水合物、醇化物(乙醇化物)等。
在用于本发明中的化合物的产生中,起始材料化合物、中间体以及各种试剂可以形成盐或溶剂化物,都取决于起始材料、所用溶剂等而变化,并且不具体受限,只要它们不抑制反应即可。并且,毋庸多言,所用溶剂取决于起始材料、试剂等而变化,并且不具体受限,只要它不抑制反应并且在一定程度上溶解起始材料即可。当获得呈游离态的化合物时,可以通过常规方法将它们转化为可接受的盐或溶剂化物。
本发明的化合物或中间体的各种异构体(如几何异构体、旋光异构体、旋转异构体、立体异构体、互变异构体等)可以使用常用分离方法进行纯化和分离,例如,重结晶、非对映异构体盐形成、酶拆分以及各种层析法(如薄层层析、柱层析以及气相层析)。
这些用于本发明中的化合物或其药学上可接受的盐可以通过常规方法来配制,并且剂型的实例包括口服配制品(如片剂、颗粒剂、粉剂、胶囊剂以及糖浆)、舌下片剂、注射液(用于静脉内给予、肌肉内给予、皮下给予、腹膜内给予、关节内给予、或硬膜外给予)以及外用制剂(如经皮吸收配制品(如软膏和贴剂)、鼻用制剂、栓剂等)。
这些固体配制品(例如片剂、胶囊剂、颗粒剂以及粉剂)通常可以包含0.001wt%至99.5wt%,优选0.01wt%至90wt%等的用于本发明中的化合物或其药学上可接受的盐。
当生产口服固体配制品时,可以通过根据需要向用于本发明中的这些化合物或其药学上可接受的盐中添加稀释剂、粘合剂、崩解剂、润滑剂、着色剂等并且通过常规方法进行处理来制备片剂、颗粒剂、粉剂以及胶囊剂。根据需要这些配制品还可以是薄膜包衣的。
稀释剂的实例包括乳糖、玉米淀粉以及微晶纤维素,粘合剂的实例包括羟丙基纤维素和羟丙基甲基纤维素,并且崩解剂的实例包括羧甲基纤维素钙和交联羧甲纤维素钠。
润滑剂的实例包括硬脂酸镁和硬脂酸钙,并且着色剂的实例包括二氧化钛。
薄膜包衣剂的实例包括羟丙基纤维素、羟丙基甲基纤维素以及甲基纤维素。
当然,对上述赋形剂没有限制。
对于注射液(用于静脉内给予、肌肉内给予、皮下给予、腹膜内给予、关节内给予或硬膜外给予)的生产,可以视情况需要将pH调节剂、缓冲剂、助悬剂、乳化剂、增溶剂、抗氧化剂、防腐剂(抗菌剂)、等渗剂等添加至该化合物或其药学上可接受的盐,而且生产可通过普通方法进行。也可以将其冷冻干燥为在使用时溶解的冷冻干燥制剂。例如,这类注射液给予至静脉、皮肤下或肌肉内。
pH值调节剂和缓冲剂的实例包括有机酸或无机酸和/或其盐,悬浮剂的实例包括甲基纤维素、聚山梨醇酯80以及羧甲基纤维素钠,乳化剂的实例包括聚氧乙烯蓖麻油、羟丙基纤维素以及卵磷脂,增溶剂的实例包括聚山梨醇酯80和聚氧化乙烯脱水山梨糖醇单月桂酸酯,抗氧化剂的实例包括α-生育酚,防腐剂的实例包括对羟基苯甲酸甲酯和对羟基苯甲酸乙酯,而等渗剂的实例包括葡萄糖、氯化钠以及甘露醇,当然并不特别地限制于这些。
这类注射液通常可以包含0.00001wt%至99.5wt%,优选0.0001wt%至90wt%的用于本发明中的化合物或其药学上可接受的盐。
对于外用制剂的生产,可以将一种基础起始材料添加至本发明的化合物或其药学上可接受的盐,并且必要时可以添加上述乳化剂、防腐剂、pH调节剂或着色剂中的任一种通过普通方法以生产例如经皮吸收制剂(软膏、医用贴剂等)、滴鼻剂或栓剂。
用于药物、准药物、化妆品等的常规使用的各种原材料可以用作基础材料,并且实例包括以下原材料,如动物和植物油、矿物油、酯油、蜡、高级醇以及净化水。
这类外用制剂通常可以包含0.00001wt%至99.5wt%,优选0.0001wt%至90wt%的用于本发明中的化合物或其药学上可接受的盐。
根据本发明的药物的剂量典型地取决于症状、年龄、性别、体重等而变化,但如果剂量足以展现出所希望的作用,就是可接受的。例如,对于成年人来说,以一天或多天一个剂量的方式或以一天2至6个分次剂量的方式每天使用约0.1至5000mg(优选0.5至1000mg,更优选1至600mg)的剂量。
实例
例如,可以通过以下产生实例中所述的方法来生产本发明中所使用的化合物,并且可以通过以下测试实例中所述的方法来证实这些化合物的作用。然而,这些方法是说明性的并且可以在不背离本发明的范围的情况下进行改变,并且本发明在任何情况下不被局限于以下具体实例。
根据公开物等生产其上附有公开物名称等的化合物。
本说明书中使用的所有缩写都是本领域的普通技术人员已知的常规缩写。将在以下实例中使用以下缩写。
BAST:双(2-甲氧基乙基)氨基三氟化硫
Bn:苄基
Boc:叔丁氧基羰基
DCM:二氯甲烷
DMF:N,N-二甲基甲酰胺
DMSO:二甲亚砜
1H-NMR:质子核磁共振光谱测定法
HPLC:高效液相层析
I.D.:内径
LC-MS:液相层析-质谱法
m-:间-
n-:正-
NBS:N-溴丁二酰亚胺
o-:邻-
p-:对-
PPTS:对甲苯磺酸吡啶鎓
SelectfluorTM:N-氟-N’-氯甲基-三亚乙基二胺-双(四氟硼酸盐)
t-:叔-
TBS:叔丁基二甲基甲硅烷基
TEA:三乙胺
THF:四氢呋喃
THP:四氢吡喃
Z(Cbz):苄氧基羰基
以下制备实例中的“室温”典型地是指约10℃至约35℃。除非另外说明,“%”表示wt%。然而,硅胶层析中的溶剂比率示出了有待混合的溶剂的体积比。
质子核磁共振光谱中的化学位移以相对于四甲基硅烷的δ单位(ppm)记录并且偶联常数以赫兹(Hz)记录。将图谱指定为s:单峰,d:双峰,t;三重峰,q:四重峰,m:多重峰,brs;宽单峰。
通过GILSON HPLC系统(泵;主泵型号305,从泵型号306,泵头50SC,动态混合器型号811D/A,测压模块型号806,UV检测器;UV/VIS检测器型号155,注射器,分馏收集器;型号215,柱;选自DAICELAD-H、IA、IB、IC、ID、IE、IF、DAICELOD-H、OJ-H,20mm I.D.×250mm)进行化合物的旋光拆分。在通过UV检测器检测馏分后,使用旋光检测器(OR-2090,JASCO,汞-氙(Hg-Xe)灯,150W)测量旋光性(+/-)。
相对于层析,如果描述了一种硅胶柱层析,则使用山善株式会社(YAMAZEN)平行制备型(柱:山善株式会社Hi-FlashTM柱(硅胶),大小;S(16×60mm),M(20×75mm),L(26×100mm),2L(26×150mm)或3L(46×130mm)),富士硅化工有限公司(FUJI SILYSIA CHEMICAL CO.,LTD.)的层析用球形硅胶PSQ 60BTM,富士硅化工有限公司的层析用硅胶BW-300TM,C-200(和光纯药工业株式会社(Wako Pure Chemical Industries,Ltd.)),或日本默克股份有限公司(Merck Ltd.Japan)的硅胶60TM(70-230目)。并且,如果描述了NH硅胶柱层析,则使用山善株式会社平行制备型(柱:山善株式会社Hi-FlashTM柱(氨基),大小;S(16×60mm),M(20×75mm),L(26×100mm),2L(26×150mm)或3L(46×130mm))或富士硅化工有限公司的NH SILICA GEL(200-350目)。
在本说明书中的化合物的命名法中,(+)-、(-)-、(R)和(S)分别表示对映异构体的(+)、(-)、(R)和(S)构型。并且,空间构型中的“*”示出了相对构型,并且除非另外表明,否则它意指某一对映异构体。
产生实例1
N-[(1S)-2,2,5,7-四氟-2,3-二氢-1H-茚-1-基]硫酰胺的合成
(1)2,5,7-三氟-2,3-二氢-1H-茚-1-酮的合成
在室温下,将SelectfluorTM(1.16g,3.27mmol)添加至MeOH(20mL)中的5,7-二氟-1-茚满酮(CAS号84315-25-3,500mg,2.97mmol)溶液中。将该混合物回流2小时并冷却至室温。然后,将溶剂在减压下蒸馏掉。将该残余物用DCM处理并且过滤掉不溶物质。然后,将溶剂在减压下蒸馏掉。将该残余物溶解于MeCN(10mL)和5N HCl(5mL)中。将该溶液在室温下搅拌1小时,并且然后在真空中浓缩。将该残余物在AcOEt与H2O之间进行分段。将该有机层用盐水洗涤,经MgSO4干燥,并且过滤。将溶剂在真空中浓缩,以提供标题化合物(547mg,2.94mmol)。
1H-NMR(400MHz,CDCl3):δppm 3.11-3.36(m,1H)3.49-3.77(m,1H)5.10-5.40(m,1H)6.82(td,J=9.0,1.9Hz,1H)6.90-7.04(m,1H)。
(2)2,2,5,7-四氟-2,3-二氢-1H-茚-1-酮的合成
在0℃下,将叔丁基二甲基甲硅烷基三氟甲磺酸酯(1.00mL,4.35mmol)添加至DCM(20mL)中的产生实例1-(1)中获得的产物(540mg,2.90mmol)和TEA(1.21mL,8.70mmol)的溶液中。将该混合物在室温下搅拌5小时。然后,向该反应混合物中添加二乙醚和饱和水性Na2CO3并将各层分离。将有机层依次用1N HCl、饱和水性Na2CO3和盐水洗涤,并用Na2SO4干燥。将溶剂在真空中蒸发并且将该残余物在减压下干燥。将该残余物溶解于MeCN(20mL)中,并且在室温下添加SelectfluorTM(1.13g,3.19mmol)。将该混合物在相同温度下搅拌11小时后,将溶剂在减压下蒸馏掉。将该残余物溶解于DCM中并且过滤掉不溶物质。将滤液在真空中浓缩。通过快速柱层析法(山善株式会社HI-FLASHTM柱硅胶L大小,20mL/min,梯度为正庚烷中的10%至50%AcOEt)纯化该残余物,以提供呈白色固体的标题化合物(532mg,2.61mmol)。1H-NMR(400MHz,CDCl3):δ(ppm)3.57(t,J=12.4Hz,2H)6.74-6.94(m,1H)6.95-7.08(m,1H)。
(3)2,2,5,7-四氟-2,3-二氢-1H-茚-1-胺的合成
在室温下,将乙酸铵(4.27g,55.4mmol)添加至异丙醇(16mL)中的产生实例1-(2)中获得的产物(377mg,1.85mmol)的溶液中并且将该混合物回流30min。将氰基硼氢化钠(348mg,5.54mmol)添加至该反应混合物中并在回流下搅拌7小时。冷却至室温后,将AcOEt和2N NaOH添加至该反应混合物中,并且将层分离。将有机层在真空中浓缩。将水添加至该残余物中,并且在AcOEt与1N HCl之间进行分段。将水层用2N NaOH碱化并用AcOEt萃取。将有机层用Na2SO4干燥,蒸发并干燥,以提供标题化合物(210mg,1.02mmol)。
ESI-MS;m/z 206[M+H]+。
1H-NMR(400MHz,CDCl3):δ(ppm)3.26-3.55(m,2H)4.59(dd,J=13.3,5.3Hz,1H)6.61-6.86(m,2H)。
(4)N-(2,2,5,7-四氟-2,3-二氢-1H-茚-1-基)氨磺酰基氨基甲酸苄酯的合成
在室温下,向在产生实例1-(3)中获得的产物(200mg,0.975mmol)的DCM溶液(10mL)中添加[(苄氧基)羰基]{[4-(二甲基亚氨基)吡啶-1(4H)-基]磺酰基}酰胺(CAS号1037211-09-8,654mg,1.95mmol,根据描述于WO 2008/083248中的方法制备)和TEA(0.55mL,3.90mmol)。将所得溶液在回流下搅拌24小时。冷却至室温后,将AcOEt和1N HCl添加至该反应混合物中。将层分离,并且将有机层用MgSO4干燥并在真空中蒸发。通过柱层析(硅胶,正庚烷中的30%AcOEt)纯化该残余物,以提供呈白色固体的标题化合物(316mg,0.755mmol)。
ESI-MS;m/z 441[M+Na]+。
1H-NMR(400МHz,CDCl3)δ(ppm):3,25-3,54(m,2H)5,14-5,38(m,3H)5,72(brs,1H)6,72(t,J=9,4Hz,1H)6,79(d,J=7,8Hz,1H)7,30-7,46(m,5H)7.51(brs,1H)。
(5)N-[(1S)-2,2,5,7-四氟-2,3-二氢-1H-茚-1-基]硫酰胺的合成
在室温下,将钯-碳(10w/w%,30mg,0.028mmol)添加至MeOH(5mL)和AcOEt(5mL)中的产生实例1-(4)中获得的产物(310mg,0.741mmol)的溶液中。将所得溶液在室温下、在H2气氛下搅拌30min。将AcOEt添加至该反应混合物中,并且通过过滤以除去钯-碳。将滤液在真空中浓缩。通过快速柱层析法(山善株式会社HI-FLASHTM柱硅胶,大小为M,10mL/min,梯度为正庚烷中的30%至70%AcOEt)纯化该残余物,以提供呈外消旋体的标题化合物(181mg,0.637mmol)。通过HPLC(CHIRALPAKTMIA,20mm I.D.×250mm,10mL/min,己烷中的15%EtOH)进行所得外消旋体(180mg,0.633mmol)的旋光拆分,以提供呈白色固体的标题化合物的S-形式(76mg,0.267mmol,98%ee),该形式是在2种异构体之中以44min的保留时间被第二洗脱的。
ESI-MS;m/z307[M+Na]+。
1H-NMR(400MHz,CDCl3)δ(ppm):3.32-3.60(m,2H),4.70(brs,2H),4.93(d,J=9.3Hz,1H),5.30(q,J=9.3Hz,1H),6.70-6.86(m,2H)。
产生实例2
(-)-N-(7-氯-2,2,5-三氟-2,3-二氢-1H-茚-1-基)硫酰胺的合成
(1)7-氯-2,5-二氟-2,3-二氢-1H-茚-1-酮的合成
在室温下,将SelectfluorTM(2.49g,7.02mmol)添加至MeOH(30mL)中的7-氯-5-氟-1-茚满酮(CAS号1260008-48-7,1.08g,5.85mmol)溶液中。将该混合物回流2小时。冷却至室温后,将溶剂在减压下蒸发。向该残余物中添加DCM并且过滤掉不溶物质。将滤液在真空中浓缩。将该残余物溶解于MeCN(20mL)和5N HCl(10mL)中并且将该溶液在室温下搅拌1小时。在真空中浓缩该溶液后,将残余物在AcOEt与H2O之间进行分段。将该有机层用盐水洗涤,经MgSO4干燥,并且过滤。将滤液在真空中浓缩,以提供标题化合物(1.13g,5.58mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):3.13-3.33(m,1H)3.47-3.71(m,1H)5.25(ddd,J=51.0,8.0,4.5Hz,1H)7.07(dt,J=7.6,2.0Hz,1H)7.14(dd,J=8.8,2.0Hz,1H)。
(2)7-氯-2,2,5-三氟-2,3-二氢-1H-茚-1-酮的合成
在0℃下,将叔丁基二甲基甲硅烷基三氟甲磺酸酯(2.56mL,11.2mmol)添加至DCM(30mL)中的产生实例2-(1)中获得的产物(1.13g,5.58mmol)和TEA(3.11mL,22.3mmol)的溶液中。将该混合物在室温下搅拌2小时。将该反应混合物用二乙醚和饱和水性Na2CO3稀释并将层分离。将有机层依次用1N HCl、饱和水性Na2CO3和盐水洗涤,并用Na2SO4干燥。过滤后,将溶剂在真空中蒸发。将该残余物溶解于MeCN(30mL)中,并且在室温下添加SelectfluorTM(2.17g,6.11mmol)。将该混合物在室温下搅拌3小时,并且然后将所得混合物在减压下蒸发。向该残余物中添加DCM并且过滤掉不溶物质。将溶剂在真空中蒸发。通过快速柱层析法(山善株式会社HI-FLASHTM柱硅胶L大小,20mL/min,梯度为正庚烷中的0%至30%AcOEt)纯化该残余物,以提供标题化合物(2)(1.11g,5.03mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):3.47-3.63(m,2H)7.06-7.13(m,1H)7.17-7.23(m,1H)。
(3)7-氯-2,2,5-三氟-2,3-二氢-1H-茚-1-胺的合成
在室温下,将乙酸铵(11.5g,150mmol)添加至异丙醇(40mL)中的产生实例2-(2)中获得的产物(1.10g,4.98mmol)的溶液中。将该混合物回流30min。将氰基硼氢化钠(940mg,15.0mmol)添加至该反应混合物中并且将该混合物在回流下加热12小时。在冷却至室温后,将该反应混合物用AcOEt稀释,并且添加2N NaOH。将层分离并且将有机层在真空中浓缩。向该残余物中添加水、AcOEt和1N HCl,并且将各层分离。将水层用2N NaOH碱化并用AcOEt萃取。将有机层经Na2SO4干燥。过滤后,将溶剂在真空中蒸发。通过快速柱层析法(山善株式会社HI-FLASHTM柱硅胶L大小,20mL/min,梯度为正庚烷中的10%至50%AcOEt)纯化该残余物,以提供标题化合物(3)(699mg,3.15mmol)。1H-NMR(400MHz,CDCl3)δ(ppm):3.24-3.41(m,1H)3.47-3.65(m,1H)4.50(d,J=14.6Hz,1H)6.85-6.93(m,1H)7.02(dd,J=9.0,2.2Hz,1H)。
(4)N-(7-氯-2,2,5-三氟-2,3-二氢-1H-茚-1-基)氨磺酰基氨基甲酸叔丁酯的合成
在室温下,将[(叔丁氧基)羰基]{[4-(二甲基亚氨基)吡啶-1(4H)-基]磺酰基}酰胺(CAS号872496-91-8,1.90g,6.31mmol,根据描述于Organic Letters(有机快报),3,2241(2001)中的方法制备)和TEA(1.76mL,12.6mmol)添加至DCM(20mL)中的产生实例2-(3)中获得的产物(699mg,3.15mmol)的溶液中。将所得混合物在回流下加热12小时。冷却至室温后,向该反应混合物中添加AcOEt和1N HCl并且将层分离。将有机层经MgSO4干燥。过滤后,将溶剂在真空中蒸发。通过硅胶柱层析(硅胶,正庚烷中的30%AcOEt)纯化该残余物,以提供标题化合物(4)(1.08g,2.69mmol)。
1H-NMR(400MHz,CDCl3)
δ(ppm):1.49(s,9H)3.28-3.55(m,2H)5.07-5.36(m,1H)5.51-5.70(m,1H)6.89(d,J=7.8Hz,1H)7.07(d,J=9.2Hz,1H)7.29(brs,1H)。
(5)(-)-N-(7-氯-2,2,5-三氟-2,3-二氢-1H-茚-1-基)硫酰胺的合成
向AcOEt(25mL)中的产生实例2-(4)中获得的产物(1.08g,2.69mmol)的溶液中添加AcOEt(26.9ml,108mmol)中的4N HCl并且将该混合物在室温下搅拌5小时。将溶剂在真空中蒸发并且通过硅胶快速柱层析法(山善株式会社HI-FLASHTM柱,大小为L,20mL/min,梯度为正庚烷中的30%至70%AcOEt)纯化该残余物,以提供呈外消旋体的标题化合物(627mg,2.09mmol)。通过HPLC(CHIRALPAKTMIB,20mm I.D.x 250mm,10mL/min,正己烷中的10%EtOH)进行所得外消旋体(200mg,0.665mmol)的旋光拆分,以提供标题(-)-形式(83mg,0.276mmol,96%ee),该形式在2种旋光异构体之中以49min的保留时间被第二个洗脱。
ESI-MS;m/z:323[M+Na]+
1H-NMR(400MHz,CDCl3)δ(ppm):3.35-3.64(m,2H),4.74(brs,2H),4.86(d,J=8.6Hz,1H),5.07-5.28(m,1H),6.83-6.95(m,1H),7.09(dd,J=8.7,2.3Hz,1H)。
产生实例3
N-[(1S)-2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺
(1)2-氟-7-甲基-2,3-二氢-1H-茚-1-酮的合成
在室温下,向MeOH(18mL)中的7-甲基-1-茚满酮(CAS号39627-61-7,513mg,3.51mmol)溶液中添加SelectfluorTM(1.49g,4.21mmol)。将该反应混合物在回流下加热2小时。冷却至室温后,将溶剂在减压下蒸发。将该残余物用DCM处理并且过滤掉不溶物质。将滤液在真空中浓缩。将该残余物溶解于MeCN(10mL)和5N HCl(5mL)中。将该溶液在室温下搅拌30min。在真空中浓缩该溶液后,将残余物在AcOEt与H2O之间进行分段。将水层用AcOEt萃取两次。将合并的有机层用盐水洗涤,用MgSO4干燥,过滤并在真空中浓缩,以提供标题化合物(555mg,3.38mmol)。
1H NMR(400MHz,CDCl3)δppm:2.64(s,3H),3.18(ddd,J=23.4,16.8,4.3Hz,1H),3.57(ddd,J=16.8,7.8,7.5Hz,1H),5.21(ddd,J=51.2,7.8,4.3Hz,1H),7.17(d,J=7.4Hz,1H),7.26(d,J=7.4Hz,1H),7.51(t,J=7.4Hz,1H)。
(2)2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-酮的合成
在0℃下,将叔丁基二甲基甲硅烷基三氟甲磺酸酯(1.55mL,6.74mmol)添加至DCM(30mL)中的产生实例3-(1)中获得的产物(555mg,3.38mmol)和TEA(1.88mL,13.49mmol)的溶液中。将该混合物在室温下搅拌1.5小时。将该反应用饱和NaHCO3淬灭,并且将层分离。将水层用DCM萃取。将合并的有机层用盐水洗涤并用MgSO4干燥。将不溶物质过滤掉并且将滤液在真空中浓缩。将该残余物溶解于MeCN(20mL)中,并且在室温下添加SelectfluorTM(1.32g,3.73mmol)。将该反应混合物在室温下搅拌1h后,将溶剂在减压下蒸发。将该残余物溶解于DCM中并且过滤掉不溶物质。将滤液在真空中浓缩。通过硅胶快速柱层析法(山善株式会社HI-FLASHTM柱,大小为L,20mL/min,梯度为正庚烷中的15%至20%AcOEt)纯化该残余物,以提供标题化合物(563mg,3.09mmol)。
1H-NMR(400MHz,CDCl3)δppm:2.66(s,3H),3.51(t,J=13.1Hz,1H),7.23(d,J=7.8Hz,1H),7.28(d,J=7.8Hz,1H),7.57(t,J=7.8Hz,1H)。
(3)2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-醇的合成
在0℃下,向MeOH(20mL)中的通过描述于产生实例3(2)中的方法制备的产物(1.09g,5.99mmol)的溶液中添加硼氢化钠(453mg,12.0mmol)。在相同温度下搅拌45分钟后,将水和AcOEt添加至该反应混合物中,并且将层分离。将分离的水层用AcOEt萃取两次。将合并的有机层用盐水洗涤,并用MgSO4干燥。过滤后,将滤液在真空中浓缩并干燥,以提供标题化合物(1.05g,5.72mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):2.23(br.s,1H),2.43(s,3H),3.26-3.39(m,1H),3.44-3.58(m,1H),5.08-5.15(m,1H),7.07(d,J=7.8Hz,1H),7.10(d,J=7.8Hz,1H),7.23-7.26(m,1H)。
(4)1-叠氮基-2,2-二氟-7-甲基-2,3-二氢-1H-茚的合成
在0℃下,将TEA(3.59ml,25.8mmol)和氯甲烷磺酰氯(1.02ml,11.4mmol)添加至DCM(25mL)中的在产生实例3-(3)中获得的产物(1.05g,5.72mmol)的溶液中。在室温下搅拌2小时后,将该反应混合物用二乙醚稀释并用饱和NaHCO3淬灭。将水层用二乙醚萃取3次。将合并的有机层用盐水洗涤,并用MgSO4干燥。将萃取物过滤并在真空中浓缩。将该残余物溶解于DMF(50mL)中,并且在室温下向该溶液中添加叠氮化钠(753mg,11.6mmol)。将该反应混合物在70℃下搅拌2小时。将该混合物冷却至室温后,添加水和二乙醚。将层分离,并且将水层用二乙醚萃取3次。将合并的有机层用水和盐水洗涤,并用MgSO4干燥。将萃取物过滤并在真空中浓缩。通过硅胶快速柱层析法(山善株式会社HI-FLASHTM柱,大小为L,20mL/min,正庚烷中的20%AcOEt)纯化该残余物,以提供标题化合物(641mg,3.06mmol)。
1H-NMR(400MHz,CDCl3)δppm:2.41(s,3H),3.30-3.43(m,1H),3.51(ddd,J=20.3,16.8,10.9Hz,1H),4.77(d,J=13.3Hz,1H),7.09(d,J=7.8Hz,1H),7.14(d,J=7.8Hz,1H),7.26-7.31(m,1H)。
(5)N-(2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-基)硫酰胺的合成
在室温下,向水(4ml)和四氢呋喃(16ml)中的在产生实例3-(4)中获得的产物(641mg,3.06mmol)的溶液中添加三苯基膦(1.21g,4.61mmol)。将该反应混合物在80℃下搅拌1小时。冷却至室温后,添加AcOEt(20mL)和1N HCl(20mL)。将分离的有机层用10mL的1N HCl萃取两次。将水层合并并用20mL的2N NaOH碱化。将该层用AcOEt萃取3次并且将合并的有机层用盐水洗涤并用MgSO4干燥。将萃取物过滤并在真空中浓缩。在室温下,向DCM(26mL)中的该残余物和TEA(1.1mL,7.89mmol)的溶液中小部分地添加氨磺酰氯(CAS号7778-42-9,915mg,7.92mmol,根据描述于US2008/96903中的方法制备)。随后将该反应混合物在室温下搅拌1小时。向该混合物中添加20mL的1N HCl,并且将水层用DCM萃取两次。将合并的有机层用MgSO4干燥,过滤并在真空中浓缩。通过硅胶快速柱层析法(山善株式会社HI-FLASHTM柱,大小为L,20mL/min,梯度为正庚烷中的50%至65%AcOEt)纯化该残余物,以提供标题化合物(348mg,1.33mmol)。
1H-NMR(400MHz,CDCl3)δppm:2.45(s,3H),3.32-3.56(m,2H),4.70-4.80(m,3H),5.17-5.26(m,1H),7.06(d,J=7.4Hz,1H),7.12(d,J=7.4Hz,1H),7.23-7.29(m,1H)。
(6)N-[(1S)-2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺的合成
通过HPLC(CHIRALPAKTMIA,20mm I.D.x 250mm,10mL/min,正己烷中的15%EtOH)进行在产生实例3-(5)中所获得的外消旋体(348mg,1.33mmol)的旋光拆分,以提供呈白色固体的标题化合物(1S)-形式(107mg,0.409mmol;>99%ee),该形式在2种旋光异构体之中以25min的保留时间被第二个洗脱。ESI-MS m/z:285[M+Na]+
1H-NMR(400MHz,CDCl3)δppm:2.45(s,3H),3.32-3.56(m,2H),4.70-4.80(m,3H),5.17-5.26(m,1H),7.06(d,J=7.4Hz,1H),7.12(d,J=7.4Hz,1H),7.23-7.29(m,1H)。
产生实例4
N-[(1S)-2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺的合成
(1)7-溴-2,2,5-三氟-2,3-二氢-1H-茚-1-酮的合成
通过如描述于产生实例1-(1)和1-(2)中的类似方法从7-溴-5-氟-1-茚满酮(CAS号1260016-95-2,4.55g,19.9mmol)获得标题化合物(5.10g,19.2mmol)。1H NMR(400MHz,CDCl3)δ(ppm):3.53(t,J=12.5Hz,2H),7.14(d,J=7.6Hz,1H),7.41(d,J=8.4Hz,1H)。
(2)7-溴-2,2,5-三氟-2,3-二氢-1H-茚-1-醇的合成
通过如描述于产生实例3-(3)中的类似方法从产生实例4-(1)中获得的产物(5.10g,19.2mmol)获得标题化合物(4.78g,17.9mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):2,50(s,1H),3,38(td,J=17.0,2.7Hz,1H),3.50-3.69(m,1H),5.06(dd,J=12.5,4.3Hz,1H),6.95(dd,J=8.0,1.0Hz,1H),7.22(dd,J=8.6,2.3Hz,1H)。
(3)2-[(7-溴-2,2,5-三氟-2,3-二氢-1H-茚-1-基)氧基]四氢-2H-吡喃的合成
在室温下,向DCM(40ml)中的在产生实例4-(2)中获得的产物(2.78g,10.4mmol)和3,4-二氢-2H-吡喃(2.18mL,23.9mmol)的溶液中添加PPTS(52mg,0.208mmol)。并且将该反应混合物在室温下搅拌86小时。将溶剂在真空中蒸发并且通过硅胶快速柱层析(山善株式会社HI-FLASHTM柱,大小为M,10mL/min,梯度为正庚烷中的10%至25%AcOEt)纯化该残余物,以提供作为外消旋非对映异构体的约1:1混合物的标题化合物(3.42g,9.74mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):1.51-1.84(m,6H),3.26-3.52(m,1H),3.52-3.68(m,2H),4.05-4.19(m,1H),5.00-5.21(m,2H),6.92(d,J=8.2Hz,1H),7.21(dt,J=8.2,2.6Hz,1H)。
(4)2-[(2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-基)氧基]四氢-2H-吡喃的合成
向1,4-二噁烷(10ml)中的在产生实例4-(3)中获得的产物(1.70g,4.84mmol)的溶液中滴加二甲基锌(4.84ml,9.68mmol)的2M正己烷溶液。然后,在添加[1,1'-双(二苯基膦基)二茂铁]二氯钯(II)(177mg,0.242mmol)后,将该反应混合物在100℃下、在氮气氛下搅拌3小时。冷却至室温后,添加水并且将该混合物用AcOEt萃取。将有机层用盐水洗涤并用无水Na2SO4干燥。将不溶物质过滤掉并且将滤液在真空中蒸发。通过硅胶快速柱层析法(山善株式会社FLASHTM柱硅胶,大小为M,10mL/min,梯度为正庚烷中的0%至25%AcOEt)纯化该残余物,以提供呈外消旋非对映异构体的约1:1混合物的标题化合物(1.06g,3.70mmol)。
ESI-MS;m/z:309[M+Na]+
1H-NMR(400MHz,CDCl3)δ(ppm):1.51-1.90(m,6H),2.35(s,1.5H),2.43(s,1.5H),3.19-3.29(m,1H),3.45-3.64(m,2H),3.98-4.11(m,1H),4.88(t,J=3.4Hz,0.5H),4.95(d,J=5.1Hz,0.5H),5.01(dd,J=11.6,2.8Hz,0.5H),5.16(d,J=11.7Hz,0.5H),6.74-6.81(m,2H)。
(5)2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-醇的合成
向MeOH(10ml)中的在产生实例4-(4)中获得的产物(1.06g,3.70mmol)的溶液中添加PPTS(46mg,0.185mmol)。并且将该反应混合物在60℃下搅拌1小时。冷却至室温后,向该反应混合物中添加饱和NaHCO3并且将该混合物用AcOEt萃取。将有机层用盐水洗涤并用无水Na2SO4干燥。将不溶物质过滤掉并且将滤液在真空中蒸发。通过硅胶快速柱层析法(山善株式会社HI-FLASHTM柱,大小为M,10mL/min,梯度为正庚烷中的5%至25%AcOEt)纯化该残余物,以提供标题化合物(692mg,3.42mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):2.23(dd,J=5.7,2.5Hz,1H),2.42(s,3H),3.30(td,J=16.8,5.2Hz,1H),3.50(td,J=16.8,11.6Hz,1H),5.05(dd,J=12.1,5.1Hz,1H),6.77(d,J=8.2Hz,1H),6.82(d,J=10.2Hz,1H)。
(6)1-叠氮基-2,2,5-三氟-7-甲基-2,3-二氢-1H-茚的合成
在0℃下,向DCM(10ml)中的在产生实例4-(5)中获得的产物(692mg,3.42mmol)和TEA(1.43ml,10.3mmol)的溶液中添加氯甲烷磺酰氯(765mg,5.13mmol)。并且将该反应混合物在室温下搅拌2小时。向该反应混合物中添加饱和NaHCO3并且将该混合物用二乙醚萃取。将有机层依次用1N HCl和盐水洗涤,然后用无水Na2SO4干燥。过滤后,将滤液在真空中蒸发。在室温下,向DMF(10ml)中的该残余物的溶液中添加叠氮化钠(442mg,6.80mmol),并且将该化合物在70℃下搅拌2小时。冷却至室温后,将该混合物在二乙醚与H2O之间进行分段。将水层用二乙醚萃取。将合并的有机层用盐水洗涤并用无水Na2SO4干燥。过滤后,将滤液在真空中蒸发。通过硅胶快速柱层析法(山善株式会社HI-FLASHTM柱,大小为M,10mL/min,梯度为正庚烷中的10%至30%AcOEt)纯化该残余物,以提供标题化合物(320mg,1.41mmol)。
1H-NMR(400MHz,CDCl3)δ(ppm):2.41(s,3H),3.30-3.56(m,2H),4.74(d,J=13.3Hz,1H),6.81(d,J=7.8Hz,1H),6.86(d,J=9.4Hz,1H)。
(7)2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-胺的合成
在室温下,向水(1ml)和THF(5ml)中的在产生实例4-(6)中获得的产物(320mg,1.41mmol)的溶液中添加三苯基膦(554mg,2.11mmol),并且将该混合物在80℃下搅拌2小时。冷却至室温后,将该混合物在AcOEt与1N HCl之间进行分段。将所得水层用5N NaOH进行碱化。将水层用AcOEt萃取3次,并将合并的萃取物经无水Na2SO4干燥。过滤后,将滤液在真空中浓缩,以提供标题化合物(180mg,0.895mmol)。
ESI-MS;m/z:202[M+H]+。
(8)N-(2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-基)氨磺酰基氨基甲酸叔丁酯的合成
在室温下,向DCM(10mL)中的在产生实例4-(7)中的产物(180mg,0.895mmol)的溶液中添加[(叔丁氧基)羰基]{[4-(二甲基亚氨基)吡啶-1(4H)-基]磺酰基}酰胺(297mg,0.984mmol)和TEA(0.374mL,2.68mmol)。将所得混合物在回流下加热65.5小时。冷却至室温后,将该混合物在AcOEt与1N HCl之间进行分段。将有机层用无水Na2SO4干燥,过滤并在真空中蒸发,以提供标题化合物(257mg,0.676mmol)。
ESI-MS;m/z:403[M+Na]+。
(9)N-[(1S)-2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺的合成
在室温下,向MeOH(4mL)中的产生实例4-(8)中的产物(257mg,0.676mmol)的溶液中添加AcOEt(3.38ml,13.5mmol)中的4N HCl并搅拌14小时。将溶剂在真空中蒸发并且通过硅胶柱层析(AcOEt)纯化该残余物,以提供呈外消旋体的标题化合物(162mg,0.578mmol)。通过HPLC(CHIRALPAKTMIF,20mm I.D.x 250mm,10mL/min,正己烷中的10%EtOH)进行所得外消旋体(162mg,0.578mmol)的旋光拆分,以提供呈白色固体的标题(S)-异构体(71mg,0.253mmol;98%ee),该异构体在2种旋光异构体之中以30min的保留时间被第二个洗脱。
ESI-MS;m/z:303[M+Na]+。
1H-NMR(400MHz,DMSO-d6)δ(ppm):2.37(s,3H),3.20-3.31(m,1H),3.38-3.64(m,1H),4.79(dd,J=14.3,8.8Hz,1H),6.77(s,2H),6.90-7.03(m,2H),7.51(d,J=9.0Hz,1H)。
(参考实例1)
N-[(1S)-2,2,5,7-四氟-2,3-二氢-1H-茚-1-基]硫酰胺的X射线晶体学分析
将在产生实例1-(5)中获得的白色固体溶解于MeOH和甲苯中,并且通过溶剂蒸发方法重结晶。使用获得的单晶进行X射线衍射分析。数据收集和晶体学分析的结果总结于表1中,并且原子坐标示于表2中。由这些结果指定标题化合物的绝对构型。
[表1]
[表2]
(参考实例2)
N-[(1S)-2,2-二氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺的X射线晶体学分析
将在产生实例3-(6)中获得的白色固体溶解于EtOH和正己烷中并且通过温度梯度重结晶,以提供微晶体。将微晶体溶解于Et2O中并且进一步通过溶剂蒸发方法重结晶。使用获得的单晶进行X射线衍射分析。数据收集和晶体学分析的结果总结于表3中,并且原子坐标示于表4中。由这些结果指定标题化合物的绝对构型。
[表3]
[表4]
(参考实例3)
N-[(1S)-2,2,5-三氟-7-甲基-2,3-二氢-1H-茚-1-基]硫酰胺的X射线晶体学分析
将在产生实例4-(9)中获得的白色固体溶解于EtOH中并且将甲苯用作储液通过蒸汽扩散法重结晶。使用如上获得的单晶进行X射线衍射分析。数据收集和晶体学分析的结果总结于表5中,并且原子坐标示于表6中。由这些结果指定标题化合物的绝对构型。
[表5]
[表6]
药理学测试实例
本发明诸位发明人进行了如下研究,分别使用小鼠热板试验以证实对急性疼痛的镇痛效果和使用大鼠慢性压迫性损伤(CCI)模型以证实对慢性疼痛的镇痛效果。此外,本发明诸位发明人进行了一项研究,使用小鼠福尔马林测试以证实对急性持久性疼痛的镇痛效果。
将专利文献1的实例1所披露的化合物(N-(苯并[b]苯硫-3-基甲基)硫酰胺)以及专利文献2的实例7所披露的化合物((2S)-(-)-N-(6-氯-2,3-二氢-苯并[1,4]二噁英-2-基甲基)-硫酰胺)根据专利文献1和2进行制备并且分别被用作参比化合物(1和2)。
(测试实例1)小鼠热板试验
进行小鼠热板试验以确定对急性疼痛的疗效。在这个模型中,将第一次疼痛响应的等待时间记录为疼痛敏感性的量度((Malmberg(马尔伯格),A.与Yaksh(雅克斯),T.,Pain(疼痛).1995,60:83-90)。
<方法>
将6周大的雄性C57BL/6NCrlCrlj小鼠(Charles River Japan(日本查尔斯河公司))(n=4至10,对于每个处理)用于本实验。将热板(模型MK 350,Muromachi Kikai Co.Ltd.(室町-机械兴业有限公司))设置为53℃。对于口服给药,将该测试化合物悬浮于含有0.45%甲基纤维素/4.5%聚氧乙烯蓖麻油(Cremophor)/10%二甲亚砜的水性溶液中,以制备具有给药体积为10ml/kg的悬浮液,并且将悬浮液在热板试验前60分钟口服给予。将吗啡用作阳性对照,并且将不含该测试化合物的混合溶剂(载体)单独用作阴性对照。
将每只小鼠置于热板上,并且立即开始秒表计时用于测量。测量第一次疼痛响应(舔爪或挣脱跳跃)的等待时间。为了避免对组织过度损害,将截止时间设置为30秒,并且在测量后立即使小鼠移离热板。所有数据均表示为平均值±SEM。使用Holm-Sidak多重比较检验进行所有统计分析。在本实验中,p-值低于0.05时判断为有统计学显著性。
<结果>
小鼠热板试验的结果示于图1中。通过预先给予测试化合物1、2、3和4使得第一次疼痛响应的等待时间显著增加。这些结果显示这些测试化合物对急性疼痛的镇痛效果。
(测试实例2)大鼠慢性压迫性损伤(CCI)模型
使用大鼠慢性压迫性损伤(CCI)模型以确定对慢性疼痛的疗效。在这个模型中,可以使用针对万福瑞(von Frey)细丝机械性刺激的响应阈值作为指标来评估触觉异常痛敏(慢性疼痛患者中的一种典型症状)(Bennett(班尼特),G.J.与Xie(谢),Y.K.,(Pain(疼痛)1988;33:p87-107)。
<方法>
将6周大的雄性SD大鼠(Charles River Japan(日本查尔斯河公司))(n=4,每个处理总计进行两次)用于本实验。根据上述班尼特与谢的方法准备大鼠慢性压迫性损伤(CCI)模型。
在异氟烷麻醉下,通过钝性解剖在大腿的中间水平处贯穿股二头肌将坐骨神经暴露出来。以大约1mm的间隔围绕坐骨神经松散地系上4条结扎线(4-0丝绸)。将股二头肌与皮肤缝合。
在手术14天后,评估这些化合物对于触觉异常痛敏的疗效。在顺应试验开始之前,将这些动物关在金属丝网下笼中30分钟。将该测试化合物悬浮于含有0.45%甲基纤维素/4.5%聚氧乙烯蓖麻油(Cremophor)/10%二甲亚砜的水性溶液中,以制备具有给药体积为10ml/kg的悬浮液,并且将悬浮液口服给予。将上述不含该测试化合物的混合溶剂(载体)单独用作阴性对照。根据Chaplan(卓别林)等人(J Neuroscience Methods(神经学方法杂志)1994;53(1):p.55-63),通过以弯曲力(0.4、0.6、1、2、4、6、8、以及15g)将万福瑞细丝施用至后脚爪的趾面来测定针对机械性刺激的响应阈值。将万福瑞细丝推至脚爪持续6秒以评估逃避反应。根据Dixon(迪克森)的倒置法(Annual Review of Pharmacology and Toxicology(药理学与毒理学年鉴)1980;20:p.441-462)确定50%响应阈值。在给药之前以及给药之后30、90以及180分钟进行该测量。所有数据均表示为平均值±SEM。使用双向重复测量ANOVA、随后邓尼特测试(Dunnett’s test)进行所有统计分析。在本实验中,p-值低于0.05时判断为有统计学显著性。
<结果>
CCI模型中触觉异常痛敏的结果在表7、8、9以及10中示出。在50mg/kg处,测试化合物1与2展现出统计学上有义意的镇痛效果。
[表7]
万福瑞细丝检验中50%响应阈值
**P<0.01vs载体(邓尼特测试)
[表8]万福瑞细丝检验中50%响应阈值
**P<0.01vs载体(邓尼特测试)
[表9]万福瑞细丝检验中50%响应阈值
[表10]万福瑞细丝检验中50%响应阈值
*P<0.05、**P<0.01vs载体(邓尼特测试)
如表7与8所示,通过预先给予测试化合物1和2使得针对机械性刺激的响应阈值以大致剂量依赖性方式增加。
这些结果显示这些测试化合物对慢性疼痛的动物模型的镇痛效果。
(测试实例3)福尔马林测试
在福尔马林测试的第二阶段检验这些化合物以确定对急性持久性疼痛的镇痛效果。在这个模型中,将由疼痛(如舔舐或咬扯注射有福尔马林的脚爪的行为)诱发的响应持续时间用作评价指标(Dubuisson(迪比松)与Dennis(丹尼斯)(Pain(疼痛),4:p.161-174(1977))。
<方法>
将5至6周大的雄性CD1(ICR)小鼠(Charles River Japan(日本查尔斯河公司))(n=6至16,对于每个处理)用于本实验。通过自动化行为分析装置(MicroAct(Neuroscience,Inc.(神经科学公司)))根据检测磁场的变化来测定由疼痛诱发的响应。在进行疼痛响应评价之前那天,在异氟烷麻醉下,将用于检测磁场变化的小磁铁植入左后肢脚背。
对于口服给药,将该测试化合物悬浮于含有0.45%甲基纤维素/4.5%聚氧乙烯蓖麻油(Ceremophor)/10%二甲亚砜的溶液中,以制备具有给药体积为10ml/kg的悬浮液,并且将悬浮液口服给予。将上述不含该测试化合物的混合溶剂(载体)单独用作阴性对照。腹膜内给予加巴喷丁作为阳性对照。通过用生理盐水(Otsuka Pharmaceutical(大冢制药))稀释甲醛溶液(Wako Pure Chemical Industries(和光纯药工业))来制备2.5%福尔马林溶液。在给予测试化合物或阳性对照之后40分钟至1小时处,将10μl的2.5%福尔马林溶液皮下注射到左后肢爪垫中。在给药之后立即将小鼠置于观察室中用于测量。
对于第二阶段中镇痛效果的评价,在注射2.5%福尔马林溶液之后15至45分钟通过自动化行为分析装置来测定如舔舐或咬扯脚爪的行为的持续时间。
所有数据均表示为平均值±SEM。使用Holm-Sidak多重比较检验进行所有统计分析。在本实验中,p-值低于0.05时判断为有统计学显著性。
<结果>
福尔马林测试的结果在图2中示出。如图2所示,通过预先给予测试化合物1、2、3和4使得疼痛响应的持续时间在统计学上显著缩短。这些结果显示这些化合物对动物模型中急性持久性疼痛的镇痛效果。
- 还没有人留言评论。精彩留言会获得点赞!