使用抗PD‑1抗体与另一抗癌剂的组合治疗肾癌的制作方法
发明领域
本发明涉及用于治疗个体中的肾癌的方法,其包括向所述个体施用作为抗程序性死亡-1(PD-1)抗体的抗癌剂与另一抗癌剂的组合。在某些实施方案中,所述其他抗癌剂是抗细胞毒性T淋巴细胞抗原-4(CTLA-4)抗体、抗血管生成酪氨酸激酶抑制剂或其组合。
发明背景
人类癌症携带许多遗传学和表观遗传学改变,生成潜在的可被免疫系统识别的新抗原(Sjoblom等人, 2006)。由T和B淋巴细胞构成的适应性免疫系统具有强力的抗癌潜力,具有响应于不同肿瘤抗原的广泛能力和敏锐特异性。另外,该免疫系统表现出相当的可塑性和记忆组分。对适应性免疫系统的所有这些属性的成功利用将使免疫疗法在所有癌症治疗形式中是独特的。
直到最近,癌症免疫疗法将实质努力聚焦于通过过继性转移(adoptive-transfer)活化的效应细胞、针对相关抗原的免疫、或提供非特异性免疫刺激剂诸如细胞因子来增强抗肿瘤免疫响应的方法。然而,在过去十年中,大力开发特异性免疫检查点途径抑制剂已经开始提供用于治疗癌症的新免疫治疗方法,包括开发结合并抑制细胞毒性T-淋巴细胞抗原-4(CTLA-4)的抗体(Ab)伊匹单抗(YERVOY®)用于治疗晚期黑色素瘤患者 (Hodi等人, 2010)和开发特异性结合程序性死亡-1(PD-1)受体并阻断抑制性PD-1/PD-1配体途径的Ab诸如nivolumab和派姆单抗(原为lambrolizumab;USAN Council Statement, 2013) (Topalian等人, 2012a, b; Topalian等人, 2014; Hamid等人, 2013; Hamid和Carvajal, 2013; McDermott和Atkins, 2013)。
PD-1是由激活的T和B细胞表达的关键免疫检查点受体并介导免疫抑制。PD-1是CD28受体家族的成员,包括CD28、CTLA-4、ICOS、PD-1和BTLA。已经识别出PD-1的两种细胞表面糖蛋白配体,程序性死亡配体-1(PD-L1)和程序性死亡配体-2(PD-L2),其在抗原递呈细胞以及许多人类癌症中表达,并且已显示在结合PD-1后下调T细胞活化和细胞因子分泌。对PD-1/PD-L1相互作用的抑制在临床前模型中介导有效的抗肿瘤活性(美国专利号8,008,449和7,943,743),且将PD-1/PD-L1相互作用的Ab抑制剂用于治疗癌症已进入临床试验(Brahmer等人, 2010; Topalian等人, 2012a; Topalian等人, 2014; Hamid等人, 2013; Brahmer等人, 2012; Flies等人, 2011; Pardoll, 2012; Hamid and Carvajal, 2013)。
Nivolumab(以前称为5C4、BMS-936558、MDX-1106或ONO-4538)是全人IgG4 (S228P) PD-1免疫检查点抑制剂Ab,其选择性防止与PD-1配体(PD-L1和PD-L2) 的相互作用,从而阻断抗肿瘤T细胞功能的下调(美国专利号8008449;Wang等人,2014)。Nivolumab已在各种晚期实体瘤(包括肾细胞癌(RCC;肾腺癌或肾上腺样瘤)、黑色素瘤和非小细胞肺癌)中显示活性(Topalian等人, 2012a; Topalian等人, 2014; Drake等人, 2013; WO 2013/173223)。
伊匹单抗(YERVOY®)是全人IgG1单克隆Ab,其阻断CTLA-4与其B7配体的结合,由此刺激T细胞活化并改善晚期黑色素瘤患者的总存活期(OS) (Hodi等人, 2010)。在1期临床试验中用nivolumab和伊匹单抗同时治疗在相当比例的晚期黑色素瘤患者中产生迅速且深度的肿瘤消退,并且比任何单独抗体明显更有效(Wolchok等人,2013;WO 2013/173223)。然而,迄今还不知道免疫调节性Ab的这种组合在其他肿瘤类型中是否有类似效果。
肾癌(也称为肾脏癌症)是源于肾脏的癌症。最常见类型的肾癌是肾细胞癌(RCC)。如果在RCC仍局限于肾脏或紧密邻近组织时(I期)诊断出并治疗,则其可以经常通过手术切除来治愈,并且根治性切除是II期以及III期RCC的接受的、经常可治愈的疗法。相反,当存在远处转移时(IV期),无疾病存活较差。此外,无论细胞类型或分期,患有进展中、反复中或复发中疾病的任何经治疗的RCC患者的预后也较差。约25%-30%的RCC患者在诊断时患有转移性疾病,并且转移性RCC的中值存活期为仅约24个月(Gupta等人, 2008; NCCN GUIDELINES®, 版本3.2014 – Kidney Cancer; Heng等人, 2009)。对于已在足够数目的患者中研究的任何方案,对细胞毒性化疗的反应通常不超过10%。然而,对RCC生物学的日渐增进的理解已导致开发且由美国食品和药品管理局(FDA)批准了七种靶向特定生长途径的新药物。经批准的靶向疗法中的两种,替西罗莫司和依维莫司,阻断雷帕霉素的哺乳动物靶标(mTOR),调节细胞生长、分裂和存活的丝氨酸/苏氨酸蛋白激酶。已经开发了靶向血管内皮细胞生长因子(VEGF)介导的促血管生成途径的若干种药剂,包括五种口服酪氨酸激酶抑制剂(TKI),帕唑帕尼、索拉非尼、舒尼替尼、阿西替尼和tivozanib,和抗VEGF单克隆抗体(mAb),贝伐单抗。这些药物显示出临床功效和改善的无进展存活 (PFS),但持久性反应是罕见的。VEGF TKI已经显示抑制免疫抑制调节性T细胞(Treg细胞)和髓源抑制细胞(MDSC),使得免疫环境更利于T细胞介导的抗肿瘤活性。此外,靶向VEGF轴可以减弱RCC-诱导的免疫抑制,当组合使用时允许肿瘤变得对免疫调节更具反应性,并由此导致更大和更持久的治疗益处。
如本文所述,已经在临床前RCC小鼠模型中研究了nivolumab与两种抗血管生成(抗VEGF) TKI,舒尼替尼(SUTENT®)或索拉非尼(NEXAVAR®)的组合的活性。本文还提供了关于nivolumab与舒尼替尼、帕唑帕尼(VOTRIENT®)或伊匹单抗的组合的临床数据。
发明概述
本公开提供了用于治疗患有肾癌的个体的方法,其包括向所述个体施用治疗有效量的以下的组合:(a)特异性结合并抑制PD-1的Ab或其抗原结合部分;和(b)另一抗癌剂。在某些实施方案中,所述其他抗癌剂是(i)特异性结合并抑制CTLA-4的Ab或其抗原结合部分,(ii)抗血管生成TKI,或(iii)其组合。在一些实施方案中,所述肾癌是RCC。在本文公开的任何治疗方法的某些实施方案中,所述抗PD-1抗体是nivolumab。在其他实施方案中,所述抗PD-1抗体是派姆单抗。在本文公开的任何治疗方法的某些其他实施方案中,所述抗CTLA-4抗体是伊匹单抗。在其他实施方案中,所述抗CTLA-4抗体是tremelimumab。在又其他实施方案中,所述TKI是舒尼替尼、帕唑帕尼、索拉非尼、阿西替尼或tivozanib。
在某些实施方案中,所述个体已经针对肾癌进行了预先治疗。在其他实施方案中,所述肾癌是晚期、转移性和/或难治性癌症。在一些实施方案中,施用Ab或其抗原结合部分与TKI的组合诱导所述个体中的持久性临床反应。
在某些实施方案中,施用抗PD-1抗体或其抗原结合部分与其他抗癌剂的组合导致肾癌组织或肿瘤的T细胞浸润相对于未经治疗的个体或用抗PD-1抗体或其抗原结合部分或其他抗癌剂的单一疗法治疗的个体增加。在其他实施方案中,所述T细胞浸润增加的特征在于CD4+ T细胞、CD8+ T细胞或两者至肾癌组织或肿瘤中的浸润增加。在仍其他实施方案中,施用抗PD-1抗体或其抗原结合部分与另一抗癌剂的组合导致T细胞增殖增加。在另一个实施方案中,施用抗PD-1抗体或其抗原结合部分与其他抗癌剂的组合导致T调节性细胞相对于未经治疗的个体或用抗PD-1抗体或其抗原结合部分或其他抗癌剂的单一疗法治疗的个体减少。
在某些实施方案中,施用抗PD-1抗体或其抗原结合部分与另一抗癌剂的组合减少单核细胞髓源抑制细胞的数目。在一些实施方案中,施用抗PD-1抗体或其抗原结合部分与另一抗癌剂的组合相对于未经治疗的个体或用抗PD-1抗体或其抗原结合部分或其他抗癌剂的单一疗法治疗的个体增加粒细胞骨髓细胞的数目。在一些实施方案中,所述单核细胞髓源抑制细胞的特征在于CD11b+/Ly6C高/Ly6G-的表达或CD11b+/Ly6C低/Ly6G-的表达。在其他实施方案中,所述粒细胞骨髓细胞的特征在于CD11b+/Ly6C-/Ly6G+的表达。
在包括使用抗PD-1抗体或其抗原结合部分与TKI的组合的方法的某些实施方案中,抗PD-1抗体或其抗原结合部分的治疗有效剂量范围为约0.1至约10.0 mg/kg体重,约每2-4周通过静脉内输注施用一次。在某些实施方案中,抗PD-1抗体或其抗原结合部分以约2 mg/kg或约5 mg/kg的剂量施用,每三周一次。
对于抗PD-1抗体与抗CTLA-4抗体的组合,在某些实施方案中,所述方法包括(a)诱导阶段,其包括以3周时间间隔施用4剂抗PD-1抗体与抗CTLA-4抗体,剂量如下:(i) 约1 mg/kg抗PD-1抗体和约3 mg/kg抗CTLA-4抗体;(ii) 约3 mg/kg抗PD-1抗体和约3 mg/kg抗CTLA-4抗体;或(iii) 约3 mg/kg抗PD-1抗体和约1 mg/kg抗CTLA-4抗体;和(b)维持阶段,其包括每2周以约3 mg/kg的剂量重复施用抗PD-1抗体。
本公开还提供了用于治疗患有肾癌的个体的药剂盒,所述药剂盒包括:(a)特异性结合并抑制PD-1的Ab或其抗原结合部分的剂量;(b) (i) 特异性结合并抑制CTLA-4的Ab或其抗原结合部分或(ii)抗血管生成TKI(例如舒尼替尼或帕唑帕尼)的剂量之一;和(c)使用抗PD-1抗体与抗CTLA-4抗体或TKI以治疗个体的说明书。
本发明的其他特征和优点从以下详细说明和实施例将是明显的,所述详细说明和实施例不应被解读为限制性的。所有引用参考文献的内容,包括整个本申请中引用的科学论文、报纸报道、GenBank条目、专利和专利申请,明确通过引用并入本文。
附图简述
图1显示用下列治疗剂治疗对平均肿瘤体积生长的影响:对照(实心圆形)、舒尼替尼(空心圆形)、索拉非尼(实心三角形)、抗PD-1(实心倒三角形)、舒尼替尼+抗PD-1(空心正方形)和索拉非尼+抗PD-1(实心菱形)。
图2显示用对照(实心圆形)、舒尼替尼(空心圆形)、索拉非尼(实心三角形)、抗PD-1(实心倒三角形)、舒尼替尼+抗PD-1(空心正方形)和索拉非尼+抗PD-1(实心菱形)治疗的肿瘤达到500 mm3的体积所花费的时间。
图3显示用下列治疗剂治疗对体重的百分比变化的影响:对照(实心圆形)、舒尼替尼(空心圆形)、索拉非尼(实心三角形)、抗PD-1(实心倒三角形)、舒尼替尼+抗PD-1(空心正方形)和索拉非尼+抗PD-1(实心菱形)。
图4显示用对照、单独的抗PD-1、单独的舒尼替尼或抗PD-1与舒尼替尼的组合治疗后CD4+和CD8+ T浸润入Renca肿瘤中。
图5显示用单独的舒尼替尼、索拉非尼或抗PD-1抗体或抗PD-1抗体与舒尼替尼或索拉非尼的组合治疗之后在Renca肿瘤模型中通过流式细胞术评估肿瘤浸润的CD4+免疫细胞的组成。
图6显示用单独的舒尼替尼、索拉非尼或抗PD-1抗体或抗PD-1抗体与舒尼替尼或索拉非尼的组合治疗之后在Renca肿瘤模型中通过流式细胞术评估肿瘤浸润的CD8+免疫细胞的组成。
图7显示用单独的舒尼替尼、索拉非尼或抗PD-1抗体或抗PD-1抗体与舒尼替尼或索拉非尼的组合治疗之后在Renca肿瘤模型中评估骨髓来源的肿瘤浸润细胞亚群(CD11b+/Ly6C高/Ly6G–、CD11b+/Ly6C低/Ly6G–或CD11b+/Ly6C–/Ly6G+)。门控:细胞→ 单体(Singlets) → Cd45+/活 → CD11b+。
图8显示抗PD-1 mAb与舒尼替尼的组合对CT26肿瘤模型中的肽特异性细胞毒性反应的影响。X轴显示细胞杀伤的百分比。各条从上到下代表(i)对照,(ii)抗PD-1 10 mg/kg, Q3D x 2 IP,(iii)舒尼替尼120 mg/kg, QD x 5, PO, 和(iv)抗PD-1 + 舒尼替尼。
图9显示抗PD-1 mAb与舒尼替尼的组合对CT26肿瘤模型中的肿瘤体积的影响。对于对照、抗PD-1 10 mg/kg Q3Dx IP、舒尼替尼120mg/kg QDx5 PO和抗PD-1 +舒尼替尼的施用,肿瘤体积(mm3)显示于y-轴。
图10A和10B显示1期研究的设计。图10A和10B分别显示患有晚期或转移性RCC (mRCC) (NCT01472081)的个体中nivolumab与舒尼替尼或帕唑帕尼的组合(A)或nivolumab与伊匹单抗的组合(B)的研究设计。*当两个剂量递增组开放时,将先前已接受针对晚期/转移性RCC的至少一个全身性治疗方案但先前没有接受过帕唑帕尼或舒尼替尼的个体以交替方式指定至两个组。如果仅一个组开放,则将这些个体均指定至开放组。**直到疾病进展(PD)或药物相关的毒性导致停药或个体撤回同意书。***当两个伊匹单抗组合组(I-1和I-3)都开放时,将先前已接受至少一次全身性疗法且当递增阶段组S和/或P开放时没有资格参加那些组的个体以交替方式指定至这些组。如果仅一个组开放,则将这些个体均指定至开放组。#如果所有组都开放,将未经治疗的个体以1:1:1:1比率随机指定至4个组(扩展组S和P,以及I-1和I-3)之一,直到所有登记都完成。##直到疾病进展(PD)或药物相关的毒性导致停药或个体撤回同意书。在经历研究者评估的临床益处和耐受研究疗法的个体中考虑进行超出最初研究者评估的RECIST 1.1定义的进展之外的治疗。当记录到进一步进展时,要求这类个体停止治疗。###对于I-1和I-3组群扩展和IN 3剂量添加组群,允许针对局部或局部晚期RCC的一种现有辅助或新辅助疗法,条件是在所述辅助或新辅助疗法的最后一剂之后≥6个月出现复发。允许针对RCC的现有基于细胞因子的治疗[例如,干扰素-α(IFN-α)或白介素-2(IL-2)]作为在前疗法。如果所有组都开放,则将这些经最小程度治疗(包括未经治疗)的个体以1:1:1比率随机指定至这3个组(I-1、I-3和IN-3)之一,直到所有登记都完成。
图11显示在nivolumab组合疗法研究的S和P组中进行的剂量递增。S组包含施用S + N2的7名预治疗的个体;施用S + N5的7名预治疗的个体;和进入S + N5扩展的19名未经治疗的患者。P组包括施用P + N2的20名预治疗的个体。P组没有扩展。
图12A-D显示用nivolumab与舒尼替尼或帕唑帕尼的组合治疗的RCC个体的临床活性。蜘蛛图显示接受各种方案的个体中的肿瘤负荷从基线的百分比变化,以所有目标病灶的垂直直径的乘积之和测量。图12A显示舒尼替尼与2 mg/kg nivolumab的方案。图12B显示舒尼替尼与5 mg/kg nivolumab的方案。图12C显示帕唑帕尼与2 mg/kg nivolumab的方案。“+”表明新病灶的第一次出现。图12D是显示S和P组中的个体的基线目标病灶的最大百分比反应的瀑布图。示出了具有基线目标病灶和至少一个基线后目标病灶评估的个体。肿瘤负荷的正向变化表明肿瘤生长;肿瘤负荷的负向变化表明肿瘤缩小。在蜘蛛图和瀑布图中,水平线代表30%减少(PR的1.1 RECIST阈值)和20%减少(PD的1.1 RECIST阈值)。并非所有从基线的30%或更大降低都是PR。
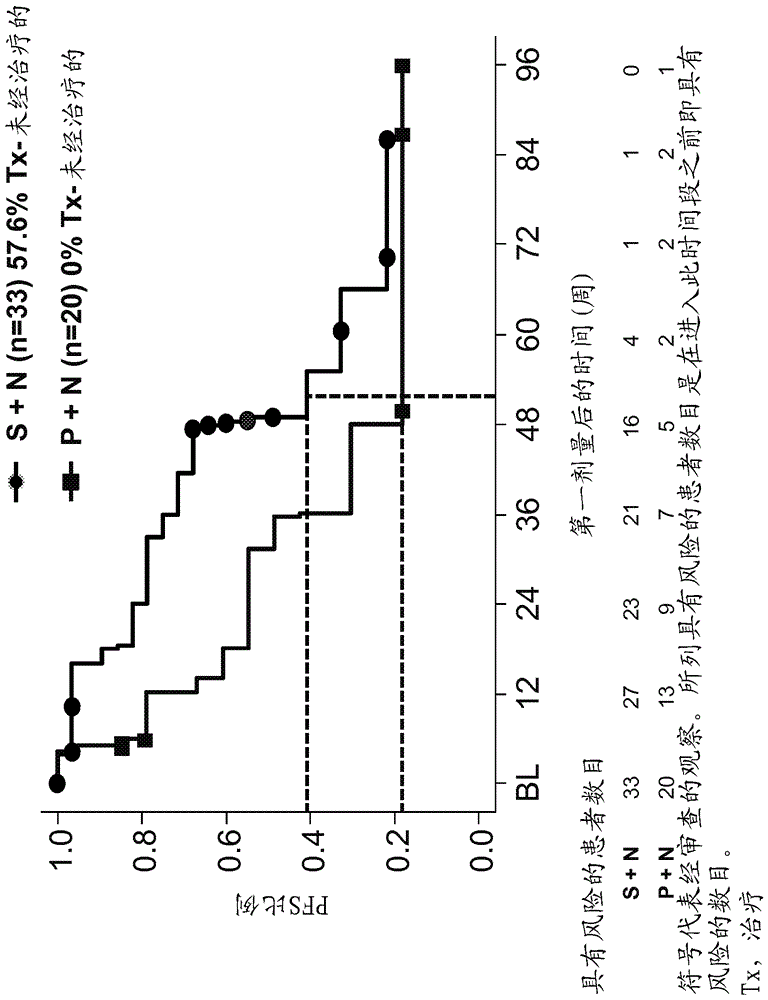
图13显示用nivolumab与舒尼替尼(S + N)或nivolumab与帕唑帕尼(P + N)治疗的个体的针对时间作图的无进展存活(PFS)的比例。符号代表经审查的观察。所列具有风险的患者的数目是在进入该时间段之前即具有风险的数目。Tx:治疗。
图14A-14D显示用伊匹单抗与nivolumab的组合治疗的RCC个体的临床活性。蜘蛛图显示接受各种方案的个体中的肿瘤负荷从基线的百分比变化,以所有目标病灶的垂直直径的乘积之和测量。图14A显示3 mg/kg伊匹单抗与1 mg/kg nivolumab的方案。图14B显示1 mg/kg伊匹单抗与3 mg/kg nivolumab的方案。图14C显示3 mg/kg伊匹单抗与3 mg/kg nivolumab的方案。“+”表明新病灶的第一次出现。图14D是显示I-1、I-3和IN-3组中的个体的基线目标病灶的最大百分比反应的瀑布图。示出了具有基线目标病灶和至少一个基线后目标病灶评估的个体。肿瘤负荷的正向变化表明肿瘤生长;肿瘤负荷的负向变化表明肿瘤缩小。在蜘蛛图和瀑布图中,水平线代表30%减少(PR的1.1 RECIST阈值)和20%减少(PD的1.1 RECIST阈值)。并非所有从基线的30%或更大降低都是PR。
图15显示S、P和I组中的个体的瀑布图的比较。示出了具有基线目标病灶和至少一个基线后目标病灶评估的个体。肿瘤负荷的正向变化表明肿瘤生长;肿瘤负荷的负向变化表明肿瘤缩小。水平线代表30%减少(PR的1.1 RECIST阈值)和20%减少(PD的1.1 RECIST阈值)。并非所有从基线的30%或更大降低都是PR。
图16显示用1 mg/kg伊匹单抗 + 3 mg/kg nivolumab (IPI1+INV3) (星号)、3 mg/kg伊匹单抗 + 1 mg/kg nivolumab (IPI3 + NIV1)(圆形)和3 mg/kg nivolumab + 3 mg/kg伊匹单抗(IPI3 + NIV3)(三角形)治疗的个体针对从第一剂量起的时间(周数)作图的PFS的比例。符号代表经审查的观察。所列具有风险的患者的数目是在进入该时间段之前即具有风险的数目。
发明详述
本发明涉及用于治疗肾癌患者的方法,其包括向所述患者施用抗PD-1抗体或其抗原结合片段与另一抗癌剂的组合。在某些实施方案中,所述其他抗癌剂是抗- CTLA-4抗体、抗血管生成TKI或其任何组合。
术语
为了可以更容易理解本公开,首先定义某些术语。如本申请中所用,除了本文另行明确提供以外,下列每个术语应当具有下述含义。额外定义记载于整个申请中。
“施用”是指使用本领域技术人员已知的任何各种方法和递送系统将包含治疗剂的组合物身体引入个体。抗PD-1抗体的优选施用途径包括静脉内、肌内、皮下、腹膜内、脊髓或其他肠胃外施用途径,例如通过注射或输注。如本文中所用的短语“肠胃外施用”是指除了肠内和局部施用以外的施用模式,通常通过注射,并且包括,但不限于,静脉内、肌内、动脉内、鞘内、淋巴内、病灶内、囊内、眼眶内、心内、真皮内、腹膜内、经气管、皮下、表皮、关节内、囊下、蛛网膜下、脊柱内、硬膜外和胸骨内注射和输注,以及体内电穿孔。TKI通常经由非肠胃外途径施用,优选口服施用。其他肠胃外途径包括局部、表皮或粘膜施用途径,例如,鼻内、阴道、直肠、舌下或局部。施用也可以进行,例如,一次,多次和/或在一个或多个延长时段内。
如本文中所用,“不良事件”(AE)是任何不利且通常无意或不期望的与药物治疗的使用相关的体征(包括异常的实验室发现)、症状或疾病。例如,不良事件可以与响应于治疗的免疫系统活化或免疫系统细胞(例如T细胞)扩增相关。药物治疗可具有一种或多种相关的AE,并且每个AE可具有相同或不同水平的严重程度。对能够“改变不良事件”的方法的提及是指减少与不同治疗方案的使用相关的一种或多种AE的发生和/或严重程度的治疗方案。
“抗体”(Ab)应当包括而不限于:特异性结合抗原且包含至少2条重(H)链和2条轻(L)链的糖蛋白免疫球蛋白或其抗原结合部分,所述重链和轻链通过二硫键互相连接。每条H链包含一个重链可变区(本文缩写为VH)和一个重链恒定区。所述重链恒定区包含三个恒定结构域,CH1、CH2和CH3。每条轻链包含一个轻链可变区(本文缩写为VL)和一个轻链恒定区。所述轻链恒定区包含一个恒定结构域,CL。VH和VL区域可以进一步细分为高变区,称为互补决定区(CDR),其间散布着更保守的区域,称为框架区(FR)。每个VH和VL包含三个CDR和四个FR,从氨基末端至羧基末端以下列顺序排列:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。重链和轻链的可变区含有与抗原相互作用的结合结构域。抗体的恒定区可以介导免疫球蛋白与宿主组织或因子(包括免疫系统的各种细胞(例如效应细胞)和经典补体系统的第一组分(C1q))的结合。
免疫球蛋白可以来源于任何通常已知的同种型,包括但不限于 IgA、分泌型IgA、IgG和IgM。IgG亚类也是本领域技术人员众所周知的,并包括但不限于IgG1、IgG2、IgG3和IgG4。“同种型”是指由重链恒定区基因编码的Ab种类或亚类(例如,IgM或IgG1)。通过实例的方式,术语“抗体”包括天然存在和非天然存在的Ab;单克隆和多克隆Ab;嵌合和人源化Ab;人或非人Ab;完全合成Ab;和单链Ab。非人Ab可通过重组方法来人源化以减少其在人体内的免疫原性。当无明确记载且除非上下文另有表示时,术语“抗体”也包括前述免疫球蛋白中任一种的抗原结合片段或抗原结合部分,且包括单价和二价片段或部分,以及单链Ab。
“分离抗体”是指基本上不含其他具有不同抗原特异性的Ab的Ab (例如,特异性结合PD-1的分离Ab基本上不含特异性结合除了PD-1以外的抗原的Ab)。然而,特异性结合PD-1的分离Ab可与其他抗原诸如来自不同物种的PD-1分子具有交叉反应性。此外,分离Ab可以基本上不含其他细胞物质和/或化学物质。
术语“单克隆抗体”(“mAb”)是指具有单分子组成的Ab分子(即其一级序列基本相同且表现出对特定表位的单一结合特异性和亲和力的Ab分子)的非天然存在的制备物。mAb是分离Ab的实例。MAb可通过杂交瘤、重组、转基因或本领域技术人员已知的其他技术来产生。
“人”抗体(HuMAb)是指具有可变区的Ab,其中所述可变区中的框架区和CDR区域都来源于人类种系免疫球蛋白序列。此外,如果该Ab含有恒定区,则所述恒定区也来源于人类种系免疫球蛋白序列。本发明的人Ab可以包含不是由人类种系免疫球蛋白序列编码的氨基酸残基(例如,通过体外随机或位点特异性诱变或通过体内体细胞突变引入的突变)。然而,如本文中所用,术语“人抗体”并非意在包括这样的Ab,其中来源于另一哺乳动物物种(诸如小鼠)的种系的CDR序列被嫁接至人框架序列上。术语“人”Ab和“全人”Ab同义使用。
“人源化抗体”是指这样的Ab,其中非人Ab的CDR结构域之外的一些、大部分或所有氨基酸被对应的来源于人免疫球蛋白的氨基酸替代。在Ab的人源化形式的一个实施方案中,CDR结构域之外的一些、大部分或所有氨基酸已被来自人免疫球蛋白的氨基酸替代,而一个或多个CDR区域内的一些、大部分或所有氨基酸不变。氨基酸的小的添加、缺失、插入、置换或修饰是允许的,只要它们没有消除Ab结合特定抗原的能力。“人源化”抗体保留与原始Ab类似的抗原特异性。
“嵌合抗体”是指其中可变区来源于一个物种、而恒定区来源于另一物种的Ab,诸如其中可变区来源于小鼠Ab、而恒定区来源于人Ab的Ab。
“抗-抗原”抗体是指特异性结合抗原的抗体。例如,抗PD-1抗体特异性结合PD-1而抗CTLA-4抗体特异性结合CTLA-4。
Ab的“抗原结合部分”(也称为“抗体结合片段”)是指Ab的一个或多个片段,其保留特异性结合完整Ab所结合的抗原的能力。
“癌症”是指特征在于体内异常细胞不受控制的生长的各种疾病的一大类。不受调节的细胞分裂和生长导致恶性肿瘤的形成,所述恶性肿瘤侵入邻近组织并还可通过淋巴系统或血流向身体的远处部分转移。“癌症”和“癌组织”可以包括肿瘤。
“细胞毒性T淋巴细胞抗原-4(CTLA-4)是指属于CD28家族的免疫抑制受体。CTLA-4在体内专门表达于T细胞上,并结合两种配体,CD80和CD86(也分别称为B7-1和B7-2)。如本文中所用,术语“CTLA-4”包括人CTLA-4(hCTLA-4),hCTLA-4的变体、同种型和物种同系物,以及与hCTLA-4具有至少一个共同表位的类似物。完整hCTLA-4序列可见于GenBank登录号AAB59385下。
术语“免疫疗法”是指通过包括诱导、增强、抑制或以其他方式改变免疫反应的方法治疗患有疾病或具有接触(contracting)或遭受疾病复发的风险的个体。
患者的“治疗”或“疗法”是指对个体进行的任何类型的干预或处理或活性剂的施用,其目标在于逆转、缓解、改善、抑制、减缓或预防与疾病相关的症状、并发症、病况或生化指标的发作、进展、发展、严重程度或复发。
“程序性死亡-1(PD-1)”受体是指属于CD28家族的免疫抑制性受体。PD-1主要在之前活化的T细胞上体内表达,并结合两种配体,PD-1和 PD-2。如本文中所用,术语“PD-1”包括人PD-1 (hPD-1),hPD-1的变体、同种型和物种同系物,以及与hPD-1具有至少一个共同表位的类似物。完整hPD-1序列可见于GenBank登录号U64863下。
“程序性死亡配体-1(PD-L1)”是PD-1的两种细胞表面糖蛋白配体之一(另一种是PD-L2),其在结合PD-1后下调T细胞活化和细胞因子分泌。如本文中所用,术语“PD-L1”包括人PD-L1 (hPD-L1),hPD-L1的变体、同种型和物种同系物,以及与hPD-L1具有至少一个共同表位的类似物。完整hPD-L1序列可见于GenBank登录号Q9NZQ7下。
“个体”包括任何人或非人动物。术语“非人动物”包括但不限于,脊椎动物诸如非人灵长类、绵羊、狗,和啮齿类诸如小鼠、大鼠和豚鼠。在一些实施方案中,个体是人。术语“个体”和“患者”在本文中可互换使用。
药物或治疗剂的“治疗有效量”或“治疗有效剂量”是药物的任何这样的剂量,当单独使用或与另一治疗剂组合使用时,其保护个体免于疾病的发作或促进疾病消退,所述疾病消退通过疾病症状的严重程度降低、疾病无症状期的频率和持续时间的增加或防止由于患有疾病导致的损伤或残疾来证明。治疗剂促进疾病消退的能力可使用技术人员已知的各种方法来评价,诸如在临床试验期间的人类个体中、在预测人类中的功效的动物模型系统中、或通过在体外测定中测定所述药剂的活性。
如本文中所用,术语“亚治疗剂量”或“亚治疗有效量”是指低于治疗化合物 (例如,抗体)在单独施用用于治疗过度增殖性疾病(例如癌症)时的通常或典型剂量的治疗化合物的剂量。例如,抗PD1抗体(nivolumab)的亚治疗剂量是小于约3 mg/kg的该抗体的单一剂量。
替代方案(例如,“或”)的使用应被理解为是指替代方案中的任一种、两种或其任何组合。如本文中所用,不定冠词“一者(a或an)”应被理解为是指任何记载或列举组分中的“一者或多者”。
通过实例的方式,“抗癌剂”促进个体中的癌症消退。在其他实施方案中,治疗有效量的药物促进癌症消退至清除癌症的点。“促进癌症消退”意味着单独或与抗肿瘤剂组合施用有效量的药物导致肿瘤生长或大小的降低、肿瘤坏死、至少一种疾病症状的严重程度减低、疾病无症状期的频率和持续时间增加、或防止由于患有疾病导致的损伤或残疾。此外,关于治疗的术语“有效”和“有效性”包括药学有效性和生理学安全性。药学有效性是指药物在患者中促进癌症消退的能力。生理学安全性是指由药物施用导致的细胞、器官和/或组织水平的毒性或其他不良生理作用(不良作用)的水平。
通过肿瘤治疗的实例的方式,治疗有效量的药物优选相对于未经治疗的个体将细胞生长或肿瘤生长抑制至少约20%,更优选至少约40%、甚至更优选至少约60%、并且仍更优选至少约80%。在本发明的其他实施方案中,可观察到肿瘤消退并持续至少约20天、更优选至少约40天或甚至更优选至少约60天的时段。尽管存在治疗有效性的这些最终测量,但对免疫治疗药物的评价也必须考虑到“免疫相关的”反应模式。
“免疫相关的”反应模式是指在用免疫治疗剂治疗的癌症患者中观察到的临床反应模式,所述免疫治疗剂通过诱导癌症特异性免疫反应或通过修饰天然免疫过程来产生抗肿瘤效果。该反应模式的特征在于在肿瘤负荷的初始增加或新病灶的出现之后的有益的治疗效果,所述肿瘤负荷的初始增加或新病灶的出现在传统化疗剂的评价中将被归类为疾病进展,并且将与药物失败同义。因此,对免疫治疗剂的正确评价可能需要长期监测这些药剂对目标疾病的效果。
药物的治疗有效量包括“预防有效量”,其是任何这样的药物量,当药物单独或与抗肿瘤剂组合向具有发展癌症(例如,具有恶性前病况的个体)或遭受癌症复发的风险的个体施用时,抑制癌症的发展或复发。在实施方案中,预防有效量完全预防癌症的发展或复发。“抑制” 癌症的发展或复发意指减小癌症发展或复发的可能性,或完全预防癌症的发展或复发。
替代方案(例如,“或”)的使用应被理解为是指替代方案中的任一种、两种或其任何组合。如本文中所用,不定冠词“一者(a或an)”应被理解为是指任何记载或列举组分中的“一者或多者”。
术语“约”或“基本上包含”是指如本领域普通技术人员所确定的特定值或组成的可接 受误差范围内的值或组成,其部分取决于如何测量或确定所述值或组成,即测量系统的限制。例如,“约”或“基本上包含”可以是指根据本领域中的实践在1个或大于1个标准偏差内。或者,“约” 或“基本上包含”可以是指多达10%或20%的范围(即,±10%或±20%)。例如,约3mg/kg可包括2.7 mg/kg和3.3 mg/kg之间(对于10%)和2.4 mg/kg和3.6 mg/kg之间(对于20%)的任何数目。此外,特别是关于生物系统或方法,所述术语可以是指该值的多达一个数量级或多达5倍。当在本申请或权利要求中提供特定值或组成时,除非另有说明,“约”或“基本上包含”的含义应假设为在该特定值或组成的可接受误差范围内。
如本文所述,任何浓度范围、百分率范围、比率范围或整数范围应理解为包括所述范围内的任何整数值,并且,当适当时,包括其分数(诸如整数的十分之一或百分之一),除非另有指明。
在以下段落中进一步详细描述了本发明的各个方面。
抗PD-1抗体
PD-1是由活化的T细胞和B细胞表达的关键免疫检查点受体并介导免疫抑制。PD-1是CD28受体家族的成员,包括CD28、CTLA-4、ICOS、PD-1和BTLA。已经识别出PD-1的两种细胞表面糖蛋白配体,程序性死亡配体-1(PD-L1)和程序性死亡配体-2(PD-L2),其在抗原递呈细胞以及许多人类癌症中表达,并且已显示在结合PD-1后下调T细胞活化和细胞因子分泌。对PD-1/PD-L1相互作用的抑制介导临床前模型中有效的抗肿瘤活性。
以高亲和力特异性结合PD-1的HuMAb已公开于美国专利号8,008,449和8,779,105中。其他抗PD-1 mAb已描述于,例如,美国专利号6,808,710、7,488,802、8,168,757和8,354,509以及PCT公开号WO 2012/145493中。美国专利号8,008,449中公开的每一种抗PD-1 HuMAb均已被证明具有以下特征中的一种或多种:(a)以1 x 10-7 M或更低的KD结合人PD-1,如通过表面等离振子共振使用Biacore生物感应器系统所测定;(b)基本上不结合人 CD28、CTLA-4或ICOS;(c)在混合淋巴细胞反应(MLR)测定中增加T细胞增殖;(d)在MLR测定中增加干扰素-γ产生;(e)在MLR测定中增加IL-2分泌;(f)结合人PD-1和食蟹猴PD-1;(g)抑制 PD-L1和/或PD-L2与PD-1的结合;(h)刺激抗原特异性记忆反应;(i)刺激Ab反应;和(j) 体内抑制肿瘤细胞生长。可用于本发明中的抗PD-1抗体包括特异性结合人PD-1并具有前述特征中的至少一种、优选至少五种的mAb。
在一个实施方案中,抗PD-1抗体是nivolumab。Nivolumab(也称为“Opdivo®”;以前称为5C4、BMS-936558、MDX-1106或ONO-4538)是全人IgG4 (S228P) PD-1免疫检查点抑制剂Ab,其选择性防止与PD-1配体(PD-L1和PD-L2) 的相互作用,从而阻断抗肿瘤T细胞功能的下调(美国专利号8008449;Wang等人,2014 Cancer Immunol Res.2(9):846-56)。
在另一个实施方案中,抗PD-1抗体是派姆单抗。派姆单抗(也称为“Keytruda®”,lambrolizumab,和MK-3475)是针对人细胞表面受体PD-1(程序性死亡-1或程序性细胞死亡-1)的人源化单克隆IgG4抗体。派姆单抗描述于,例如,美国专利号8,354,509和8,900,587;还参见(最后登录:2014年12月14日)。派姆单抗已经由FDA批准用于治疗复发或难治性黑色素瘤。
在其他实施方案中,抗PD-1抗体是MEDI0608(原为AMP-514),其为单克隆抗体。MEDI0608描述于,例如,美国专利号8,609,089B2或(最后登录:2014年12月14日)。
在一些实施方案中,抗PD-1抗体是Pidilizumab (CT-011),其为人源化单克隆抗体。Pidilizumab描述于美国专利号8,686,119 B2或WO 2013/014668 A1中。
可用于本公开方法中的抗PD-1抗体还包括特异性结合人PD-1并与nivolumab交叉竞争结合人PD-1的分离Ab (参见,例如,美国专利号8,008,449和8,779,105;WO 2013/173223)。Ab交叉竞争结合抗原的能力表明这些Ab结合抗原的相同表位区,并空间阻碍其他交叉竞争性Ab与该具体表位区域的结合。预期这些交叉竞争性Ab凭借其与PD-1 的相同表位区域的结合而具有与nivolumab非常类似的功能特性。可以在标准PD-1结合测定诸如Biacore分析、ELISA测定或流式细胞术中基于其与nivolumab交叉竞争的能力来容易地识别交叉竞争性Ab(参见,例如,WO 2013/173223)。
在某些实施方案中,与nivolumab交叉竞争结合人PD-1或结合人PD-1的相同表位区域的Ab是mAb。对于施用于人类个体,这些交叉竞争Ab优选为嵌合Ab,或更优选为人源化或人Ab。这样的嵌合、人源化或人mAb可以通过本领域众所周知的方法制备和分离。
可用于本公开发明的方法中的抗PD-1抗体还包括上述Ab的抗原结合部分。已充分证明,Ab的抗原结合功能可通过全长Ab的片段来进行。术语Ab的“抗原结合部分”所涵盖的结合片段的实例包括(i)Fab片段,由VL、VH、CL和CH1结构域组成的单价片段;(ii) F(ab’)2片段,包含通过二硫键在铰链区连接的两个Fab片段的二价片段;(iii)由VH和CH1结构域组成的Fd片段;和(iv)由Ab的单一臂的VL和VH结构域组成的Fv片段。
抗CTLA-4抗体
本发明的抗CTLA-4抗体结合人CTLA-4从而破坏CTLA-4与人B7受体的相互作用。因为CTLA-4与B7的相互作用转导导致携带CTLA-4受体的T细胞失活的信号,所以相互作用的破坏有效地诱导、增强或延长此类T细胞的活化,从而诱导、增强或延长免疫反应。
以高亲和力特异性结合CTLA-4的HuMAb已公开于美国专利号6,984,720和7,605,238中。其他抗PD-1 mAb已描述于,例如,美国专利号5,977,318、6,051,227、6,682,736和7,034,121。美国专利号6,984,720和7,605,238中公开的抗PD-1 HuMAb已被证明具有以下特征中的一种或多种:(a)特异性结合人CTLA-4,其结合亲和力反映为至少约107 M-1或约109M-1或约1010 M-1至1011 M-1或更高的平衡结合常数(Ka),如通过Biacore分析所测定;(b)至少约103、约104或约105 m-1 s-1的动力学结合常数(ka);(c)至少约103、约104或约105 m-1 s-1的动力学解离常数(kd);和(d)抑制CTLA-4与B7-1(CD80)和B7-2(CD86)的结合。可用于本发明中的抗CTLA-4抗体包括特异性结合人CTLA-4并具有前述特征中的至少一种、优选至少三种的mAb。示例性临床抗CTLA-4抗体是人mAb 10D1 (现在称为伊匹单抗且作为YERVOY®销售),如美国专利号6,984,720中所公开。
示例性临床抗CTLA-4抗体是人mAb 10D1 (现在称为伊匹单抗且作为YERVOY®销售),如美国专利号6,984,720中所公开。伊匹单抗是用于本文公开的方法中的抗CTLA-4抗体。伊匹单抗是全人IgG1单克隆Ab,其阻断CTLA-4与其B7配体的结合,由此刺激T细胞活化并改善晚期黑色素瘤患者的总存活期(OS)。
可用于本方法中的另一种抗CTLA-4抗体是tremelimumab(也称为CP-675,206)。Tremelimumab是人IgG2单克隆抗CTLA-4抗体。Tremelimumab描述于WO/2012/122444、美国公开号2012/263677或WO公开号2007/113648 A2。
可用于本公开方法中的CTLA-4抗体还包括特异性结合人PD-1并与伊匹单抗或tremelimumab交叉竞争结合人CTLA-4或与伊匹单抗或tremelimumab结合相同的人CTLA-4的表位区域的分离Ab。在某些实施方案中,与伊匹单抗或tremelimumab交叉竞争结合人CTLA-4或结合人PD-1的相同表位区域的Ab是包含人IgG1同种型的重链的Ab。对于施用于人个体,这些交叉竞争Ab优选为嵌合Ab,或更优选为人源化或人Ab。可用的抗CTLA-4抗体还包括上述Ab的抗原结合部分,诸如Fab、F(ab’)2、Fd或Fv片段。
肾癌的护理标准疗法
本发明还包括抗PD-1抗体与用于治疗肾癌的护理标准疗法的组合疗法。可以在抗PD1抗体治疗之前、期间或之后的任何时间对个体进行护理标准疗法。在一些情况下,护理标准疗法和PD-1抗体治疗也与额外治疗组合。不同癌症类型的护理标准疗法是本领域技术人员众所周知的。例如,美国国立综合癌症网络(National Comprehensive Cancer Network,NCCN),美国21个主要癌症中心的联盟, 出版了NCCN肿瘤学临床实践指南(NCCN GUIDELINES®),其提供了关于多种癌症的护理标准治疗的详细最新信息(参见NCCN GUIDELINES®, 2014)。
RCC是成人中最常见类型的肾癌,占肾肿瘤的约90%,而这些中的80-90%具有透明细胞组织学(NCCN GUIDELINES®, 版本3.2014 – Kidney Cancer)。RCC占美国所有癌症的约3%,并且2014年在美国,估计63,920例患者将被诊断患有肾癌,而13,860例将死于该疾病(Siegel等人,2014)。约30%的个体在诊断时患有转移性疾病。
对于临床上局部RCC(IA期和IB期),手术切除,包括根治性肾切除术和肾单位保留手术(nephron-sparing surgery),是有效的疗法。部分肾切除术通常不适合于局部晚期肿瘤(II期和III期)患者,在该情况下优选根治性肾切除术。当肿瘤局限于肾实质时,5年存活率为60-70%,但在其中转移已经扩散的IV期疾病中,则大幅降低。IV期RCC对RT和化疗相对耐受,但患者可能从手术中获益,而全身性疗法前的减瘤性肾切除术(cytoreductive nephrectomy)被推荐用于具有潜在手术可切除的原发性和多处可切除转移的患者。
直到最近,细胞因子IL-2和IFNα,例如,IFN-2b和PegIFN-2b,依然是晚期或转移性RCC(mRCC)的唯一有效的全身性治疗,两者都提供5-27%的报道的客观反应率(ORR)。IL-2基于15%可持久反应率而被批准。然而,由于大量毒性,包括低血压(71%)、腹泻(67%)、呼吸困难(43%)、皮疹(42%)、室上性心动过速(12%)和各种代谢和血液紊乱(Fyfe等人,1995),IL-2仅施用于相对年轻和格外适合医疗的个体。此外,由于严重的急性毒性,其施用仅限于三级医疗设施中的重症护理设置。IFN-α未被批准用于治疗RCC,并且由于IL-2和IFN-α的有限的临床益处和大量毒性概况,更新的靶向药物已在晚期或转移性肾细胞癌的治疗中大量取代细胞因子。
透明细胞RCC的发病机理中低氧诱导因子α(HIFα)信号传导的重要性的认识已导致两类靶向疗法(酪氨酸激酶抑制剂TKI和mTOR抑制剂)在1L和2L治疗中的广泛 研究(Mulders, 2009)。靶向血管生成是合理的,因为组成型HIFα活化导致若干种蛋白(包括血管内皮生长因子(VEGF))的上调或活化,其可以随后导致肿瘤增殖和新生血管系统形成。此外,对VEGF活性的阻断可以调节免疫环境并刺激抗肿瘤反应。靶向mTOR途径是重要的,因为上游PI3K/Akt/mTOR信号传导途径的活化是发生组成型HIFα活化或上调的一种方法(Mulders, 2009)。
靶向血管生成的药剂包括 VEGF受体(VEGFr) TKI(例如索拉非尼、舒尼替尼、帕唑帕尼、阿昔替尼和tivozanib)以及VEGF结合mAb (例如,贝伐单抗),而靶向mTOR 途径的药剂包括mTOR抑制剂(例如,依维莫司和替西罗莫司)(Mulders, 2009;NCCN GUIDELINES®, 版本3.2014 –肾癌)。然而,持久性反应是罕见的,因为大多数患者发展出耐药性和最终的进行性疾病,并且仅在低风险患者中的一项3期试验中显示出OS改善:替西罗莫司(TORISEL®)在晚期RCC患者中显示出相比于IFNα统计学显著的OS的益处(10.9个月相比于7.3个月)(Hudes等人, 2007)。依维莫司(AFINITOR®)也已在中值无进展存活(PFS)中显示出相比于安慰剂的2.1个 月的改善,但无OS改善(Motzer等人, 2008)。在五种批准的抗血管生成剂(索拉非尼、舒尼替尼、帕唑帕尼、阿昔替尼和贝伐单抗)以及两种批准的mTOR抑制剂(替西罗莫司、依维莫司)中,仅依维莫司被批准专门在抗血管生成疗法的治疗失败后使用。在美国,依维莫司用于在舒尼替尼或索拉非尼的第一线治疗失败后的晚期RCC 的治疗,而在欧洲,依维莫司被更广泛地用于晚期RCC患者,其疾病在用VEGF靶向疗法的治疗中或治疗后已进展。对于正在使用mTOR抑制剂却依然进展的患者尚无任何推荐。
肾癌的免疫疗法
明确需要用于已经进行多线靶向疗法的患者的有效药剂,以及相比于目前标准治疗使存活延长更长时间的疗法。在IL-2和IFN-α之外,更新的免疫治疗方法最近已显示出前景。最初在mRCC患者中测试了免疫疗法,因为传统化疗和放疗已证明是令人失望的,已知肾肿瘤能够激发免疫应答,且RCC是最常见的显示自发消退的肿瘤之一(Elhilali等人, 2000; Inman等人, 2013)。目前,用于治疗RCC的各种免疫治疗策略正处于临床开发中,包括T细胞调节、免疫引发和先天免疫(Rosenblatt和McDermott, 2011; Inman等人, 2013)。使用靶向PD-1受体或其PD-L1配体的Ab逆转T细胞免疫抑制的方法也已被证明在治疗RCC中非常有希望(Topalian等人, 2012a; Brahmer等人; 2012; WO 2013/173223)。
肾癌的组合免疫疗法
涉及药剂组合(例如抗PD-1抗体与抗CTLA-4抗体)的免疫治疗方法已被证明在治疗黑色素瘤中高度有效(Wolchok等人, 2013; WO 2013/173223)。通过类比,RCC患者能够得益于不同免疫治疗药物的组合或此类药物与靶向药物或其他治疗(包括外科手术、放疗、标准癌症化疗或疫苗)的组合。然而,当免疫治疗剂与其他抗癌剂组合时,有时已观察到令人惊讶和意料之外的并发症。例如,用抗PD-1药剂(分别为nivolumab和MK-3475)对两例携带BRAF V600E突变的黑色素瘤患者的一线疗法没有引起显著毒性,但在疾病进展后用vemurafenib (ZELBORAF®)治疗导致了严重超敏性药疹,其中在其vemurafenib疗程早期具有多器官损伤(Johnson等人,2013)。一例患者随后发生急性炎性脱髓鞘性多发性神经病,另一例在低剂量vemurafenib再攻击后发生过敏反应。
具体涉及RCC,迄今批准用于治疗mRCC的所有药剂均已被批准作为单一疗法,除了贝伐单抗(AVASTIN®),其仅被批准与IFN-α组合。该组合基于PFS的改善并结合可接受的安全性概况而被批准,但是其没有导致相比于仅接受单独IFN-α的患者的中值OS的显著改进(Escudier等人,2010; Rini等人,2010),并引起≥3级高血压和蛋白尿的更高发生率。抗血管生成疗法加上免疫疗法的许多其他组合已在临床试验中评价,但已经经常观察到显著毒性,包括在舒尼替尼加上IFN-α (Motzer等人,2009)、舒尼替尼加上IL-21(Grunwald等人,2011)、舒尼替尼加上tremelimumab(Rini等人,2011)、索拉非尼加上IFN-α(Escudier等人,2007;Ryan等人,2007;Gollob等人,2007)和索拉非尼加上IL-2(Procopio等人,2011)的试验中。
在研究未经治疗的mRCC个体(n = 25)中舒尼替尼加上IFN-α的使用的1期试验中,所有个体都经历3/4级治疗出现的AE(不良事件),最常见的中性粒细胞减少症、疲劳和血小板减少症(Motzer等人,2009)。尽管更低起始剂量有更好的耐受性,但临床活性降低。用舒尼替尼加上IFN-α看到的12%反应率低于用单独的舒尼替尼看到的27.5%的历史反应率。还在未经治疗的个体(n = 9)中的1期临床试验中研究了舒尼替尼加上IL-21(Procopio等人,2011)。全剂量舒尼替尼与10 mcg/kg的IL-21的组合基于所得血液学剂量限制毒性(DLT)被认为毒性太强,而与更低剂量IL-21的组合不被认为是治疗相关的。在先前已接受≤ 1次全身性疗法的患有mRCC的28名个体中的1期剂量递增试验中研究了舒尼替尼加上tremelimumab(抗CTLA-4抗体; Ribas, 2010; 美国专利号6,682,736) (Rini等人, 2011)。在接受每天37.5 mg舒尼替尼与每12周10 mg/kg或15 mg/kg tremelimumab的组合的4名个体中观察到快速发作的肾衰竭的意料之外的毒性。尽管观察到43%部分反应率,但在MTD下的组合(每天37.5 mg舒尼替尼加上每12周10 mg/kg tremelimumab)的毒性被认为是不可接受的。
与索拉非尼的组合也已被证明毒性太强或有效性不超过索拉非尼单一疗法。尽管在包括12个mRCC个体的群体中的索拉非尼加上IFN-α的1期临床试验得出的结论是全剂量索拉非尼加上IFN 9 MIU被良好耐受且仅遇到一例三级虚弱的DLT(Escudier等人,2007),但当使用该组合时,两个随后的2期试验发现了增加的毒性(Ryan等人,2007;Gollob等人,2007)。在患有未经治疗的mRCC的62名个体中的索拉非尼加上IFN-α的2期试验中,该组合的经证实的反应率(19%)高于任一单独药剂的历史反应率,但毒性增加,包括77%的个体中的3级或更坏的毒性,限制了进一步开发(Ryan等人,2007)。在40名第一线或第二线个体中的索拉非尼加上IFN-α的2期试验中,反应率为33%,且包括2例完全反应(Gollop等人,2007)。然而,该组合的毒性比任一单独药物更强,且疗法仅在疗程间休息和剂量减少的情况下是可耐受的(65%的个体)。在患有未经治疗的mRCC的128名个体中的随机化2期试验中研究了索拉非尼加上皮下IL-2相比于单独的索拉非尼(Procopio等人,2011)。在治疗40名个体之后由于毒性而将IL-2剂量从4.5 MIU降低至3 MIU。尽管组合组中的反应率高于索拉非尼单一治疗组(分别为27%相比于15%),但两个治疗组之间的中值PFS没有显著不同。作者的结论是:使用索拉非尼加上IL-2的组合没有改善功效。
因此,当相比于用任一药物的单一疗法时,包括抗血管生成TKI的组合方案没能够(在无增加的且通常不可接受的毒性的同时)展示出对RCC的改善功效。黑色素瘤和RCC中的上述实例清楚地表明,免疫疗法(包括免疫检查点抑制剂药物诸如抗CTLA-4或抗PD-1抗体)与靶向抗癌疗法(诸如抗血管生成TKI)的组合是不可预测的,并且必须在临床试验中小心地评估安全性以及功效。尽管nivolumab(抗PD-1)与伊匹单抗(抗CTLA-4)的组合已在治疗黑色素瘤中高度有效且具有可管控的毒性(Wolchok等人,2013),但迄今尚不知道该组合在人类个体中是否比用单独药剂治疗RCC和其他癌症明显更有效。
药物组合物和剂量
本发明的治疗剂可以构成组合物,例如含有Ab或TKI和药学上可接受的载体的药物组合物。如本文中所用,“药学上可接受的载体”包括,生理上相容的任何和所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗和吸收延迟剂等。优选地,用于含有Ab的组合物的载体适合于静脉内、肌内、皮下、肠胃外、脊椎或表皮施用(例如通过注射或输注),而用于含有TKI的组合物的载体适合于非肠胃外(例如,口服)施用。本发明的药物组合物可以包含一种或多种药学上可接受的盐、抗氧化剂、水和非水载体、和/或佐剂诸如防腐剂、润湿剂、乳化剂和分散剂。
调整给药方案以提供最佳所需反应,例如最大治疗反应和/或最小不良效果。对于抗PD-1抗体(包括与另一抗癌剂组合)的施用,剂量范围可为约0.01至约20 mg/kg,约0.1至约10 mg/kg个体体重。例如,剂量可以是约0.1、约0.3、约1、约2、约3、约5或约10 mg/kg体重和约0.3、约1、约2、约3或约5 mg/kg体重。通常设计给药方案以实现这样的暴露,其导致基于Ab的典型药代动力学特性的持续受体占用(RO)。代表性治疗方案需要每周一次、约每2周一次、约每3周一次、约每4周一次、约1个月一次、约每3-6个月或更长一次施用。在某些实施方案中,约每2周一次向个体施用抗PD-1抗体,诸如nivolumab。在其他实施方案中,约每3周一次施用Ab。剂量和时间表在疗程期间可变化。例如,抗PD-1单一疗法的给药时间表可包括如下施用Ab:(i)在6周周期中每2周;(ii)每4周,持续6个剂量,然后每3个月;(iii)每3周;(iv)3-10 mg/kg一次,随后每2-3周1 mg/kg。考虑到IgG4抗体通常具有2-3周的半衰期,本发明的抗PD-1抗体的剂量方案包括经由静脉内施用约0.3-约10 mg/kg体重,优选约1-约5 mg/kg体重,更优选约1-约3 mg/kg体重,其中在最长达6周或12周周期中每14-21天给予Ab,直至完全反应或证实疾病进展。
当与其他抗癌剂组合使用时,抗PD-1抗体的剂量可以相比于单一疗法剂量降低。例如,显著低于典型的每3周约3 mg/kg的nivolumab的剂量,例如约0.1 mg/kg或少于约每3或4周,被视为亚治疗剂量。在一些实施方案中,所述亚治疗剂量为约2 mg/kg、约1 mg/kg、约0.1 mg/kg或约0.01 mg/kg。来自接受0.3 mg/kg至10 mg/kg剂量nivolumab的15名个体的受体占用数据表明PD-1占用似乎在此剂量范围内是非剂量依赖的。在所有剂量中,平均占用率为85% (范围,70%至97%),其中平均平台占用率为72%(范围,59%至81%)(Brahmer等人, 2010)。因此,约0.3 mg/kg的剂量可以允许充分暴露,以导致最大生物活性。
尽管已实现高达每两周10 mg/kg的更高nivolumab单一疗法剂量而未达到最大耐受剂量(MTD),但在检查点抑制剂加上抗血管生成疗法的其他试验中报道的显著毒性(参见,例如,Johnson等人, 2013; Rini等人, 2011)支持选择低于10 mg/kg的nivolumab剂量。
伊匹单抗(YERVOY®)被批准每3周一次静脉内给予3 mg/kg(持续4个剂量)而用于治疗黑色素瘤。因此,在实施方案中,约3 mg/kg是与抗PD-1抗体组合使用的伊匹单抗的最高剂量,但在某些实施方案中,抗CTLA-4抗体诸如伊匹单抗,当与nivolumab组合时,可以在约每两或三周约0.3-10 mg/kg体重的范围内给予。显著低于批准的每3周3 mg/kg的伊匹单抗的剂量,例如约每3或4周约0.3 mg/kg或更低,被视为亚治疗剂量。在一些实施方案中,所述亚治疗剂量为约2 mg/kg、约1 mg/kg、约0.1 mg/kg或约0.01 mg/kg。
已经证明,3 mg/kg的nivolumab与3 mg/kg的伊匹单抗的组合超过黑色素瘤群体中的最大耐受剂量(MTD),而据发现,1 mg/kg的nivolumab加上3 mg/kg的伊匹单抗或者3 mg/kg的nivolumab加上1 mg/kg的伊匹单抗的组合在黑色素瘤患者中是可耐受的(Wolchok等人, 2013)。因此,尽管每2周静脉内给予的高达10 mg/kg的nivolumab是耐受的,但在实施方案中,当与伊匹单抗组合时,抗PD-1抗体的剂量不超过约3 mg/kg。在某些实施方案中,基于风险-效益和PK-PD评估,所用剂量包含约1 mg/kg的nivolumab加上约3 mg/kg的伊匹单抗、约3 mg/kg的nivolumab加上约1 mg/kg的伊匹单抗或约3 mg/kg的nivolumab加上约3 mg/kg的伊匹单抗的组合,其各自以约每2-4周一次、约每3周一次的给药频率施用。在某些其他实施方案中,nivolumab以约0.1、约0.3、约1、约2、约3或约5 mg/kg的剂量施用,相组合地,伊匹单抗以约0.1、约0.3、约1、约2、约3或约5 mg/kg的剂量施用,其每2周施用一次,每3周施用一次,或每4周施用一次。在某些实施方案中,在诱导阶段约每2或3周向个体静脉内施用抗PD-1抗体与抗CTLA-4抗体的组合,持续2、3或4次施用。在某些实施方案中,在诱导阶段约每3周静脉内施用nivolumab与伊匹单抗的组合,持续4次施用。诱导阶段随后为维持阶段,在其期间每两或三周仅向个体以约0.1、约0.3、约1、约2、约3、约5或约10 mg/kg的剂量施用抗PD-1抗体,只要治疗证明有效或者直到不可管控的毒性或疾病进展出现。在某些实施方案中,在维持阶段约每2周以约3 mg/kg体重的剂量施用nivolumab。
对于nivolumab与其他抗癌剂的组合,在一些实施方案中,这些药剂可以其批准的剂量施用。继续治疗,只要观察到临床效果或直到不可接受的毒性或疾病进展发生。尽管如此,在某些实施方案中,施用的这些抗癌剂的剂量显著低于批准的剂量,即,将亚治疗剂量的药剂与抗PD-1抗体组合施用。可以已证明在临床试验中作为单一疗法产生最高功效的剂量,例如约每三周一次施用约3 mg/kg nivolumab,或以显著更低剂量,即,以亚治疗剂量,施用抗PD-1抗体。
TKI的给药时间表针对不同TKI而变化。例如,舒尼替尼的通常剂量包括每天一次随或不随食物向成人口服施用50 mg的固定剂量,其中以约12.5-mg增量或基于个体患者安全性和耐受性推荐的增量进行剂量调整。以四周使用治疗、随后两周停用治疗的时间表给予舒尼替尼。在一些实施方案中,以约12.5、约25、约37.5、约50、约62.5、约75、约87.5或约90mg的剂量每天一次施用舒尼替尼。在进食之前至少一小时或进食之后两小时向成年患者以400 mg的剂量每天两次口服施用索拉非尼。在一些实施方案中,以约100、约200、约300、约400、约500、约600或约800 mg的剂量每天两次施用索拉非尼。不随食物(在进食之前至少1小时或进食之后2小时)向患者以800 mg的剂量每天一次口服施用帕唑帕尼。在一些实施方案中,以约200、约400、约600、约700、约800、约900或约1000mg的剂量每天一次施用帕唑帕尼。在随或不随食物向患者以约5 mg的剂量间隔约12小时每天两次口服施用阿西替尼(INLYTA®),但必要时可以进行剂量调整至每天两次口服施用约2、约3、约7或约10mg。Tivozanib尚未被FDA批准,但推荐口服每日剂量为约1或1.5 mg,优选为1.5 mg,使用28天,随后停用14天(Eskens等人, 2011),或者使用21天,随后停用7天(Nosov等人, 2012)。在一些实施方案中,以约0.5、约0.75、约1、约1.25、约1.5、约1.75或约2 mg的剂量每天一次施用tivozanib。对于TKI与抗PD-1抗体的组合治疗,在一些实施方案中,将TKI以其批准或推荐的剂量施用。继续治疗,只要观察到临床效果或直到不可接受的毒性或疾病进展发生。
在某些实施方案中,施用的TKI的剂量是批准或推荐的剂量。在其他实施方案中,将显著低于批准剂量的剂量(即,亚治疗剂量)的TKI与抗PD-1抗体组合施用。可以已证明在临床试验中作为单一疗法产生最高功效的剂量,例如约每三周一次施用约3 mg/kg nivolumab (Topalian等人, 2012a; Topalian等人, 2012),或以更低剂量,即,以亚治疗剂量,施用抗PD-1抗体。在一些实施方案中,以约0.1、约0.3、约1、约2、约3或约5 mg/kg的剂量施用抗PD-1抗体,并且以约0.1 mg、约0.5、约1、约2、约3、约5、约7.5、约10、约12.5、约15、约20、约30、约40、约50或约75 mg/kg的剂量施用TKI。
剂量和频率根据Ab在个体中的半衰期而变化。通常,人Ab具有最长的半衰期,随后为人源化Ab、嵌合Ab和非人Ab。施用的剂量和频率可以根据治疗是预防性还是治疗性的而变化。在预防性应用中,通常在长时间内以相对较低频率时间间隔施用相对低剂量。一些患者在其余生继续接受治疗。在治疗性应用中,有时需要相对短时间间隔的相对高剂量,直到疾病进程减少或终止,优选直到患者显示疾病症状的部分或完全改善。其后,可以向患者施用预防性方案。
本发明的药物组合物中活性成分的实际剂量水平可以变化,以便获得一定量的活性成分,其对于具体患者、组合物和施用模式实现所需治疗反应是有效的,且对患者没有过度毒性。所选剂量水平取决于各种药代动力学因素,包括本发明采用的具体组合物的活性、施用途径、施用时间、采用的具体化合物的分泌速度、治疗持续时间、与采用的具体组合物组合使用的其他药物、化合物和/或材料、所治疗的患者的年龄、性别、体重、状况、总体健康和既往病史、以及医疗领域众所周知的类似因素。本发明的组合物可以经由一种或多种施用途径使用一种或多种各种本领域众所周知的方法来施用。如技术人员将理解,施用途径和/或模式将根据所需结果而变化。
本发明的方法
本公开提供治疗患有肾癌的个体的方法。在某些实施方案中,本发明包括治疗肾癌或患有肾癌的个体的方法,其包括组合疗法。在一些实施方案中,所述方法包括向个体施用治疗有效量的以下的组合:(a)作为特异性结合PD-1受体并抑制PD-1活性的Ab或其抗原结合部分的抗癌剂;和(b)另一抗癌疗法。在一个实施方案中,本发明涉及治疗有此需要的个体中的肾癌或患有肾癌的个体的方法,所述方法包括向个体施用治疗有效量的以下的组合:(a)抗PD-1抗体或其抗原结合部分;和(b)另一抗癌剂。在另一个实施方案中,本发明涉及治疗肾癌或患有肾癌的个体的方法,所述方法包括向个体施用治疗有效量的以下的组合:(a)抗PD-1抗体或其抗原结合部分;和(b)本文别处公开的护理标准疗法。由于RCC占肾肿瘤的约90%,在一些实施方案中,所述肾癌即是RCC。在其他一些实施方案中,所述个体是人类患者。
在某些实施方案中,本发明涉及治疗有此需要的个体中的肾癌或患有肾癌的个体的方法,所述方法包括向个体施用抗PD-1抗体或其抗原结合部分,其中所述个体同时用另一抗癌剂治疗。在另一个实施方案中,本发明涉及治疗有此需要的个体中的肾癌或患有肾癌的个体的方法,所述方法包括向个体施用抗PD-1抗体或其抗原结合部分,其中所述个体同时用本文别处公开的护理标准疗法治疗。由于RCC占肾肿瘤的约90%,在一些实施方案中,所述肾癌即是RCC。在其他一些实施方案中,所述个体是人类患者。
在一些实施方案中,PD-1抗体或其抗原结合部分和/或抗CTLA-4抗体或其抗原结合部分与一种或多种其他抗癌剂组合施用,所述其他抗癌剂选自VEGF-受体(VEGFr) TKI (例如,索拉非尼、舒尼替尼、帕唑帕尼、阿西替尼和tivozanib)、VEGF-结合mAb和抑制剂(例如,贝伐单抗、阿柏西普和ziv-阿柏西普)、mTOR抑制剂(例如,依维莫司和替西罗莫司)、细胞因子(例如,IFN-α、IFN-b2、聚乙二醇化IFNb2 (PegIFN-b2)和IL-2)和有丝分裂抑制剂(例如,紫杉醇、多西他赛、长春新碱、艾瑞布林、雌莫司汀、依托泊苷、伊沙匹隆、卡巴他赛、长春新碱脂质体、长春瑞滨、长春新碱、长春碱和替尼泊苷)。在一些实施方案中,所述抗癌疗法是本文描述的任何其他药剂。在某些实施方案中,本发明的疗法(例如,施用抗PD-1抗体或施用抗PD-1抗体与另一抗癌疗法)有效地增加个体存活的持续时间。例如,当与仅用另一疗法(例如,贝伐单抗或替莫唑胺)或在组合疗法的情况下仅单独使用组合疗法的两个成员之一(例如,单独的抗PD-1抗体)治疗的另一个体相比时,所述个体存活的持续时间增加至少约1个月、至少约2个月、至少约3个月、至少约4个月、至少约5个月、至少约6个月、至少约7个月、至少约8个月、至少约9个月、至少约10个月、至少约11个月或至少约1年或更长。在一些实施方案中,存活的持续时间增加至少约2个月。在某些实施方案中,本发明的疗法有效地增加个体的无进展存活的持续时间。例如,当与未经治疗的个体或仅用另一疗法(例如,贝伐单抗或替莫唑胺)或在组合疗法的情况下仅单独用组合疗法的两个成员之一(例如,单独的抗PD-1抗体)治疗的个体相比时,所述个体的无进展存活增加至少约1个月、至少约2个月、至少约3个月、至少约4个月、至少约5个月、至少约6个月、至少约7个月、至少约8个月、至少约9个月、至少约10个月、至少约11个月或至少约1年。在一些实施方案中,无进展存活增加至少约2个月。在某些实施方案中,本发明的疗法有效地增加一组个体中的反应率。例如,当与仅用另一疗法(例如,贝伐单抗或替莫唑胺)或在组合疗法的情况下仅单独用组合疗法的两个成员之一(例如,单独的抗PD-1抗体)(即,单一疗法)治疗的另一组个体相比时,该组个体中的反应率增加至少约2%、至少约3%、至少约4%、至少约5%、至少约10%、至少约15%、至少约20%、至少约25%、至少约30%、至少约35%、至少约40%、至少约45%、至少约50%、至少约55%、至少约60%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约95%、至少约99%或至少约100%。
适用于本公开的方法中的抗PD-1和抗PD-L1抗体
适用于本公开的方法中的抗PD-1抗体是以高特异性和亲和力结合PD-1、阻断PD-L1和或PD-L2的结合并抑制PD-1信号传导途径的免疫抑制效果的Ab。在任何本文公开的治疗方法中,抗PD-1或抗CTLA-4“抗体”包括分别结合PD-1或CTLA-4受体并在抑制配体结合和上调免疫系统方面表现出类似于完整Ab的功能特性的抗原结合部分或片段。在某些实施方案中,抗PD-1抗体或其抗原结合部分与nivolumab交叉竞争结合人PD-1。在其他实施方案中,抗PD-1抗体或其抗原结合部分是嵌合、人源化或人单克隆Ab或其部分。在用于治疗人类个体的某些实施方案中,所述Ab是人源化Ab。在用于治疗人类个体的其他实施方案中,所述Ab是人Ab。可以使用IgG1、IgG2、IgG3或IgG4同种型的Ab。
在某些实施方案中,抗PD-1抗体或其抗原结合部分包含人IgG1或IgG4同种型的重链恒定区。在某些其他实施方案中,抗PD-1抗体或其抗原结合部分的IgG4重链恒定区的序列包含S228P突变,其用IgG1同种型抗体的相应位置处通常存在的脯氨酸残基替代铰链区中的丝氨酸残基。Nivolumab中存在的该突变防止与内源IgG4抗体的Fab臂交换,同时保留低亲和力用于活化与野生型IgG4抗体相关的Fc受体(Wang等人, 2014)。在又其他实施方案中,所述Ab包含作为人κ或λ恒定区的轻链恒定区。在其他实施方案中,所述抗PD-1抗体或其抗原结合部分是mAb或其抗原结合部分。在包括施用抗PD-1抗体的本文描述的任何治疗方法的某些实施方案中,所述抗PD-1抗体是nivolumab。在其他实施方案中,所述抗PD-1抗体是派姆单抗。在其他实施方案中,抗PD-1抗体选自美国专利号8,008,449中描述的人抗体17D8、2D3、4H1、4A11、7D3和5F4。在仍其他实施方案中,所述抗PD-1抗体是MEDI0608(原为AMP-514)、AMP-224或Pidilizumab (CT-011)。
在其他实施方案中,本发明包括治疗有此需要的个体中的肾癌或患有肾癌的个体的方法,其包括施用抗PD-1拮抗剂或抗PD-1拮抗剂与本文描述的一种或多种抗癌剂的组合以治疗癌症。在一些实施方案中,所述肾癌是RCC。如本文提及的“抗PD-1拮抗剂”包括抑制PD-1(受体)和PD-L1(配体)之间的相互作用、使得PD-1/PD-L1的信号通路被阻断的任何分子。PD-L1和PD-L2是已经识别出的PD-1的两种配体。在一个实施方案中,抗PD-1拮抗剂是抗PD-L1抗体。PD-L1和PD-L2已显示在结合PD-1后下调T细胞激活。PD-L1和PD-L2均是结合PD-1、但不结合其他CD28家族成员的B7同系物。已经显示PD-L1在各种人类癌症中是富集的。PD-1和PD-L1的相互作用导致肿瘤浸润淋巴细胞的减少、T细胞受体介导的增殖的减少和癌细胞的免疫逃逸。免疫抑制可通过抑制PD-1与PD-L1的局部相互作用而被逆转,并且当PD-1与PD-L2的相互作用同样被阻断时,其效果是累加的。在一个实施方案中,抗PD-L1抗体可以取代本文公开的任何治疗方法中的抗PD-1抗体。在某些实施方案中,所述抗PD-L1抗体是BMS-936559(原为12A4或MDX-1105)(参见,例如,美国专利号7,943,743;WO 2013/173223)。在其他实施方案中,抗PD-L1抗体是MPDL3280A(也称为RG7446)(参见,例如,Herbst等人2013;美国专利号8,217,149)或MEDI4736 (Khleif,2013)。
在另一个实施方案中,抗PD-1拮抗剂是可溶性PD-1蛋白。在其他实施方案中,抗PD-1拮抗剂是PD-1-Fc融合蛋白。在一些实施方案中,所述PD-1-Fc融合蛋白是本文描述的任何PD-1抗体与Ig Fc结构域融合的结果。在一些实施方案中,所述Ig Fc结构域是IgG、IgA或IgM结构域。在一些实施方案中,可以通过化学合成或通过生成编码所需融合蛋白的多核苷酸而生成融合蛋白。
在某些实施方案中,抗PD-1拮抗剂包括抑制或阻止PD-1和PD-L1之间的相互作用的抗PD-1融合蛋白、反义分子、小分子、核酶或纳米抗体(nanobody)。
用于治疗RCC的抗PD-1抗体与抗血管生成TKI的组合
在公开的用于治疗RCC的方法的某些实施方案中,其他抗癌剂,即除了抗PD-1抗体以外与抗PD-1抗体组合使用的抗癌剂是抗血管生成TKI。已批准用于治疗RCC或已在临床试验中证明功效的各种TKI,包括索拉非尼、舒尼替尼、帕唑帕尼、阿西替尼和tivozanib,可以与抗PD-1抗体组合用于所公开的组合治疗方法中。然而,直到这些组合的功效和安全性在临床前或临床试验中得到评估之前,依然存在可导致意料之外的毒性或相对于单一疗法缺乏显著增强的功效的可能性。本文公开的Renca小鼠RCC模型中的临床前数据表明,抗PD-1抗体与舒尼替尼协同相互作用以抑制RCC肿瘤生长,而抗PD-1抗体和索拉非尼的组合没有显著增强索拉非尼在该小鼠RCC模型中的低水平的抗肿瘤活性(图1和2)。除了证明抗PD-1抗体与舒尼替尼的组合在小鼠模型中治疗RCC方面是高度有效的,已经在临床研究中验证了该组合以及nivolumab与帕唑帕尼的组合在人类RCC患者中的功效和安全性。
实施例5提供mRCC患者中的nivolumab与舒尼替尼或帕唑帕尼的组合的正在进行的1期试验的中期结果(NCT01472081;参见实施例4)。数据显示,nivolumab与任一这些TKI的组合表现出在这些患者中的显著更高的抗肿瘤活性和可管控的安全性概况(参见图12)。因此,在本组合治疗方法的某些实施方案中,所述抗血管生成TKI是舒尼替尼。在其他实施方案中,所述TKI是帕唑帕尼。在另外的实施方案中,所述TKI是阿西替尼。阿西替尼是VEGF受体1、2和3的高选择性、有效的抑制剂,其具有有利的毒性概况(Sonpavde等人, 2008)且已经由FDA批准用于在一种先前全身性疗法失败后治疗晚期RCC。在其他实施方案中,所述TKI是索拉非尼。在又另外的实施方案中,所述TKI是tivozanib。Tivozanib是VEGFR-1、-2和-3的有效、选择性、长半衰期的抑制剂,其正在进行用于治疗RCC的临床试验。阿西替尼和tivozanib还没有在本文的临床前或临床研究中测试。然而,基于其对所有三种VEGF受体的高度选择性的抑制(其与舒尼替尼、帕唑帕尼和索拉非尼的VEGFR抑制活性重叠,同时使脱靶毒性最小化),阿西替尼和tivozanib两者均为在公开的治疗方法中与抗PD-1抗体组合的优异候选。
在某些实施方案中,施用抗PD-1抗体或其抗原结合部分与抗血管生成TKI导致相比于未经治疗的个体或用抗PD-1抗体或其他抗癌剂的单一疗法治疗的个体T细胞对癌组织的浸润增加。如实施例2中所述,组合疗法可导致例如CD4+和CD8+ T细胞在肾肿瘤中定位的增加。T细胞浸润的这种增加可以比未经治疗的个体或用抗PD-1抗体或其他抗癌剂的单一疗法治疗的个体中的T细胞浸润水平高至少约10%、至少约20%、至少约30%、至少约40%、至少约50%、至少约60%、至少约70%、至少约80%、或至少约90%。公开的方法可以进一步导致相比于未经治疗的个体或用抗PD-1抗体或其他抗癌剂的单一疗法治疗的个体T细胞增殖增加。T细胞增殖可以增加至少约10%、至少约20%、至少约30%、至少约40%、至少约50%、至少约60%、至少约70%、至少约80%或至少约90%。在某些实施方案中,公开的方法也可以导致相比于未经治疗的个体或用抗PD-1抗体或其他抗癌剂的单一疗法治疗的个体T调节性细胞减少。
在其他实施方案中,公开的用于治疗RCC的方法相比于未经治疗的个体或用抗PD-1抗体或其他抗癌剂的单一疗法治疗的个体降低单核细胞髓源抑制细胞的数目或增加粒细胞骨髓细胞的数目。在一个实施方案中,所述单核细胞髓源抑制细胞的特征在于CD11b+/Ly6C高/Ly6G-表达或CD11b+/Ly6C低/Ly6G-表达。在另一个实施方案中,所述粒细胞骨髓细胞的特征在于CD11b+/Ly6C-/Ly6G+表达。单核细胞髓源抑制细胞的数目可以减少至少约5%、至少约10%、至少约15%、至少约20%、至少约25%、至少约30%、至少约40%、至少约50%、至少约60%、至少约70%、至少约80%或至少约90%。粒细胞骨髓细胞的数目可以增加至少约5%、至少约10%、至少约20%、至少约30%、至少约40%、至少约50%、至少约60%、至少约70%、至少约80%或至少约90%。
组合疗法中使用的抗PD-1抗体和抗血管生成TKI的剂量
在本组合治疗方法的某些实施方案中,抗PD-1抗体或其抗原结合部分的治疗有效剂量包括每周约一次、每2周约一次、每3周约一次或约每个月一次通过静脉内输注与抗血管生成TKI组合施用约0.1至约10.0 mg/kg体重的剂量。在某些实施方案中,以2 mg/kg的剂量每3周一次施用抗PD-1抗体。在其他实施方案中,以约5 mg/kg的剂量约每3周一次施用抗PD-1抗体。
舒尼替尼的剂量可以包括,例如,在42天周期中向成年RCC患者施用约25至约50mg、在一些实施方案中50mg的剂量,所述42天周期包括28天每天施用,14天未施用,并且重复周期,只要观察到临床益处或直到不可管控的毒性或疾病进展出现。或者,在28天每天施用25至50mg和14天未施用之后,可以每天以37.5 mg施用舒尼替尼,只要观察到临床益处或直到不可管控的毒性或疾病进展出现。在一些实施方案中,以约5、约10、约15、约20、约25、约30、约35、约40、约45、约50、约55、约60、约65、约70或约75 mg/kg体重的剂量施用舒尼替尼。
帕唑帕尼以每天一次口服约50、约100、约200、约300、约400、约500、约600、约700或约800 mg的剂量与抗PD-1抗体组合施用。在一些实施方案中,帕唑帕尼每天一次以其800 mg的推荐剂量不随食物而施用。
与抗PD-1抗体组合的还没有在临床试验中测试的除了舒尼替尼和帕唑帕尼以外的TKI的剂量基于已知的治疗剂量。因此,在一些实施方案中,索拉非尼以其每天两次口服400 mg的推荐剂量施用。在一些实施方案中,阿西替尼(INLYTA®)以其间隔约12小时每天两次口服5 mg的推荐剂量向患者施用。在一些实施方案中,tivozanib以其1.5 mg的推荐口服每日剂量施用,使用28天,随后停用14天(Eskens等人, 2011),或者使用21天,随后停用7天(Nosov等人, 2012)。
在实施方案中,nivolumab在每6周(42天)周期中在第1天和第22天通过静脉内输注以约2或约5 mg/kg与TKI组合施用,例如在每42天周期的第1 – 28天口服施用约50 mg舒尼替尼,或者在每42天周期的第1 – 42天口服施用约800 mg帕唑帕尼。本方法中采用的其他TKI根据其批准或推荐的剂量方案施用。继续治疗,只要观察到临床效果或直到不可接受的毒性或疾病进展发生。
在例如抗PD-1和舒尼替尼之间的临床前模型中观察到的协同相互作用表明,这些治疗剂中的一种或两种可以亚治疗剂量(即,显著低于当作为单一疗法施用用于治疗癌症时的通常或FDA批准的剂量的治疗剂的剂量)向患者施用。在公开发明的某些实施方案中,抗PD-1抗体或其抗原结合部分以亚治疗剂量向RCC患者施用。在其他实施方案中,TKI以本文描述的亚治疗剂量施用。在另外的实施方案中,抗PD-1抗体或其抗原结合部分与TKI各自以本文描述的亚治疗剂量施用。
抗PD-1抗体或其抗原结合部分与其他抗癌剂的组合疗法可以同时或依次施用(例如,首先施用抗PD-1抗体或其抗原结合部分,然后施用其他抗癌剂,或者首先施用其他抗癌剂,然后施用抗PD-1抗体或其抗原结合部分)。
用于治疗RCC的抗PD-1抗体与抗CTLA-4抗体的组合
本公开还提供用于治疗RCC的组合治疗方法,其中将抗PD-1抗体与作为特异性结合CTLA-4并抑制CTLA-4活性的Ab或其抗原结合部分的另一抗癌剂组合。本文已经证明(参见实施例6),抗PD-1抗体 nivolumab与抗CTLA-4抗体伊匹单抗的组合产生一定水平的临床活性,如通过ORR测量,其显著高于用单独的nivolumab或伊匹单抗观察到的活性。因此,在某些实施方案中,与抗PD-1抗体组合使用的抗CTLA-4抗体是伊匹单抗。在实施方案中,所述抗CTLA-4抗体是tremelimumab。在其他实施方案中,所述抗CTLA-4抗体或其抗原结合部分是与伊匹单抗交叉竞争结合人CTLA-4的Ab或其部分。在某些其他实施方案中,所述抗CTLA-4抗体或其抗原结合部分是嵌合、人源化或人mAb或其部分。在又其他实施方案中,所述抗CTLA-4抗体或其抗原结合部分包含人IgG1或IgG4同种型的重链恒定区。在实施方案中,所述抗CTLA-4抗体包含人IgG1同种型的重链恒定区。
对于抗PD-1抗体与抗CTLA-4抗体的组合,给药方案包括诱导期(本文中也称为诱导阶段),其中向患者施用抗PD-1抗体与抗CTLA-4抗体的一个或多个,优选约四个,组合剂量,随后为维持期或阶段,其包括用单独的抗PD-1抗体给药,即,不包括抗CTLA-4抗体。在某些实施方案中,所述方法包括:(a)诱导阶段,其中以2、4、6、8或10个剂量组合施用抗PD-1抗体或其抗原结合部分与抗CTLA-4抗体或其抗原结合部分,每次剂量为至少约每2周一次、约每3周一次或约每4周一次施用约0.1至约10.0 mg/kg体重,随后是(b)维持阶段,其中没有施用抗CTLA-4抗体或其抗原结合部分,而至少约每2周一次、约每3周一次或约每4周一次以约0.1至约10 mg/kg的剂量重复施用抗PD-1抗体或其抗原结合部分。
在某些实施方案中,抗PD-1抗体或其抗原结合部分以亚治疗剂量施用。在某些其他实施方案中,抗CTLA-4抗体或其抗原结合部分以亚治疗剂量施用。在另外的实施方案中,抗PD-1抗体或其抗原结合部分与抗CTLA-4抗体或其抗原结合部分均各自以亚治疗剂量施用。在一些实施方案中,抗PD-1抗体或其抗原结合部分与/或抗CTLA-4抗体或其抗原结合部分以治疗剂量施用。
在某些实施方案中,(a)所述诱导阶段包括以3周时间间隔施用至少4个剂量,其中抗PD-1抗体与抗CTLA-4抗体以下列剂量施用:(i) 0.1 mg/kg 抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(ii) 0.3 mg/kg 抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(iii) 1 mg/kg 抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(iv) 3 mg/kg 抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(v) 5 mg/kg 抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(vi) 10 mg/kg 抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(vii) 0.1 mg/kg 抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(viii) 0.3 mg/kg 抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(ix) 1 mg/kg 抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(x) 3 mg/kg 抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(xi) 5 mg/kg 抗PD-1抗体与1 mg/kg抗CTLA-4抗体;或(xii) 10 mg/kg 抗PD-1抗体与1 mg/kg抗CTLA-4抗体,并且(b)所述维持阶段包括以每2周3 mg/kg的剂量重复施用抗PD-1抗体。
由于先前用免疫疗法通过抑制免疫检查点所展示出的临床效果的持久性(参见,例如,WO 2013/173223),所以在替代实施方案中,所述维持阶段可以包括,有限数目的剂量,例如,1-10个剂量,或者可以涉及以长时间间隔给药,例如,约每3-6个月施用一次或约每1-2年或更长时间间隔施用一次。可以继续维持阶段,只要观察到临床效果或直到不可管控的毒性或疾病进展发生。
考虑到经过12周的伊匹单抗施用是否有助于黑色素瘤中的临床益处的不确定性以及美国食品和药品管理局(FDA)和欧洲药品管理局(EMA)批准的YERVOY®的时间表为每3周共4个剂量的事实,在实施方案中,抗CTLA-4抗体在诱导阶段每3周施用一次,共4 个剂量。因此,在某些实施方案中,所述方法包括(a)由以3周时间间隔施用的4个组合剂量组成的诱导阶段,其中(i)以3 mg/kg体重施用抗PD-1抗体或其抗原结合部分,且以1 mg/kg体重施用抗CTLA-4抗体或其抗原结合部分;(ii)以1 mg/kg体重施用抗PD-1抗体或其抗原结合部分,且以3 mg/kg体重施用抗CTLA-4抗体或其抗原结合部分;或(iii)以3 mg/kg体重施用抗PD-1抗体或其抗原结合部分,且以3 mg/kg体重施用抗CTLA-4抗体或其抗原结合部分;并且(b)维持阶段包括以每2周3 mg/kg的剂量重复施用抗PD-1抗体或其抗原结合部分。在这些方法的另外的实施方案中,继续维持阶段,只要观察到临床效果或直到不可接受或不可管控的毒性或疾病进展发生。
在本方法的某些实施方案中,所述抗PD-1抗体是nivolumab。在其他实施方案中,其为派姆单抗。在又其他实施方案中,所述抗CTLA-4抗体是伊匹单抗。在另外的实施方案中,所述抗CTLA-4抗体是tremelimumab。通常,抗PD-1抗体与抗CTLA-4抗体被配制用于静脉内施用。在一些实施方案中,抗PD-1抗体或其抗原结合部分与抗CTLA-4抗体或其抗原结合部分作为单一组合物或作为单独的组合物同时施用。在一些实施方案中,抗PD-1抗体与抗CTLA-4抗体依次施用。在一些实施方案中,抗PD-1抗体与抗CTLA-4抗体在诱导阶段依次施用。在某些实施方案中,当抗PD-1抗体与抗CTLA-4抗体组合施用时, 它们在彼此的30分钟内施用。可以首先施用任一Ab,也就是说,在某些实施方案中,抗PD-1抗体在抗CTLA-4抗体之前施用,而在其他实施方案中,抗CTLA-4抗体在抗PD-1抗体之前施用。通常,每种Ab在60分钟内通过静脉内输注施用。在某些实施方案中,抗 PD-1抗体与抗CTLA-4抗体混合于药学上可接受的制剂中作为单一组合物同时施用,或各Ab分别在药学上可接受的制剂中作为单独的组合物同时施用。
本发明方法的某些实施方案包括(a)由以下组成的诱导阶段:每3周通过静脉内输注施用nivolumab,随后通过静脉内输注施用伊匹单抗,共4个组合剂量,随后是(b)在诱导疗法的第4个剂量之后或第113天之后(如果由于治疗延迟而还没有施用诱导疗法的第4个剂量)3周开始每2周通过静脉内输注施用nivolumab维持给药。
在某些实施方案中,可以向个体施用抗PD-1抗体或其抗原结合部分、抗血管生成TKI与抗CTLA-4抗体或其抗原结合部分的组合用于治疗RCC。在一些实施方案中,所述抗血管生成TKI是索拉非尼、舒尼替尼、帕唑帕尼、阿西替尼或tivozanib。在某些实施方案中,所述抗血管生成TKI是舒尼替尼或帕唑帕尼。在另一个实施方案中,所述抗CTLA-4抗体是伊匹单抗或tremelimumab。在一个具体实施方案中,所述抗PD-1抗体或其抗原结合部分、抗血管生成TKI与抗CTLA-4抗体或其抗原结合部分的组合以选自以下的剂量施用:(i) 0.1 mg/kg抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(ii) 0.3 mg/kg抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(iii) 1 mg/kg抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(iv) 3 mg/kg抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(v) 5 mg/kg抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(vi) 10 mg/kg抗PD-1抗体与3 mg/kg抗CTLA-4抗体;(vii) 0.1 mg/kg抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(viii) 0.3 mg/kg抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(ix) 1 mg/kg抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(x) 3 mg/kg抗PD-1抗体与1 mg/kg抗CTLA-4抗体;(xi) 5 mg/kg抗PD-1抗体与1 mg/kg抗CTLA-4抗体;或(xii) 10 mg/kg抗PD-1抗体与1 mg/kg抗CTLA-4抗体,其中TKI以约其推荐剂量施用。例如,舒尼替尼可以约25至约50 mg、在一些实施方案中50 mg的剂量施用;帕唑帕尼可以约200、约400或约800 mg的剂量施用;索拉非尼可以每天两次以约400 mg的剂量施用;而tivozanib可以其每天约1.5 mg的推荐口服每日剂量施用。
用于治疗RCC的抗PD-1抗体与其他已知抗癌药物的组合
本领域技术人员将认识到,所述用抗PD-1抗体或其抗原片段治疗RCC的方法可以与RCC的其他已知治疗组合。例如,抗PD-1抗体或其抗原性片段可以与有丝分裂抑制剂诸如紫杉醇、多西他赛、长春新碱、艾瑞布林、雌莫司汀、依托泊苷、伊沙匹隆、卡巴他赛、长春新碱脂质体、长春瑞滨、长春新碱、长春碱或替尼泊苷组合。各种细胞因子也已知可用于治疗癌症,包括如上所讨论的IFN-α或IL-2。抗PD-1抗体或其抗原性片段可以与细胞因子诸如如IFN-α、IFN-b2、聚乙二醇化IFNb2(PegIFN-b2)或IL-2组合。本领域中已知的其他小分子治疗也可以是合适的,包括但不限于,长春氟宁。在一些实施方案中,本文描述的抗PD-1抗体,包括抗PD-1抗体与TKI或抗CTLA-4抗体的组合治疗与本文描述的任何其他治疗(包括SOC治疗)组合使用。
抗PD-1抗体可以与一种或多种本文描述的其他已知抗癌药物组合施用,其剂量为特定抗癌药物的标准。例如,帕唑帕尼可以约400至约800mg的剂量施用;阿西替尼可以约1 mg、约2 mg、约3 mg、约4 mg、约5 mg、约6 mg、约7 mg、约8 mg、约9 mg或至少约10 mg的剂量施用;紫杉醇和/或多西他赛可以约10 mg/m2、约25 mg/m2、约30 mg/m2、约35 mg/m2、约40 mg/m2、约45 mg/m2、约50 mg/m2、约75 mg/m2、约100 mg/m2、约125 mg/m2、约150 mg/m2、约175 mg/m2、约200 mg/m2、约225 mg/m2、约250 mg/m2、约275 mg/m2或至少约300 mg/m2的剂量施用;而长春氟宁可以约50 mg/m2、约75 mg/m2、约100 mg/m2、约125 mg/m2、约150 mg/m2、约175 mg/m2、约200 mg/m2、约225 mg/m2、约250 mg/m2、约275 mg/m2、约300 mg/m2、约325 mg/m2、约350 mg/m2、约375 mg/m2、约400 mg/m2、约425 mg/m2、约450 mg/m2、约475 mg/m2或至少约500 mg/m2的剂量施用。
药剂盒
用于治疗用途的包括抗PD-1抗体与TKI的药剂盒也在本发明的范围内。药剂盒通常包括表明药剂盒内容物的预期用途的标签和使用说明书。术语标签包括在药剂盒上提供或与药剂盒一起提供或另外伴随药剂盒的任何书面或录制的材料。因此,本公开提供了用于治疗患有肾癌的个体的药剂盒,所述药剂盒包括:(a)剂量为0.1至10 mg/kg体重的抗癌剂,其为特异性结合PD-1受体并抑制PD-1活性的Ab或其抗原结合部分;(b)一定剂量的另一抗癌剂,其为(ⅰ)抗血管生成TKI,例如,以治疗四周、随后停止治疗两周的时间表每天一次12.5、25或50 mg的剂量的舒尼替尼,每天一次200、400或800 mg的剂量的帕唑帕尼,或每天一次或两次400 mg的剂量的索拉非尼;或(ii)剂量为0.1至10 mg/kg体重的特异性结合并抑制CTLA-4的抗体或其抗原结合部分;和(c)用于在本文描述的任何组合治疗方法中使用抗PD-1抗体与其他抗癌剂的说明书。在某些实施方案中,抗PD-1、抗CTLA-4抗体和/或TKI可以共同包装在单位剂型中。在用于治疗人类患者的某些实施方案中,所述药剂盒包括本文公开的抗人PD-1抗体,例如,nivolumab或派姆单抗。在其他实施方案中,所述药剂盒包括本文公开的抗人CTLA-4抗体,例如,伊匹单抗或者tremelimumab。
本发明通过以下实施例进一步说明,所述实施例不应被解释为进一步限制性的。整个本申请中引用的所有参考文献的内容通过引用明确地并入本文。
实施例1
在小鼠模型中用抗PD-1与TKIs治疗肾癌
材料和方法
动物
在所有研究中都使用雌性8-12周龄Balb/c和C57/BL6小鼠(Harlan Laboratories, Federick, MD)。将小鼠饲养于无菌微隔离笼中,并随意施用无菌食物和水,除非另有规定。
药剂
舒尼替尼(SUTENT®)是靶向若干种受体酪氨酸激酶(包括VEGFR-1、VEGFR-2 和VEGFR-3、PDGFR-α 和-β、c-Kit、FLT-3、CSF-1R 和RET)的可口服的、小分子、多激酶抑制剂,其由FDA批准用于治疗肾细胞癌。舒尼替尼已报道具有免疫调节作用,包括减少髓源抑制细胞((Ko等人, 2009; Ko等人, 2010)和减少T调节性抑制细胞(Hipp等人, 2008; Ozao-Choy等人, 2009)。
索拉非尼(NEXAVAR®)是可口服的、小分子多激酶抑制剂(cRAF、bRAF、KIT、FLT-3、VEGFR-2、VEGFR-3和PDGFR-β),其由FDA批准用于治疗原发性肾癌(晚期肾细胞癌)、晚期原发性肝癌和放射性碘耐受的晚期甲状腺癌。索拉非尼已报道通过诱导细胞凋亡来抑制T细胞增殖(Houben等人, 2009; Molhoek等人, 2009)、减少抗原特异性T细胞的诱导和减少树突细胞成熟(Hipp等人, 2008)。
抗PD-1 mAb,4H2,是嵌合大鼠-小鼠抗小鼠PD-1抗体(美国专利号8,008,449),其被纯化和证明具有< 0.5 EU/mg的内毒素水平、> 95%的纯度和<5%的高分子量物质。
将小鼠肾癌(Renca)和小鼠结肠腺癌(CT-26)肿瘤细胞系在体外维持少于10代。
功效研究
皮下(SC)植入肿瘤细胞。当平均肿瘤体积达到约100 mm3时,将小鼠以8只为一组随机分组。每日一次口服施用媒介物(对照)、舒尼替尼(120 mg/kg)或索拉非尼(200 mg/kg),持续14天(QDx14, PO)。每4天以10 mg/kg腹膜内(IP)施用MAb 4H2,总共4次治疗(Q4Dx4)。每周两次测量肿瘤,并将肿瘤体积计算为长度 x (宽度2/2)。协同作用被定义为超过最高单药剂的量(EOHSA),即,如果组合的抗肿瘤效果显著高于用单一疗法观察到的效果,则该组合被认为是协同的(P < 0.05, Wilcoxon符号秩检验)。
结果
用抗PD-1与舒尼替尼或索拉非尼作为单一疗法或组合疗法治疗对植入的RCC小鼠肿瘤的中值体积的影响显示于图1中。舒尼替尼单一疗法显示在该Renca小鼠RCC模型中到治疗结束时(第33天;参见表1)产生85%的肿瘤生长抑制(TGI)的活性;然而,在疗法停止之后,肿瘤逐步生长(参见图1)。当作为单一疗法施用时,抗PD-1 mAb在该模型中无活性,但抗PD-1与舒尼替尼的组合产生显著的抗肿瘤活性(101%的TGI),导致完全肿瘤消退(表1)或肿瘤生长的显著延迟,直到植入后50天监测结束时(图1)。在相同模型的两次独立研究中观察到的这些结果显示小鼠RCC (Renca)肿瘤模型中的nivolumab小鼠(代表抗PD-1 mAb)与舒尼替尼之间的协同相互作用,因为该组合在抑制肿瘤生长中的作用明显大于每种治疗剂单独表现出的抑制水平的总和。
相反,尽管索拉非尼单一疗法在RCC模型中有活性(662% TGI;表1),用抗PD-1 mAb与索拉非尼的组合治疗在该鼠模型中没有显著增强索拉非尼的抗肿瘤活性,并且没有导致协同效应(图1)。
图2显示肿瘤达到500 mm3的体积所花费的时间(以天数计)。与图1中的数据一致,用抗PD-1/舒尼替尼组合治疗的肿瘤的生长被显著抑制,并且这些肿瘤花费约63天达到500 mm3,显著长于用对照、抗PD-1抗体、舒尼替尼、索拉非尼或抗PD-1/索拉非尼组合治疗的肿瘤所花费的19-34天。
在植入后33天测量的百分比肿瘤生长抑制显示于表1中。这些数据与图1和2显示的数据一致,证实肿瘤生长被抗PD-1与舒尼替尼的组合完全抑制,而其他治疗引起更低水平的肿瘤生长抑制。抗PD-1与舒尼替尼的组合相比于抗PD-1和索拉非尼的组合表现出的优越功效与舒尼替尼和索拉非尼分别介导的不同免疫调节作用一致,这表明,舒尼替尼,而非索拉非尼,适于与用于治疗癌症患者的免疫治疗方法进行组合(参见Hipp等人, 2008)。然而,抗PD-1与索拉非尼的组合在人类个体中的作用不能完全反映小鼠RCC模型中观察到的作用,并且该组合在治疗人患者中可能是非常有效的。
表 1.植入后33天测试的治疗剂介导的肿瘤生长抑制(TGI)和客观反应率
CR = 完全消退;PR = 部分消退。
各种治疗对小鼠的总体重的影响显示于图3中。抗PD-1与舒尼替尼的组合被良好耐受,如缺乏小鼠体重的降低所证明。抗PD-1与索拉非尼的组合也被良好耐受,但明显略逊于抗PD-1与舒尼替尼的组合,因为索拉非尼组合引起了小的、但可察觉的体重降低。
实施例2
用抗PD-1与舒尼替尼治疗后肿瘤的免疫细胞浸润
材料和方法
使用免疫组织化学和流式细胞术分析评估肿瘤的免疫细胞浸润。
免疫组织化学
将冷冻切片固定于丙酮/甲醇混合物中,并在磷酸盐缓冲盐水(PBS)中清洗。针对内源性过氧化物酶封闭切片,并添加一抗(抗CD8、抗CD4、抗FoxP3和抗PD-L1),并温育过夜。使用基于棕色聚合物的检测系统(Biocare Medical)进行显色,其中苏木精用作蓝色复染剂用于显现细胞核。
通过流式细胞术的免疫分型
分离肿瘤浸润性免疫细胞并制备为单细胞悬浮液,然后染色,并经由流式细胞术分析T-细胞子集(调节性和激活的)和骨髓细胞的表达(即,CD3、CD4、CD8、FoxP3、CD25和CD69的表达)。
结果
在对照小鼠中,免疫组织化学(IHC)分析检测到肿瘤周边的CD4+和CD8+ T细胞(图4)。当给动物施用舒尼替尼时,存在CD4+和CD8+ T细胞向周边和沿血管的主要流入。相反,当给小鼠施用抗PD-1 mAb与舒尼替尼的组合时,在整个肿瘤中观察到CD4+和CD8+ T细胞的更大浸润。与单独的每种药剂或对照媒介物相比,抗PD-1 mAb与舒尼替尼的组合导致产生更大数量的肿瘤浸润性CD4+和CD8+ T细胞(图4)。相反,与对照组相比,用单独的抗PD-1 mAb或索拉非尼或抗PD-1 mAb与索拉非尼的组合没有观察到增加的T-细胞浸润入肿瘤中(数据未显示)。已证明舒尼替尼和索拉非尼表现出免疫调节作用,这可能同当这些药剂与抗PD-1抗体免疫检查点抑制剂组合时所观察到的不同的治疗效果密切相关。
肿瘤浸润性免疫细胞的流式细胞术分析显示用与舒尼替尼或索拉非尼组合的抗PD-1 mAb治疗的组内的显著差异(图5-7)。与抗PD-1 mAb与索拉非尼的组合相比,抗PD-1 mAb与舒尼替尼的组合导致(i)增殖中的CD4+/Ki67+、CD8+和CD8+/Ki67+的更大频率(P < 0.05;图5和6);(ⅱ)单核细胞髓源抑制细胞(如CD11b+/Ly6C高/Ly6G-所定义(P < 0.05))、表达CD11b+/Ly6C低/Ly6G-的单核细胞或两者的减少百分比(P < 0.05;图7);和(iii)粒细胞骨髓细胞CD11b+/Ly6C-/Ly6G+的增加百分比(P < 0.05;图7)。因此,用舒尼替尼与抗PD-1 mAb的组合治疗诱导肿瘤微环境的组成的变化,这可以支持有效的抗肿瘤免疫应答。在所有实验组中均检测到低数目的T-调节性细胞(CD45+/CD4+/CD25+/FoxP3+),使得难以确定治疗效果(数据未显示)。
实施例3
抗原特异性免疫应答的体内细胞毒性的增强
材料和方法
细胞毒性测定
将表达AH1抗原的CT26小鼠结肠癌模型用于实验中以确定治疗对抗原特异性T细胞是否具有任何作用。用CT26细胞SC植入后五天,将小鼠用媒介物或抗PD-1 mAb 10 mg/kg治疗,间隔3天IP给予2个剂量(Q3Dx2),用或不用舒尼替尼120 mg/kg,每天一次,持续5天(QDx5)。在最后一次治疗后第2和4天确定细胞杀伤。
测定前一天,将来自未经实验的(naive) BALB/c供体的脾细胞与或不与 CT26肽([H] SPSYVYHQF [OH]; Sigma-Genosys)一起温育,并用5(6)-羧基荧光素二乙酸酯N-琥珀酰亚胺基酯(CFSE, Sigma-Aldrich)标记。将抗原致敏和不致敏的细胞合并(1:1)并注入经处理的小鼠。24小时后经由流式细胞术测定百分比细胞杀伤。
结果
在CT26肿瘤模型中,抗PD-1 mAb与舒尼替尼的组合导致针对抗原特异性免疫应答的体内细胞毒性增强(图8)。该增强的细胞毒性显示增加的免疫应答组分。它也与肿瘤体积的减少相关(图9)。
实施例4
用与舒尼替尼、帕唑帕尼或伊匹单抗组合的Nivolumab治疗转移性RCC的1期临床试验
总而言之,实施例1-3中公开的数据支持对抗PD-1抗体诸如nivolumab或派姆单抗与抗血管生成的TKI诸如舒尼替尼、帕唑帕尼、索拉非尼、阿西替尼或tivozanib的组合用于治疗患有RCC的个体的研究。这些数据表明,舒尼替尼的免疫调节特性可以导致肿瘤微环境的变化,其支持有效的抗肿瘤免疫应答,这可以由抗PD-1抗体扩增和增强。尽管抗PD-1 mAb与索拉非尼的组合疗法没有显著增强小鼠RCC模型中的索拉非尼的抗肿瘤活性,并且没有导致协同效应(图1),但该组合被良好耐受,并且在人类患者中是有效的。目前在患有mRCC的个体中在正在进行的1期研究(NCT01472081)中评估与舒尼替尼、帕唑帕尼或伊匹单抗组合的nivolumab的安全性和功效。帕唑帕尼是可口服的多激酶抑制剂(VEGFR-1、VEGFR-2、VEGFR-3、PDGFR-α和-β以及c-KIT),并且与用nivolumab的单一疗法相比,nivolumab与伊匹单抗的组合已经在黑色素瘤的治疗中显示显著增强的效力(Wolchok等人, 2013; WO 2013/173223)。
该1期研究的主要目标是评估nivolumab加上舒尼替尼、帕唑帕尼或伊匹单抗的总体安全性和耐受性,以确定RCC患者中每种组合的MTD和推荐的2期剂量。次要目标是评价组合的初步抗肿瘤活性,而探索性目标是确定响应是否与基线PD-L1表达相关。
方法
研究设计
患有mRCC的个体中的nivolumab加上舒尼替尼、帕唑帕尼或伊匹单抗的1期开放标签研究正在进行。该试验包括5个平行治疗组:nivolumab加上舒尼替尼(组S)、nivolumab加上帕唑帕尼(组P)、nivolumab 3 mg/kg加上伊匹单抗1 mg/kg (组I-1)、nivolumab 1 mg/kg加上伊匹单抗3 mg/kg (组I-3)以及nivolumab 3 mg/kg加上伊匹单抗3 mg/kg (组IN-3)。在下列两个部分中正在进行组S和组P的治疗:剂量递增(经先前治疗的患者),随后是剂量扩大(未经治疗的患者)。I-1和I-3组的治疗是每组中由经先前治疗的个体以及未经治疗的个体两者组成的单个初始组群。组I-1和I-3以及组IN-3的扩大组群由经最小程度治疗(包括未经治疗)的个体组成。组S和组P的研究设计总结于图10A中,并且组I-1、I-3和IN-3的研究设计总结于图10B中。
研究群体
研究群体由患有病理证实的转移性肾细胞癌(具有透明细胞组成)的≥18岁的个体组成,其具有至少一个可测量病灶,如实体瘤(RECIST) 1.1的反应评价标准所定义。仅在组S和组P的剂量递增组群中允许具有如RECIST 1.1所定义的可测量病灶的非透明细胞RCC患者。对于组S、P以及I-1 和I-3初始组群,还需要有利或中等风险MSKCC (Memorial Sloan Kettering Cancer Center)预后评分和Karnofsky表现状态(KPS) ≥ 80%,(对于I-1和I-3扩展以及IN-3,任何MSKCC预后评分是可接受的)。
组S和组P
在晚期/转移性设置中,个体必须先前已接受至少一个全身治疗方案,才有资格参加研究的剂量递增部分,而个体必须是未经治疗的,才有资格参加研究的剂量扩展部分。指定先前接受帕唑帕尼的参加剂量递增阶段的个体接受nivolumab加上舒尼替尼,而指定先前接受舒尼替尼的个体接受nivolumab加上帕唑帕尼。已接受先前帕唑帕尼和舒尼替尼的个体没有资格。还从研究中排除用全身性类固醇或任何其他免疫抑制剂长期治疗的个体,或先前用抗PD-1、抗PD-L1或抗PD-L2、抗CD137、抗CTLA-4或类似药剂治疗的那些。
先前没有接受舒尼替尼或帕唑帕尼的个体可登记参加剂量递增组,或者当两个组开放时,以交替方式被指定至这些组。预期约36名个体(每组18名个体)在研究的剂量递增部分中进行治疗。在递增组群完成时,基于改良的毒性概率区间(mTPI)设计评估MTD。如果在组S或组P中,nivolumab的MTD被确定为< 5 mg/kg,该组将不被进一步扩展以收集未经治疗个体中的安全性数据。如果与舒尼替尼或帕唑帕尼组合的nivolumab的MTD被确定为5 mg/kg或更高,则相应组中的单独扩展组群将以5 mg/kg的剂量开放登记。在组S和组P的扩展组群中,仅登记未经治疗的患者,使得每组治疗约20名未经治疗的个体。每个治疗周期的持续时间为6周,其中在第1和22天给予nivolumab,并且根据舒尼替尼(每天50 mg;使用4周,停用2周)和帕唑帕尼(每天800 mg)的批准产品标签给予抗血管生成疗法。
组S和组P的剂量递增
剂量水平定义于表2和3中。Nivolumab的患者内剂量递增是不允许的。
表 2.nivolumab加上舒尼替尼的剂量递增的剂量水平
a 仅在剂量水平1需要递减(de-escalation)时,才登记剂量水平-1。
表 3.nivolumab加上帕唑帕尼的剂量递增的剂量水平
a 仅在剂量水平1需要递减(de-escalation)时,才登记剂量水平-1。
各个剂量递增组的nivolumab的开始剂量均为2 mg/kg。最初,在剂量水平1治疗每组7名有资格的个体。关于额外个体是否将在相同剂量水平治疗或递增至更高剂量水平(5 mg/kg)的决定将由DLT观察时段期间观察到的剂量限制性毒性(DLTs)的数目来指导。由于除了剂量限制性毒性以外的原因而未完成DLT时段的个体将被替换。如果2 mg/kg的安全性和耐受性概况是不可接受的,可考虑剂量水平-1 (0.3 mg/kg)。
作为组S和组P的第1阶段剂量递增试验,这些组中每个剂量的样品大小取决于观察到的毒性和后验推断。预期每个组中在剂量递增阶段期间治疗总共18名个体。然而,每组可能由于替换而治疗多于18名个体,或者每组可能由于在最低剂量水平的毒性而治疗少于18名个体。
组S和组P的剂量扩展
一旦已经表征测试的所有剂量的安全性概况和耐受性且对于给定治疗组已经定义nivolumab加上舒尼替尼和nivolumab加上帕唑帕尼的组合施用的MTD,则仅当在MTD的nivolumab剂量为5 mg/kg或更高时,对于该组在5mg/kg起始组群扩展。如果在任何组中,nivolumab的MTD被确定为< 5 mg/kg,该组将不被进一步扩展。对于与舒尼替尼或帕唑帕尼组合的nivolumab,以5 mg/kg剂量水平治疗额外20名个体/治疗组,以获得额外的安全性信息。
对于组I-1和I-3(第一组群)
先前治疗和未经治疗的个体都有资格被登记,其中每个组治疗约20个个体,但排除先前用抗PD-1、抗PD-L1或抗PD-L2、抗CD137、抗CTLA-4或其他类似药剂治疗的个体。I-1、I-3和IN-3组的剂量详述于下表4中。对于任一药物均不允许患者内剂量递增或递减。
表 4.nivolumab加上伊匹单抗的组合的剂量水平
。
每个治疗周期的持续时间为6周。每3周给予Nivolumab和伊匹单抗,持续四个剂量,然后在诱导疗法的第4个剂量之后或第113天之后(如果由于治疗延迟还没有施用诱导疗法的第4个剂量)3周开始每2周给予nivolumab。
将登记约20名个体/组,并在I-1和I-3中在个体的初始组群中进行治疗。将这些组扩展至每组治疗的总共45名个体(在每组扩展组群中治疗额外25名个体),以更好地评估安全性和效力。
对于组 I-1、I-3 (扩展组群)和组IN-3
经最小程度治疗(包括未经治疗)的个体有资格被登记,其中每个组治疗约25个个体。每个治疗周期的持续时间为6周。每3周给予Nivolumab和伊匹单抗,持续四个剂量,然后在诱导疗法的第4个剂量之后或第113天之后(如果由于治疗延迟还没有施用诱导疗法的第4个剂量)3周开始每2周给予nivolumab。
将先前治疗的个体指定至治疗组
将在晚期或转移性设置中先前已接受治疗的个体指定至如上所述的组S和P的剂量递增组群中。当两个伊匹单抗组合组(I-1和I-3)都开放时,将先前已接受至少一次全身性疗法的个体(条件是当递增阶段组S和/或P开放时,它们没有资格参加那些组),以交替方式指定至这两个组。如果仅一个组开放,则将这些个体均指定至开放组。
将未经治疗的个体指定至治疗组
如果所有四组(扩展组S和P;I-1和I-3)均开放登记,则将未经治疗的个体以1:1:1:1比率随机指定至开放组之一,直到登记完成。
将经最小程度治疗的个体指定至治疗组
对于I-1和I-3扩展以及IN-3,将经最小程度治疗(包括未经治疗)的个体以1:1:1比率随机指定至开放组之一,直到登记完成。根据NCI CTCAE v4.0分级不良事件。疾病评估从研究药物的首次剂量起每6周(±1周)进行一次(对于前四次评估),然后每12周(±1周)进行一次,直到疾病进展。根据RECIST 1.1进行反应评估。对于组S和组P的剂量递增阶段,治疗个体,直到毒性不可接受、疾病进展或撤回知情同意书。
研究评估
安全结果测量
组合的安全性/耐受性结果测量和MTD的测定是本研究的主要终点。重点安全性评估包括:(1)直至研究药物的最后一次剂量之后100天(或者对于还没有消除、稳定、回复至基线或到最后一次剂量之后100天被认为不可逆的药物相关的不良事件(AE)而言更长)发生的AE的发生率,(2)直至研究药物的最后一次剂量之后100天(或者对于还没有消除、稳定、回复至基线或到最后一次剂量之后100天被认为不可逆的药物相关的严重不良事件(SAE)而言更长)发生的SAE的发生率,和(3)通过最坏毒性分级(包括血液学、血清化学和尿分析测试)的实验室测试的频率(其在组S和组P的筛选、基线、周期1-4的第1、8、22和29天、周期5的第1和22天时评估;在组I-1和组I-3的筛选、基线、周期1-2的第1、8、22和29天、周期3+的第1、15和29天时评估;在所有组治疗结束后和最后一次剂量之后100天随访时评估)。
功效结果测量
抗肿瘤活性是本研究的次要终点,并且通过客观反应率(ORR)和反应持续时间(DOR)基于RECIST 1.1进行测量。肿瘤反应基于筛选时、从研究药物的首次剂量起每6周(±1周)(对于前4次评估)、之后每12周(±1周)直到疾病进展的肿瘤评估。
实施例5
S和P组中表现出的抗肿瘤活性
组S N2和S N5中各自治疗七名个体,其接受与S (50 mg, 使用4周,停用2周)组合的nivolumab (分别为2或5 mg/kg),直到疾病进展或毒性不可接受。没有观察到DLT,且未达到MTD。因此,基于耐受性,将S N5组扩展19名未经治疗的患者(对于S组,总共n=33)。组P在N2登记20名个体,其接受与P (每天800 mg)组合的2 mg/kg nivolumab。观察到四例DLT(ALT/AST升高[n=3],疲劳[n=1]),导致终止该组。图11中举例说明进行的剂量递增。
S N2组中的七名个体、S N5组中的26名个体和P N2组中的20名个体可通过2014年3月数据库锁(database lock)评价反应。在所有组中都观察到临床活性(OR:证实的完全[CR]或部分[PR]反应),如表5中所示。用舒尼替尼与2 mg/kg nivolumab治疗的可评价个体的数目较小(7名患者),但这些中的1名(14%)显示完全反应,5名(71%)显示部分反应,而1名(14%)具有稳定的疾病。总体而言,在用舒尼替尼与两种浓度的nivolumab (2 mg/kg和5 mg/kg)治疗的33名可评价个体中,18 名(55%)显示证实的客观反应(OR)。在用帕唑帕尼与2 mg/kg nivolumab治疗的20名可评价个体中,9 名(45%)显示证实的OR。在反应个体中的41% (组S)和56% (组P)中,反应于首次评价(6周)前发生。通过Clopper和Pearson的方法计算ORR和对应的双侧精确95% CI。
表 5.用nivolumab与舒尼替尼或帕唑帕尼治疗的RCC患者中的肿瘤反应
BOR:最佳总体反应;CR:完全反应;PR:部分反应;SD:疾病稳定;PD:疾病进展;UD:不能确定;CI:置信区间;(A):仅CR;(B):CR + 未证实的反应;未证实的反应:在最后一次肿瘤评估时表明的正在进行的个体具有肿瘤反应;DOR:反应持续时间。
组S中的反应持续时间为18.1-118++周,而组P中的反应持续时间为12.1至117.1++周。组S中的稳定疾病率为30% (n=10),而组P中的稳定疾病率为35% (n=7)。使用Kaplan-Meier方法测定中值反应持续时间和相应双侧 95% CI。
通过蜘蛛图(图12A-C)和瀑布图(图12D)举例说明对S和P组中的可评估个体的肿瘤负荷的影响。
表5中显示的证实ORR(范围为PAZ + NIV2组的45%至SUN + NIV2组的86%)显著高于用nivolumab或舒尼替尼或帕唑帕尼单一疗法看到的ORR。例如,WO 2013/173223公开了在1期研究中单独的nivolumab (1或10 mg/kg)用于治疗RCC的约28-31%的ORR。在更新的分析中,Motzer等人(2014)报道了在2期试验中用分别以0.3、2和10.mg/kg施用于患者的nivolumab治疗转移性RCC的20、22和20%的ORR。Choueiri等人(2014)报道了,以0.3、2和10 mg/kg施用的nivolumab在用1-3种先前抗血管生成疗法预治疗的转移性RCC患者中分别产生9、23和22%的ORR,而10 mg/kg nivolumab在未经治疗的患者中产生13%的ORR。对于TKI,已用单独的舒尼替尼观察到约27.5%的历史ORR(参见,例如,Motzer等人, 2007; 2013),并且用单独的帕唑帕尼观察到约30%的历史ORR(参见,例如,Sternberg等人, 2010; Motzer等人, 2013)。
在第一次评估(6周)时,S组中的17名个体中的7名(41.2%)是反应者,而P组中的9名个体中的5名(55.6%)是反应者。正在进行的反应者包括S组中的17名个体中的10名(58.8%)和P组中的9名个体中的3名(33.3%)。
S和P组中的无进展存活
无进展存活(PFS)的比例相对于时间的图显示于图13中。组S的24周时的PFS率为79%,而组P的24周时的PFS率为55%。S组(S + N; n=33)的中值PFS为55.3周(95% CI 47.9-72.4周),而P组(P + N; n=20)的中值PFS为31.4周(95% CI 12.1-48.1周)。
通过基线PD-L1表达的客观反应率
肿瘤组织收集是回顾性的(S组有33个样品可用,而P组有20个样品可用)。通过 Dako免疫组织化学测定使用兔抗人PD-L1 mAb, 28-8 (WO 2013/173223)测量表面PD-L1表达。阳性PD-L1信号的截止值为1%或5%的肿瘤膜染色。在根据PD-L1表达状态而分类的两组患者中观察到的ORR显示于表6中。
表 6.用nivolumab与舒尼替尼或帕唑帕尼的组合治疗的RCC患者中的客观反应率(ORR)和PD-L1肿瘤表达状态
aPD-L1可评价患者的ORR;ORR包括RECIST确定的完全或部分反应者。
这些数据显示,RCC患者对nivolumab与舒尼替尼或帕唑帕尼的组合的反应与PD-L1的肿瘤表达不相关。
S和P组中的不良事件
在组S的27/33名患者(82%)和组P的14/20名患者(70%)中观察到3-4级相关的不良事件(AE)。大多数毒性与TKI的已知概况一致。最通常相关的3-4级AE包括组S中的升高的ALT和高血压(各自为18%)、低钠血症和减少的淋巴细胞计数(各自为15%),以及组P中的升高的ALT和AST以及腹泻(各自为20%)和疲劳(15%)。1名患者(组S、N5)中发生3级肺炎。在组S的11/33名患者(33%; 3 N2, 8 N5)和组P的4/20名患者(20%)中,3-4级相关的AE导致疗法中断。没有观察到5级治疗相关的AE。肝毒性是使用治疗算法可管控的。
总而言之,临床数据表明,nivolumab与舒尼替尼的组合在患有mRCC的患者中具有令人鼓舞的抗肿瘤活性和可管控的安全性概况。组P由于DLT而关闭。
实施例6
伊匹单抗组中表现出的抗肿瘤活性
将mRCC患者(有利或中间的MSKCC评分;Karnofsky表现状态≥ 80%;未治疗或除了先前免疫-肿瘤治疗以外的任何数目的先前疗法)随机化至不同的治疗组。用1 mg/kg伊匹单抗与3 mg/kg nivolumab (I-1组)治疗的四十七名个体、用3 mg/kg伊匹单抗与1 mg/kg nivolumab (I-3组)治疗的47名个体和用3 mg/kg伊匹单抗与3 mg/kg nivolumab (IN-3)治疗的六名个体可针对反应进行评估。大多数患者(n=54; 54%)具有先前的全身性疗法(I-1:22; I-3:26;IN-3:3)。在I-1和I-3组中观察到OR,如表7中所示。通过Clopper和Pearson的方法计算ORR和对应的双侧精确95% CI。总而言之,I-1组中的16名(34%)患者显示证实的OR,而I-3组中的17名(36%)患者显示证实的OR。
这些ORR显著高于用单独的nivolumab(WO 2013/173223; Motzer等人, 2014; Choueiri等人, 2014; 参见实施例5)或单独的伊匹单抗看到的ORR。在2期试验中用伊匹单抗单一疗法治疗转移性RCC患者导致组群A (3 mg/kg负荷剂量,随后1 mg/kg,每3周一次)中的5%的ORR和组群B(对于所有剂量,3 mg/kg,每3周一次)中的12.5%的ORR,但这2组的特征存在差异,因而无法直接比较功效(Yang等人, 2007)。
中值反应持续时间(DOR)在I-3组中为53.9周,而在I-1组中没有达到。在19名(40%) (I-1)和18名(38%) (I-3)患者中观察到稳定疾病(SD)作为最佳总体反应。
通过蜘蛛图(图14A-C)和瀑布图(图14D)举例说明对可评价个体的肿瘤负荷的影响。S、P和I组中治疗的个体的瀑布图的比较显示于图15中。
表 7.用nivolumab与舒尼替尼或帕唑帕尼治疗的RCC患者中的肿瘤反应
BOR: 最佳总体反应;CR:完全反应;PR:部分反应;SD:疾病稳定;PD:疾病进展;UD:不能确定;CI:置信区间;(A):仅CR;(B):CR + 未证实的反应;未证实的反应:在最后一次肿瘤评估时表明的正在进行的个体具有肿瘤反应;PFS:无进展存活,DOR:反应持续时间;NR:未达到;a由于正在进行的反应的高百分比,中值DOR可能是误导的。反应持续时间被定义为第一次反应的日期和疾病进展或死亡的日期之间的时间(以先发生者为准)。
I-1和I-3组中的无进展存活
PFS的比例相对于时间的图显示于图16中。中值PFS在I-1组中为30.3周,而在I-3组中为36.0周。通过Kaplan-Meier方法估计PFS率,其具有基于Greenwood公式推导的对应的95% CI。
通过基线PD-L1表达的客观反应率
回顾性收集肿瘤组织样品,并且通过 Dako免疫组织化学测定使用mAb, 28-8 (WO 2013/173223)测量表面PD-L1表达。阳性PD-L1信号的截止值为1%或5%的肿瘤膜染色。在根据PD-L1表达状态而分类的不同患者中观察到的ORR显示于表8中。
表 8.用nivolumab与伊匹单抗的组合治疗的RCC患者中的客观反应率(ORR)和PD-L1肿瘤表达状态
aPD-L1可评价患者的ORR;ORR包括RECIST确定的完全或部分反应者。
这些数据显示,RCC患者对nivolumab与伊匹单抗的组合的反应与PD-L1的肿瘤表达不相关。
I-1和I-3组中的不良事件
在I1、I3和IN-3组的16/47(34%)、30/47(64%)和5/6(83%)中分别观察到3-4级相关的不良事件(AE)。I1和I3组中的五名(11%)和10名(21%)分别由于相关AE而停止治疗。IN-3组中没有患者由于不良事件而停止,然而所有六名患者均需要类固醇用于治疗免疫介导的不良事件,并且在IN3组中没有登记额外患者。最常见的3-4级相关的不良事件包括升高的脂肪酶(20%, n=20)、升高的ALT (11%, n=11)、腹泻(9%, n=9)和升高的AST (6%, n=6)。没有看到3-4级肺炎。
总而言之,nivolumab与伊匹单抗显示可接受的安全性、在确立的治疗指导内可管控的3-4级事件,和mRCC中的显著抗肿瘤活性的令人鼓舞的证据,其中大多数反应正在进行。ORR表明在RCC中比先前用nivolumab或伊匹单抗单一疗法报道的更大的活性(Topalian等人, 2012a; Motzer等人, 2014; Choueiri等人, 2014; Yang et al., 2007)。I-1组中还没有达到中值反应持续时间。一部分患者在停止治疗后继续反应,表明对该方案的免疫记忆组分。初始和扩展组群与IN-3组一起继续进行评估。用nivolumab + 伊匹单抗组合报道的令人鼓舞的抗肿瘤活性是所计划的第一线mRCC中的III期组合试验的基础。
参考文献
Amin等人. (2014) Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC). J Clin Oncol 32:5s, (增刊;摘要5010, Clinical Science Symposium).
Blank等人. (2005) Cancer Immunol. Immunother. 54:307-314.
Brahmer等人. (2010) J Clin Oncol 28:3167-75.
Brahmer等人. (2012) N Engl J Med 366:2455-65.
Carter等人. (2002) Eur J Immunol 32:634-43.
Choueiri等人. (2014) Immunomodulatory activity of nivolumab in previously treated and untreated metastatic renal cell carcinoma (mRCC): Biomarker-based results from a randomized clinical trial. J Clin Oncol 32:5s, (增刊;摘要5012, Clinical Science Symposium).
Dong等人. (2002) Nat. Med. 8:787-9.
Dong等人. (2003) J. Mol. Med. 81:281-7.
Drake等人. (2013) BJU Int 112(增刊3):1-17.
Elhilali等人. (2000) BJU Int 86:613-8.
Escudier等人. (2007) Clin Cancer Res 13(6):1801-1809.
Escudier等人. (2010) J Clin Oncol 28(13):2144-50.
Eskens等人. (2011) Clin Cancer Res 17(22):7156-63.
Flies等人. (2011) Yale J Biol Med 84:409-21.
Freeman等人. (2000) J Exp Med 192:1027-34.
Fyfe等人. (1995) J Clin Oncol 13(3):688-96.
Gollob等人. (2007) J Clin Oncol 25(22): 3288-95.
Grunwald等人. (2011) Acta Oncol 50(1):121-126.
Gupta等人. (2008) Cancer Treat Rev 34(3):193-205.
Hamid和Carvajal (2013) Expert Opin Biol Ther 13(6):847-61.
Hamid等人. (2013) N Engl J Med 369:134-144.
Hammers等人. (2014) Phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC). J Clin Oncol 32:5s, (增刊;摘要4504, Oral Abstract Session).
Heng等人. (2009) J Clin Oncol. 27:5794-9.
Herbst等人. (2013) J Clin Oncol 31(增刊;摘要3000).
Hipp等人. (2008) Blood 111:5610-20.
Hodi等人. (2010) N Engl J Med 363:711-23.
Houben等人. (2009) Mol Cancer Ther 28:433-40.
Hudes等人. (2007) N Engl J Med 356(22):2271-81.
Inman等人. (2013) Eur Urol 63:881-9.
Konishi等人. (2004) Clin. Cancer Res. 10:5094-100.
Johnson等人. (2013) Cancer Immunol Res 1:373-77.
Khleif (2013) : Proceedings from the European Cancer Congress 2013; September 27-October 1, 2013; Amsterdam, The Netherlands. Abstract 802.
Ko等人. (2009) Clin Cancer Res 15:2148-57.
Ko等人. (2010) Cancer Res 70:3526-36.
Konishi等人. (2004) Clin. Cancer Res. 10:5094-100.
Latchman等人. (2001) Nat Immunol 2:261-8.
McDermott和Atkins (2013) Cancer Med 2(5):662-73.
Molhoek等人. (2009) Cancer Immunol Immunother 58:867-76.
Motzer等人. (2007) N Engl J Med 356:115-24.
Motzer等人. (2008) Lancet 372:449-56.
Motzer等人. (2009) Clin Genitourin Cancer 7(1):28-33.
Motzer等人. (2013) N Engl J Med 369:722-31.
Motzer等人. (2014) Nivolumab for metastatic renal cell carcinoma (mRCC): Results of a randomized, dose-ranging phase II trial. J Clin Oncol 32:5s, (增刊;摘要5009, Clinical Science Symposium).
Mulders (2009) BJU Int 104:1585-89.
NCCN GUIDELINES® (2014), 可得自: http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site,2014年5月30日最后登录。
Nosov等人. (2012) J Clin Oncol 30(14):1678-85.
Ozao-Choy等人. (2009) Cancer Res 69:2514-22.
Pardoll (2012) Nat Rev Cancer 12:252-64.
PCT公开号WO 2012/145493,由Amplimmune, Inc.于2012年10月26日公开。
PCT公开号WO 2013/173223,由Bristol-Myers Squibb Co.于2013年11月21日公开。
Procopio等人. (2011) Br J Cancer 104(8):1256-1261.
Ribas (2010) Semin Oncol 37(5):450-4.
Rini等人. (2010) J Clin Oncol 28(13): 2137-43.
Rini等人. (2011) Cancer 117:758-67.
Rosenblatt和McDermott (2011) Hematol Oncol Clin North Am 25:793-812.
Ryan等人. (2007) J Clin Oncol 25(22):3296-301.
Siegel等人. (2014) CA Cancer J Clin 64(1):9-29.
Sjoblom等人. (2006) Science 314:268-74.
Sonpavde等人. (2008) Expert Opin Investig Drugs 17(5):741-8.
Sternberg等人. (2010) J Clin Oncol 28(6):1061-8.
Topalian等人. (2012a) N Engl J Med 366:2443-54.
Topalian等人. (2012b) Curr Opin Immunol 24:207-12.
Topalian等人. (2014) J Clin Oncol 32(10):1020-30.
USAN Council Statement (2013) Pembrolizumab: Statement on a nonproprietary name adopted by the USAN Council (ZZ-165),2013年11月27日。
1999年11月2日授权给Linsley等人的美国专利号5,977,318。
2000年4月18日授权给Allison等人的美国专利号6,051,227。
2004年1月27日授权给Hanson等人的美国专利号6,682,736。
2004年10月26日授权给Wood等人的美国专利号6,808,710。
2006年1月10日授权给Korman等人的美国专利号6,984,720。
2006年4月25日授权给Carreno等人的美国专利号7,034,121。
2009年10月20日授权给Korman等人的美国专利号7,605,238。
2009年2月10日授权给Collins等人的美国专利号7,488,802。
2011年5月17日授权给Korman等人的美国专利号7,943,743。
2011年8月30日授权给Korman等人的美国专利号8,008,449。
2012年5月1日授权给Finnefrock等人的美国专利号8,168,757。.
2012年7月10日授权给Irving等人的美国专利号8,217,149。
2013年1月15日授权给Carven等人的美国专利号8,354,509。
Yang等人. (2007) J Immunother 30(8):825-30.
Wang等人. (2014) In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates, Cancer Imm Res, 出版中。
Wolchok等人. (2013) N Engl J Med 369(2):122-33。
- 还没有人留言评论。精彩留言会获得点赞!