细胞重编程诱导用组合物的制作方法
本专利申请要求2014年7月1日提交的韩国专利申请第10-2014-0081889号的优先权,所述专利申请的公开内容通过参考并入本文。本发明涉及一种细胞重编程诱导用组合物。
背景技术:
:随着老龄化社会的到来,很多人由于退行性疾病已被遭受了痛苦,且退行性疾病患者数有持续增加的趋势。包括帕金森氏病、卢格里格病的无法治愈的多种退行性疾病的最理想的治疗法就是将源自患者细胞的分化全能性干细胞或从其分化的功能细胞移植到患处。最近,世界各国为了基于细胞重编程(reprogranmming)或去分化技术主导细胞治疗剂和新药研发而正在进行激烈的技术竞争,以便实现无法治愈的退行性疾病的根本治疗。去分化技术是比较简单的方法,其几乎没有对患者带来的身体损伤和不便。其确保自体细胞,由此能制备如人类胚胎干细胞等特性的诱导多能干细胞,从而实现了从患者体细胞确保定制自多能干细胞系的战略划时代的进化。例如,为了诱导未分化干细胞而可以利用组合有特定基因的病毒,通过将所述病毒传达到细胞中来以基因被插入到细胞基因组的形式表达相关蛋白质的方法被许多研究者提出。另外,虽然通过这种方法制备的重编程的细胞被发现具有与胚胎干细胞几乎同等的细胞特性和分化潜能,但由于以病毒载体为媒介传达基因而存在导致细胞的基因组上包括大量的病毒来源的遗传物质的问题。并且,在体内实验出现由诱导多能干细胞形成癌的副作用,这就意味着诱导多能干细胞的实际临床应用仍然是很危险的。如上所述的去分化干细胞存在在重编程过程中利用癌基因(oncogene)的问题,因此,最近对摆脱现有重编程方式而将体细胞直接分化成患者所需的细胞系的直接重编程(directreprogramming)的研究活跃地进行。另外,由于在现有化合物中如逆转素、Aurora激酶抑制剂等可用作细胞重编程用途的化合物具有细胞毒性,因而不适合用作细胞重编程组合物。为了克服上述问题,本发明人合成出无细胞毒性并能使分化的细胞直接重编程成具有不同特性的细胞的化合物,且确定通过利用该化合物可以实现安全有效的细胞重编程。在本说明书全文中,参照了多篇论文及专利文献,并表示了其引用。所引用的论文及专利文献的公开内容全部插入于本说明书作为参照,从而更加明确说明本发明所属
技术领域:
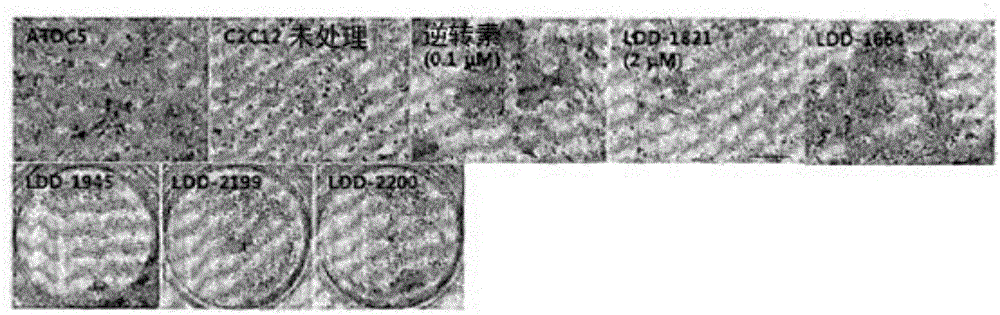
的水平及本发明的内容。技术实现要素:发明要解决的问题本发明人努力研究开发了与现有吲唑衍生物化合物相比在呈现改善的生物特征谱(biologicalprofile)的同时能够进行细胞重编程的化合物。其结果,本发明人通过分子设计合成出具有细胞重编程诱导能力的新颖的吲唑衍生物化合物,得知这些化合物与如激酶抑制剂等现有吲唑衍生物不同地,不呈现细胞毒性(cytotoxicity)并发挥优异的细胞重编程能力,从而完成了本发明。因此,本发明的目的在于提供一种细胞重编程诱导用组合物。本发明的另一个目的在于提供一种新颖的吲唑衍生物。本发明的其它目的及优点将在下面的详细说明、权利要求书及附图得到进一步阐释。用于解决问题的方案根据本发明的一实施方式,本发明提供一种细胞重编程诱导用组合物,其包括由下述化学式A表示的化合物:化学式A在化学式A中,R1为氢、硝基、硝基氧基或Y-羰基,所述Y为氢、羟基、卤、C1-30烷氧基、作为杂原子包含氮、氧或硫的C2-30杂环烷基、作为杂原子包含氮、氧或硫的C2-20杂环烯基、C6-30芳基、C1-30烷基或C2-30烯基,R2为氢、羟基、卤、C1-30芳基、C1-30烷氧基、C1-30烷基或C2-30烯基,R3为氢、羟基、卤、C1-30芳基、C1-30烷氧基、C1-30烷基或C2-30烯基,X为胺基、酰氨基、磺酰氨基、羰基、肟或酰亚胺酰氨。根据本发明的另一实施方式,本发明提供一种从分化的细胞制备重编程的细胞的方法,其特征在于,包括如下步骤:(a)使本发明的细胞重编程组合物与分化的细胞接触;及(b)培养所述步骤(a)的细胞。本发明人努力研究开发了与现有吲唑衍生物化合物相比可以呈现改善的生物特征谱并进行细胞重编程的化合物,其结果,通过分子设计合成具有细胞重编程诱导能力的新颖吲唑衍生物化合物,得知这些化合物与如激酶抑制剂等现有吲唑衍生物不同地,不呈现细胞毒性并发挥优异的细胞重编程能力。由化学式A表示的本发明的化合物为将分化的细胞重编程成新特性的细胞的化合物,其以吲唑为母核,经化学合成得到。现有吲唑衍生物化合物作为细胞重编程的用途完全未被知道,在本发明的化合物的情况下,与现有吲唑衍生物化合物相比,没有细胞毒性或细胞毒性显著低,且细胞重编程能力非常优异。现有直接重编程方法为利用病毒导入基因的方法,其具有如下缺点:难以直接适用于患者,诱导过程的效率性较低,在免疫方面有问题。在本发明的利用低分子物质的直接重编程的情况下,替代通过病毒的基因导入方法,由于仅通过低分子物质的处理诱导重编程,因而稳定性、经济性及效率性非常优越。本发明的组合物非常有效于从分化的细胞诱导重编程的细胞。根据本发明,本发明的组合物可以从源自哺乳动物的分化细胞诱导重编程的成骨(osteogeniclineage)细胞或成脂(adipogeniclineage)细胞等各种类型的细胞系。根据本发明的一实施例,本发明的组合物诱导成肌细胞(myoblast)分化成成骨细胞(osteoblast)或脂肪细胞(adipocyte)。分化的细胞没有特别限制,例如,包括体细胞或体细胞干细胞。体细胞是指作为构成成体的细胞,具有有限的分化能力和自我生产能力的细胞,所述体细胞可以是构成人的皮肤、毛发、脂肪或骨的体细胞。所述分化的细胞可以是源自如人、猴、猪、马、牛、羊、狗、猫、鼠及兔等不同哺乳动物的细胞,优选源自人的细胞。如本文所用,术语“重编程(Reprogramming)”是指使以如无分化能力的细胞或有部分分化能力的细胞等具有不同特征的状态存在的分化的细胞,最终恢复或转化成新型细胞或具有新型分化能力的细胞的过程。根据本发明,分化的细胞通过与本发明的组合物接触来可以被重编程为0%至100%的呈现不同特性的细胞。本说明书中,为了定义化学式A的吲唑衍生物而使用的术语“硝基”是指卤族元素,例如,包括氟、氯、溴和碘。术语“硝基氧基”是指-O-N=O官能团。术语“羰基”是指-(C=O)-官能团。术语“羟基”是指-OH官能团。术语“卤”是指卤族元素,例如,包括氟、氯、溴和碘。术语“烷基”是指包括直链或支链的、取代或未取代的饱和烃基,例如,包括甲基、乙基、丙基、异丁基、戊基、己基、庚基、半基、壬基、癸基、十一烧基、十二烧基、十五烧基及十七烧基。C1-30烷基是指包括具有1至30个碳原子数的烷基单元的烷基,当C1-30烷基被取代时,其不包括取代产物的碳原子数。根据本发明,位于化学式A的Y位的C1-30烷基为C1-15烷基,而位于化学式A的R2位的C1-30烷基优选为C1-15烷基。术语“烯基”是指具有预定碳原子数的直链或支链的、取代或未取代的不饱和烃基,例如,包括次乙基、乙烯基、丙烯基、烯丙基、异丙烯基、丁烯基、异丁烯基、叔丁烯基、正戊烯基及正己烯基。C2-30烯基是指包括具有2至30个碳原子数的烯基单元的烯基,当C2-30烯基被取代时,其不包括取代产物的碳原子数。根据本发明,位于化学式A的Y位的C2-30烯基为C2-15烯基,而位于化学式A的R2位的C2-30烯基优选为C2-15烯基。术语“烷氧基”是指与1个氧原子结合的烷基或芳基。例如,其包括甲氧基、乙氧基、丙氧基、芳基氧基等。C1-30烷氧基是指包括具有1至30个碳原子数的烷氧基单元的烷氧基,当C1-30烷氧基被取代时,其不包括取代产物的碳原子数。术语“烷氧基烷基”是指被烷氧基取代的烷基。C2-5烷氧基烷基是指包括具有2至5个碳原子数的烷氧基烷基单元的烷氧基烷基,当C2-5烷氧基烷基被取代时,其不包括取代产物的碳原子数。术语“烷氧基羰基”是指被烷氧基取代的羰基。C2-5烷氧基羰基是指包括具有2至5个碳原子数的烷氧基羰基单元的烷氧基羰基,当C2-5烷氧基羰基被取代时,其不包括取代产物的碳原子数。术语“作为杂原子包含氮、氧或硫的杂环烷基”是指包括碳、氢及至少1个杂原子(氮、氧或硫)的非芳族环状碳化氢。所述杂原子优选为氮或氧,最优选为氮。根据本发明,杂原子的个数为1-4、1-3或1-2个。C2-30杂环烷基是指形成环结构的碳的个数为2-30个的杂环烷基。另外,所述杂环烷基是被全部或部分取代或未被取代的碳环,所述杂环烷基可以被羟基、卤、取代或未取代的、直链或支链的C1-5烷基、直链或支链的C1-5烷氧基、直链或支链的C1-5烯基、C2-8烷氧基烷基、C2-8烷氧基羰基、作为杂原子包含氮的C2-8杂环烯基烷基、或其组合取代。术语“作为杂原子包含氮、氧或硫的杂环烯基”是指包括碳、氢、至少1个杂原子(氮、氧或硫)及至少1个双键的非芳族环状碳化氢。所述杂原子优选为氮或氧,最优选为氮。根据本发明,杂原子的个数为1-4、1-3或1-2个。C2-30杂环烯基是指形成环结构的碳的个数为2-30个的杂环烯基。术语“杂环烯基烷基”是指被杂环烯基取代的烷基。C2-8杂环烯基是指包括具有2至5个碳原子数的杂环烯基烷基单元的杂环烯基烷基,当C2-5杂环烯基烷基被取代时,其不包括取代产物的碳原子数。术语“芳基”是指全部或部分不饱和的取代或未取代的单环或多环碳环。C6-30芳基是指具有6至30个碳原子数的碳环原子的芳基,当C6-30芳基被取代时,其不包括取代产物的碳原子数。优选地,芳基为单芳基或非芳基。单芳基优选具有5-6个碳原子数,而非芳基优选具有9-10个碳原子数。根据本发明,所述芳基为取代或未取代的苯基。当单芳基被取代时,例如,当苯基被取代时,可以在不同位置被不同取代基取代,例如,可以被卤、羟基、硝基、氰基、取代或未取代的直链或支链C1-5烷基、直链或支链C1-5烷氧基、直链或支链C1-5烯基或其组合取代。特别优选的芳基部位包括苯基、苄基、萘基、吡咯基、吡咯烷基、吡啶基、嘧啶基、嘌呤基、喹啉基、异喹啉基、呋喃基、苯硫基、咪唑基、恶唑基、噻唑基、吡唑基及噻吩基,但不限于此。术语“胺(amine)”是指包括具有未共用电子对(lonepair)的碱性氮原子的官能团。C1-10胺是指包括具有-NH2或1至10个碳原子数的胺单元的胺,当C1-10胺被取代时,其不包括取代产物的碳原子数。根据本发明,位于化学式A的X位的C1-10胺为C1-5胺或-NH2。术语“酰胺”是指由结合到氮原子的酰基构成的官能团。C1-10酰胺是指包括具有1至10个碳原子数的酰胺单元的酰胺,当C1-10酰胺被取代时,其不包括取代产物的碳原子数。根据本发明,所述酰胺为R-CO-NH-R'、磺胺或磷酰胺。术语“肟(oxime)”是指包括RR'C=N-OH的亚胺(imines)的官能团。术语“酰亚胺酰胺”是指包括N-CR=N'H2的官能团。根据本发明,在化学式A中,所述Y为氢、羟基、卤、C1-15烷氧基、作为杂原子包含氮、氧或硫的C2-15杂环烷基、作为杂原子包含氮、氧或硫的C2-15杂环烯基、C6-15芳基、C1-15烷基或C2-15烯基。根据本发明,在所述Y中,杂环烷基可以被羟基、卤、C1-5烷基、C2-5烯基、C1-5烷氧基、C2-8烷氧基烷基、C2-8烷氧基羰基、作为杂原子包含氮的C2-8杂环烯基烷基、或其组合取代。根据本发明的一实施例,所述Y的杂环烯基烷基为(1H-咪唑-1-ly)烷基。根据本发明,所述R2为氢、卤、C1-15芳基、C1-15烷氧基或C1-15烷基或C2-15烯基,并且,在所述R2中,芳基可以被羟基、卤、C1-5烷氧基、C1-5烯基、C2-5烯基或其组合取代。另外,在化学式A中,当所述X为酰亚胺酰氨时,所述R2可以与所述酰亚胺酰氨的C或N'结合。本发明的化合物可以具有1个或多个手性中心及/或几何异构中心,且本发明包括由化学式A表示的所有立体异构体,即,光学异构体、非对映异构体及几何异构体。根据本发明,本发明的细胞重编程诱导用组合物为由选自由下述化学式1至化学式45组成的组中的化学式表示的化合物:化学式1化学式2化学式3化学式4化学式5化学式6化学式7化学式8化学式9化学式10化学式11化学式12化学式13化学式14化学式15化学式16化学式17化学式18化学式19化学式20化学式21化学式22化学式23化学式24化学式25化学式26化学式27化学式28化学式29化学式30化学式31化学式32化学式33化学式34化学式35化学式36化学式37化学式38化学式39化学式40化学式41化学式42化学式43化学式44化学式45根据本发明,本发明的细胞重编程诱导用组合物为由选自由下述化学式1、化学式25、化学式30、化学式31、化学式33、化学式34、化学式35及化学式45组成的组中的化学式表示的化合物:化学式1化学式25化学式30化学式31化学式33化学式34化学式35化学式45根据本发明的另一实时方式,本发明提供一种化合物,作为吲唑衍生物,由下述化学式A表示:化学式A在化学式A中,R1为氢、硝基、硝基氧基或Y-羰基,所述Y为氢、羟基、卤、C1-30烷氧基、作为杂原子包含氮、氧或硫的C2-30杂环烷基、作为杂原子包含氮、氧或硫的C2-20杂环烯基、C6-30芳基、C1-30烷基或C2-30烯基,R2为氢、羟基、卤、C1-30芳基、C1-30烷氧基、C1-30烷基或C2-30烯基,R3为氢、羟基、卤、C1-30芳基、C1-30烷氧基、C1-30烷基或C2-30烯基,X为胺基、酰氨基、磺酰氨基、羰基、肟或酰亚胺酰氨,但,在所述R1为硝基且所述X为酰氨基时,R2不为苯基,在所述R1和R3为氢,所述X为N-羟基-酰亚胺酰氨,所述R2为苯基时,R2与酰亚胺酰氨的碳原子不结合(或R2与酰亚胺酰氨的N'原子结合)。根据本发明,所述Y为氢、羟基、卤、C1-15烷氧基、作为杂原子包含氮、氧或硫的C2-15杂环烷基、作为杂原子包含氮、氧或硫的C2-15杂环烯基、C6-15芳基、C1-15烷基或C2-15烯基。根据本发明,在化学式A的所述Y中,杂环烷基可以被羟基、卤、C1-5烷基、C2-5烯基、C1-5烷氧基、C2-8烷氧基烷基、C2-8烷氧基羰基、作为杂原子包含氮的C2-8杂环烯基烷基、或其组合取代。根据本发明的一实施例,杂环烯基烷基为(1H-咪唑-l-ly)烷基。根据本发明,在化学式A中,所述R2为氢、卤、C1-15芳基、C1-15烷氧基或C1-15烷基或C2-15烯基,而且,在所述R2中,芳基可以被羟基、卤、C1-5烷氧基、C1-5烷基、C2-5烯基或其组合取代。根据本发明的一实施例,在化学式A中,杂原子为氮。本发明的吲唑衍生物化合物由选自由下述化学式1、化学式2及化学式4至化学式45组成的组中的化学式表示:化学式1化学式2化学式4化学式5化学式6化学式7化学式8化学式9化学式10化学式11化学式12化学式13化学式14化学式15化学式16化学式17化学式18化学式19化学式20化学式21化学式22化学式23化学式24化学式25化学式26化学式27化学式28化学式29化学式30化学式31化学式32化学式33化学式34化学式35化学式36化学式37化学式38化学式39化学式40化学式41化学式42化学式43化学式44化学式45根据本发明,本发明的吲唑衍生物化合物由选自由下述化学式1、化学式25、化学式30、化学式31、化学式33、化学式34、化学式35及化学式45组成的组中的化学式表示:化学式1化学式25化学式30化学式31化学式33化学式34化学式35化学式45根据本发明的再一实施方式,本发明提供一种通过使包括由下述化学式A表示的化合物的组合物与细胞接触来诱导细胞重编程的方法:化学式A在化学式A中,R1为氢、硝基、硝基氧基或Y-羰基,所述Y为氢、羟基、卤、C1-30烷氧基、作为杂原子包含氮、氧或硫的C2-30杂环烷基、作为杂原子包含氮、氧或硫的C2-20杂环烯基、C6-30芳基、C1-30烷基或C2-30烯基,R2为氢、羟基、卤、C1-30芳基、C1-30烷氧基、C1-30烷基或C2-30烯基,R3为氢、羟基、卤、C1-30芳基、C1-30烷氧基、C1-30烷基或C2-30烯基,X为胺基、酰氨基、磺酰氨基、羰基、肟或酰亚胺酰氨。由于本发明的细胞重编程(re-programming)诱导方法利用上述本发明的细胞重编程诱导用组合物,因而为了避免反复记载使说明书过于复杂,省略两者之间共同的内容的记载。发明的效果本发明特征及优点的摘要如下:(a)本发明涉及细胞重编程诱导组合物。(b)本发明的吲唑衍生物化合物呈现改善的生物特征谱并能够进行有效的细胞重编程。(c)本发明的吲唑衍生物化合物与如逆转素或BIO等现有低分子细胞重编程诱导化合物不同地,不呈现细胞毒性,因此,当临床应用时,能够期待在细胞治疗剂或细胞重编程诱导剂市场高速增长。(d)另外,现有吲唑衍生物化合物作为细胞重编程的用途完全未被知道,且由于非特异性细胞毒性而无法进行高浓度处理。(e)在本发明的化合物的情况下,与现有吲唑衍生物化合物相比,没有细胞毒性或细胞毒性显著低,并且细胞重编程能力非常优异,具有新颖的作用机理。附图说明图1及图2示出在使C2C12小鼠成肌细胞(myoblast)和本发明的化合物相接触后,重编程为成骨细胞的结果。在与本发明的化合物即LDD-1821、LDD-1664、LDD-1945、LDD-2199或LDD-2200接触后,染成茜素红色,进行观察(ATDC5:软骨祖细胞)。图3示出在使C2C12小鼠成肌细胞和本发明的化合物相接触后,进行茜素红试验的结果。图表示出当对各个化合物进行处理时的吸光度。图4示出C2C12小鼠成肌细胞的MTT比色试验的结果。具体实施方式下面,通过实施例更详细地说明本发明。这些实施例只用于更详细地说明本发明,根据本发明的要旨,本发明的范围并不局限于这些实施例,这对于本发明所属
技术领域:
的普通技术人员来说是显而易见的。合成例合成示意图1下面,在合成例所记载的反应过程和取代基指代所述合成示意图中的反应过程和取代基。一般合成过程步骤(a)的一般过程在正丁醇(20mL)中溶解起始材料(1.0g,6.02mmol)。然后,向溶液添加水合肼(420uL,7.22mmol)。在110℃下搅拌所述混合物2小时,放凉,然后向反应物添加亚甲基氯(MC),对所产生的沉淀物进行过滤,从而获得目标化合物。合成例1:5-硝基-1H-吲唑-3-基胺(化合物1)在正丁醇(20mL)中溶解2-氟-5-硝基苄腈(1.0g,6.02mmol)。然后,向溶液添加水合肼(420uL,7.22mmol)。在110℃下搅拌所述混合物2小时,放凉,然后向反应物添加亚甲基氯,对所产生的沉淀物进行过滤,从而获得目标化合物(收率:84%)。1HNMR(400MHz,DMSO-d6)δ8.89(d,1H),8.05(dd,1H),7.34(d,2H),5.98(s,2H).MASS=178.15合成例2:3-氨基-1H-吲唑-5-羧酸(化合物2)在正丁醇(20mL)中溶解3-氰基-4-氟苯甲酸(1.0g,6.02mmol)。然后,向溶液添加水合肼(420uL,7.22mmol)。在110℃下搅拌所述混合物2小时,放凉,对所产生的沉淀物进行过滤,用MeOH进行清洗,从而获得目标化合物(收率:55%)。1HNMR(400MHz,DMSO-d6)δ11.35(s,1H),8.45(s,1H),7.80(d,2H),7.22(d,1H),5.59(s,2H).MASS=177.16步骤(b-2)的一般过程合成示意图1,将化学结构式2的化合物(50mg,0.28mmol)溶解于吡啶(0.3mL)中。然后,向反应混合物添加R2-苯甲酰氯(16.5uL,0.28mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。通过盐水(brine)清洗结合的有机层,用无水Na2SO4进行干燥,在真空浓缩后,通过柱层析进行精制,从而获得目标化合物(化合物3至化合物6)。合成例3(LDD-1664):N-(5-硝基-1(2)H-吲唑-3-基)-苯甲酰胺(化合物3)将5-硝基-1H-吲唑-3-基胺(50mg,0.28mmol)溶解于吡啶(0.3mL)中。接着,向反应混合物添加苯甲酰氯(16.5uL,0.28mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空浓缩后,通过柱层析进行精制,从而获得目标化合物(收率:42%)。1HNMR(400MHz,DMSO-d6)δ8.95(d,1H),8.22(dd,1H),8.12(m,2H),8.12(m,2H),7.69(m,4H).MASS=282.26合成例4(LDD-1663):3-溴-N-(5-硝基-1H-吲唑-3-基)苯甲酰胺(化合物4)将5-硝基-1H-吲唑-3-基胺(0.14mmol)溶解于吡啶(0.4mL)中。接着,向反应混合物添加3-溴苯甲酰氯(19uL,0.14mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。向所述提取物添加二氯甲烷(DCM)和核酸,以便形成沉淀物,对此进行过滤,从而获得目标化合物(收率:37%)。1HNMR(400MHz,DMSO-d6)δ11.32(s,1H),8.93(d,1H),8.26(s,1H),8.19(dd,1H).MASS=361.16合成例5(LDD-2348):4-甲氧基-N-(5-硝基-1H-吲唑-3-基)苯甲酰胺(化合物5)将5-硝基-1H-吲唑-3-基胺(25mg,0.14mmol)溶解于吡啶(0.3mL)中。接着,向反应混合物添加4-甲氧基苯甲酰氯(24mg,0.14mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空浓缩。通过柱层析进行精制,从而获得目标化合物(收率:40%)。1HNMR(400MHz,DMSO-d6)δ8.93(d,1H),8.20(dd,1H),8.12(d,2H),7.67(d,1H),7.11(d,2H),3.86(s,3H).MASS=312.28合成例6(LDD-1665):3-苯甲酰氨基-IH-吲唑-5-羧酸(化合物6)将3-氨基-1H-吲唑-5-羧酸(25mg,0.14mmol)溶解于吡啶(0.5mL)中。接着,向反应混合物添加苯甲酰氯(16.5uL,0.14mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。利用二氯甲烷形成沉淀物,对此进行过滤,从而获得目标化合物(收率:31%)。1HNMR(400MHz,DMSO-d6)δ10.98(s,1H),8.11(d,2H),7.94(dd,1H),7.66(t,1H),7.58(t,3H).MASS=281.27步骤(c-3)的一般过程合成示意图1,将化学结构式3的化合物(25mg,0.14mmol)溶解于二甲基甲酰胺(DMF,1mL)中。然后,向反应混合物添加二甲氨基吡啶(DMAP,0.03mmol),哌啶(0.17mmol)及二氯化乙烯(EDC,0.28mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物。合成例7(LDD-1820):N-(5-(哌啶-1-羰基)-1H-吲唑-3-基)苯甲酰胺(化合物7)将3-苯甲酰氨基-1H-吲唑-5-羧酸(25mg,0.14mmol)溶解于二甲基甲酰胺(1mL)中。然后,向反应混合物添加二甲氨基吡啶(0.03mmol),哌啶(0.17mmol)及二氯化乙烯(0.28mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:33%)。1HNMR(400MHz,CDC13)δ11.02(s,1H),9.26(s,1H),8.15(s,1H),8.04(d,2H),7.59(m,3H),7.37(d,1H),7.21(d,1H),3.59(m,4H),1.75(m,6H).MASS=348.41步骤(d-4)的一般过程合成示意图1,将化学结构式3的化合物(25mg,0.14mmol)溶解于二甲基甲酰胺(1mL)中。然后,向反应混合物添加二甲氨基吡啶(0.03mmol),叔丁基4-氨基哌啶-1-羧酸(0.17mmol)及二氯化乙烯(0.28mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物。合成例8(LDD-1690):叔丁基4-(3-苯甲酰氨基1H-吲唑-5-羰基)哌嗪-1-羧酸盐(化合物8)将3-苯甲酰氨基-1H-吲唑-5-羧酸(25mg,0.14mmol)溶解于二甲基甲酰胺(1mL)中。然后,向反应混合物添加二甲氨基吡啶(0.03mmol),叔丁基4-氨基哌啶-1-羧酸(0.17mmol)及二氯化乙烯(0.28mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:33%)。1HNMR(400MHz,DMSO-d6)δ10.82(s,1H),8.23(s,2H),8.07(d,2H),7.85(d,1H),7.60(m,4H),3.96(m,3H),2.79(m,2H),1.74(m,2H),1.41(m,11H).MASS=449.51步骤(e-5)的一般过程合成示意图1,将化学结构式5的化合物(30mg,0.065mmol)溶解于二氯甲烷(0.7mL)中。然后,在0℃下添加三氟乙酸(TFA,0.2mL),在0℃下搅拌30分钟。在真空下进行浓缩后,通过利用以氨饱和的氯仿和甲醇的硅胶层析进行精制,从而获得目标化合物。合成例9(LDD-1691):N-(5-(哌嗪-1-羰基)-1H-吲唑-3-基)苯甲酰胺(化合物9)将叔丁基4-(3-苯甲酰氨基1H-吲唑-5-羰基)哌嗪-1-羧酸盐(30mg,0.065mmol)溶解于二氯甲烷(0.7mL)中。接着,在0℃下添加三氟乙酸(0.2mL),在0℃下搅拌30分钟。在真空下进行浓缩后,通过利用以氨饱和的氯仿和MeOH的硅胶层析进行精制,从而获得目标化合物(收率:20%)。1HNMR(400MHz,DMSO-d6)δ10.86(s,1H),8.40(d,1H),8.24(s,1H),8.07(d,2H),7.85(d,1H),7.63(m,4H).MASS=349.39合成示意图2下面,在合成例所记载的反应过程和取代基指代所述合成示意图中的反应过程和取代基。一般合成过程步骤(a)的一般过程合成示意图2,将化学结构式2的化合物(50mg,0.28mmol)溶解于吡啶(0.9mL)中。接着,向反应混合物添加R2-苯磺酰氯(36uL,0.28mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。利用二氯甲烷形成沉淀物,然后进行过滤,从而获得目标化合物。合成例10(LDD-1667):N-(5-硝基-1H-吲唑-3-基)苯磺酰胺(化合物10)将5-硝基-1H-吲唑-3-基胺(50mg,0.28mmol)溶解于吡啶(0.9mL)中。接着,向反应混合物添加苯磺酰氯(36uL,0.28mmol),搅拌1小时,然后用醋酸盐和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。利用亚甲基氯形成沉淀物,然后进行过滤,从而获得目标化合物(收率:43%)。1HNMR(400MHz,DMSO-d6)δ11.14(s,1H),8.70(d,1H),8.12(dd,1H),7.78(d,2H),7.55(m,4H).MASS=318.31合成例11(LDD-1666):3-溴-N-(5-硝基-1H-吲唑-3-基)苯磺酰胺(化合物11)将5-硝基-1H-吲唑-3-基胺(0.28mmol)溶解于吡啶(0.9mL)中。接着,向反应混合物添加3-溴苯磺酰氯(40uL,0.28mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。向所述提取物添加二氯甲烷和核酸,以便形成沉淀物,对此进行过滤,从而获得目标化合物(收率:50%)。1HNMR(400MHz,DMSO-d6)δ10.99(s,1H),8.72(d,1H),8.19(m,1H),7.99(t,1H),7.87(d,1H),7.81(d,1H),7.63(d,1H),7.54(t,1H).MASS=397.20合成例12(LDD-1668):3-(苯基磺酰氨基)-1H-吲唑-5-羧酸(化合物12)将3-氨基-1H-吲唑-5-羧酸(25mg,0.14mmol)溶解于吡啶(0.7mL)中。接着,向反应混合物添加苯磺酰氯(20uL,0.14mmol),搅拌1小时,然后用乙酸乙酯和1NHCl进行提取。利用甲醇形成沉淀物,对此进行过滤,从而获得目标化合物(收率:20%)。1HNMR(400MHz,DMSO-d6)δ10.88(s,1H),8.42(s,1H),7.89(dd,3H),7.63(m,4H).MASS=317.32步骤(b-2)的一般过程合成示意图2,将化学结构式7的化合物(25mg,0.08mmol)溶解于二甲基甲酰胺(0.8mL)中。向反应混合物添加二甲氨基吡啶(0.02mmol),叔丁基4-氨基哌啶-1-羧酸(0.09mmol)及二氯化乙烯(0.16mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物。合成例13(LDD-1692):叔丁基4-(3-(苯基磺酰氨基)-1H-吲唑-5-羰基)哌嗪-1-羧酸(化合物13)将3-(苯基磺酰氨基)-1H-吲唑-5-羧酸(25mg,0.08mmol)溶解于二甲基甲酰胺(0.8mL)中。向反应混合物添加二甲氨基吡啶(0.015mmol),叔丁基4-氨基哌啶-1-羧酸(0.09mmol)及二氯化乙烯(0.16mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:50%)。1HNMR(400MHz,DMSO-d6)δ10.77(s,1H),8.31(d,2H),7.83(m,3H),7.62(m,1H),7.57(t,2H),7.44(d,1H).MASS=485.56步骤(c-3)的一般过程合成示意图2,将化学结构式8的化合物(20mg,0.038mmol)溶解于二氯甲烷(0.8mL)中。在0℃下添加三氟乙酸(0.15mL),然后在0℃下搅拌30分钟。在真空下进行浓缩后,通过利用以氨饱和的氯仿和MeOH的硅胶层析进行精制,从而获得目标化合物。合成例14(LDD-1693):N-(5-(哌嗪-1-羰基)-1H-吲唑-3-基)苯磺酰胺(化合物14)将叔丁基4-(3-(苯基磺酰氨基)-1H-吲唑-5-羰基)哌嗪-1-羧酸(20mg,0.038mmol)溶解于二氯甲烷(0.4mL)中。接着,在0℃下添加三氟乙酸(0.15mL),然后在0℃下搅拌30分钟。在真空下进行浓缩后,通过利用以氨饱和的氯仿和甲醇的硅胶层析进行精制,从而获得目标化合物(收率:20%)。1HNMR(400MHz,DMSO-d6)δ11.92(s,1H),8.28(d,1H),8.22(s,1H),7.80(m,2H),7.68(d,1H),7.38(d,3H),7.20(d,1H),3.95(m,1H),3.16(d,3H),2.78(t,2H),1.85(m,2H),1.57(m,2H).MASS=385.44合成示意图3下面,在实施例所记载的反应过程和取代基指代所述合成示意图中的反应过程和取代基。一般合成过程步骤(a)的一般过程向起始材料(1.0g,6.2mmol)添加水(40mL),然后对此添加溶解于水中的亚硝酸钠(4.3g,62mmol)。接着,向反应混合物逐渐滴加3NHCl20分钟。在常温下搅拌8小时,对所产生的沉淀物进行过滤。在完全干燥后,获得目标化合物。合成例15:5-硝基-1H-吲唑-3-碳醛(化合物15)向5-硝基-1H-吲哚(1.0g,6.2mmol)添加水(40mL),然后对此添加溶解于水中的亚硝酸钠(4.3g,62mmol)。接着,向反应混合物逐渐滴加3NHCl(21mL)20分钟。在常温下搅拌8小时,对所产生的沉淀物进行过滤。在真空状态下完全干燥后,获得目标化合物(收率:40%)。1HNMR(400MHz,DMSO-d6)δ11.98(s,1H),9.75(s,1H),8.87(s,1H),8.56(m,2H).MASS=191.15合成例16:3-甲酰基-1H-吲唑-5-羧酸(化合物16)向1H-吲哚-5-羧酸(1.0g,6.02mmol)添加水(40mL),然后对此添加溶解于水中的亚硝酸钠(4.3g,62mmol)。向反应混合物逐渐滴加3NHCl(21mL)20分钟。在常温下搅拌8小时,对所产生的沉淀物进行过滤。在真空状态下完全干燥后,获得目标化合物(收率:84%)。1HNMR(400MHz,DMSO-d6)δ10.20(s,1H),8.74(s,1H),8.02(d,1H),7.75(d,2H).MASS=190.16步骤(b-2)的一般过程合成示意图3,将化学结构式11的化合物(500mg,2.63mmol)溶解于二甲基甲酰胺(13mL)中。接着,向反应混合物添加盐酸羟胺(550mg,7.89mmol)和三乙胺(110uL,7.89mmol),在80℃下搅拌12小时后,利用乙酸乙酯和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。不经过另外的精制过程,获得用于进行下述反应的目标化合物。合成例17:5-硝基-1H-吲唑-3-羧醛肟(化合物17)将5-硝基-1H-吲唑-3-碳醛(500mg,2.63mmol)溶解于二甲基甲酰胺(13mL)中。向反应混合物添加盐酸羟胺(550mg,7.89mmol)和三乙胺(110uL,7.89mmol),在80℃下搅拌12小时后,利用乙酸乙酯和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。不经过另外的精制过程,获得用于进行下述反应的目标化合物(收率:93%)。1HNMR(400MHz,DMSO-d6)δ11.98(s,1H),11.12(s,1H),8.87(s,1H),8.56(m,2H),7.95(s,1H).MASS=206.16合成例18:3-((肟基)甲基)-1H-吲唑-5-羧酸(化合物18)将3-甲酰基-1H-吲唑-5-羧酸(500mg,2.63mmol)溶解于二甲基甲酰胺(13mL)中。向反应混合物添加盐酸羟胺(550mg,7.89mmol)和三乙胺(110uL,7.89mmol),在80℃下搅拌12小时后,利用乙酸乙酯和1NHCl进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。不经过另外的精制过程,获得用于进行下述反应的目标化合物(收率:93%)。1HNMR(400MHz,DMSO-d6)δ11.61(s,1H),8.75(s,1H),8.38(s,1H),7.96(d,2H),7.62(d,1H).MASS=205.17步骤(c-3)的一般过程合成示意图3,将化学结构式12的化合物(500mg,2.44mmol)溶解于二甲基甲酰胺(12mL)中。接着,向反应混合物添加N-氯琥珀酰亚胺(320mg,2.44mmol),然后在40℃下搅拌混合物1小时。利用乙酸乙酯和水对所获得的残余物进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。不经过另外的精制过程,利用所获得的目标化合物进行下述反应。合成例19:N-羟基-5-硝基-1H-吲唑-3-carimidoyl氯化物(化合物19)将5-硝基-1H-吲唑-3-羧基醛(500mg,2.44mmol)溶解于二甲基甲酰胺(12mL)中。向反应混合物添加N-氯琥珀酰亚胺(320mg,2.44mmol),然后在40℃下搅拌混合物1小时。利用乙酸乙酯和水对所获得的残余物进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。不经过另外的精制过程,获得目标化合物(收率:80%)。1HNMR(400MHz,DMSO-d6)δ11.98(s,1H),11.12(s,1H),8.87(s,1H),8.56(m,2H).MASS=240.60合成例20:3-(氯(肟基)甲基)-1H-吲唑-5-羧酸(化合物20)将3-(肟基)甲基)-1H-吲唑-5-羧酸(500mg,2.44mmol)溶解于二甲基甲酰胺(12mL)中。向反应混合物添加N-氯琥珀酰亚胺(320mg,2.44mmol),然后在40℃下搅拌混合物1小时。利用乙酸乙酯和水对所获得的残余物进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。不经过另外的精制过程,获得目标化合物(收率:86%)。1HNMR(400MHz,DMSO-d6)δ10.98(s,1H),8.78(s,1H),7.99(d,1H),7.68(d,1H).MASS=239.62步骤(d-4)的一般过程合成示意图3,将化学结构式13的化合物(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加R2-苯胺(35μL,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物。合成例21(LDD-2369):Ν'-羟基-5-硝基-N-苯基-1H-吲唑-3-羧基酰亚胺酰胺(化合物21)将N-羟基-5-硝基-1H-吲唑-3-carimidoyl氯化物(30mg,0.13mmol)溶解于乙醇(1.5mL)中。添加苯胺(35μL,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:33%)。1HNMR(400MHz,DMSO-d6)δ10.97(s,1H),8.90(d,1H),8.47(s,1H),8.23(dd,1H),7.75(dd,1H).MASS=297.27合成例22(LDD-2370):N-(4-氟苯基)-Ν'-羟基-5-硝基-1H-吲唑-3-羧基酰亚胺酰胺(化合物22)将N-羟基-5-硝基-1H-吲唑-3-carimidoyl氯化物(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加4-氟苯胺(37μL,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:33%)。1HNMR(400MHz,DMSO-d6)δ10.95(s,1H),8.92(d,1H),8.49(s,1H),8.23(dd,1H),7.75(dd,1H),6.94(m,2H),6.75(m,2H).MASS=315.26合成例23(LDD-2371):Ν'-羟基-N-(4-羟基苯基)-5-硝基-1H-吲唑-3-羧基酰亚胺酰胺(化合物23)将N-羟基-5-硝基-1H-吲唑-3-carimidoyl氯化物(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加4-羟基苯胺(41mg,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:33%)。1HNMR(400MHz,DMSO-d6)δ10.65(s,1H),8.92(s,1H),8.84(d,1H),8.21(dd,1H),8.09(s,1H),7.72(d,1H),6.60(m,2H),6.50(m,2H).MASS=313.27合成例24(LDD-2372):Ν'-羟基-N-(4-甲氧基苯基)-5-硝基-1H-吲唑-3-羧基酰亚胺酰胺(化合物24)将N-羟基-5-硝基-1H-吲唑-3-carimidoyl氯化物(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加4-甲氧基苯胺(41mg,47mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:33%)。1HNMR(400MHz,DMSO-d6)δ10.74(s,1H),8.87(d,H),8.23(s,1H),8.21(dd,1H),7.72(d,1H),6.68(m,4H).MASS=327.30合成例25(LDD-1986):3-(Ν'-羟基-N-苯基碳亚氨基)-1H-吲唑-5-羧酸(化合物25)将3-(氯(肟基)甲基)-1H-吲唑-5-羧酸(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加苯胺(35μL,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:75%)。1HNMR(400MHz,DMSO-d6)δ10.82(s,1H),8.66(s,1H),8.38(s,1H),7.95(dd,1H),7.60(d,1H),7.07(t,2H),6.78(t,1H),6.69(d,2H).MASS=296.29合成例26(LDD-2197):3-N-(4-氟苯基)-Ν'-羟基碳亚氨基-1H-吲唑-5-羧酸(化合物26)将3-(氯(肟基)甲基)-1H-吲唑-5-羧酸(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加4-氟苯胺(37μL,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:29%)。1HNMR(400MHz,DMSO-d6)δ10.79(s,1H),8.67(s,1H),8.40(s,1H),7.95(dd,1H),7.60(dd,1H),6.93(m,2H),6.714(m,2H).MASS=314.28合成例27(LDD-2349):3-(Ν'-羟基-N-(4-羟基苯基)碳亚氨基)-1H-吲唑-5-羧酸(化合物27)将3-(氯(肟基)甲基)-1H-吲唑-5-羧酸(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加4-羟基苯胺(41mg,0.38mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:31%)。1HNMR(400MHz,DMSO-d6)δ13.23(s,1H),10.45(s,1H),8.93(s,1H),8.90(s,1H),7.92(m,2H),7.47(d,1H),6.64(d,2H),6.40(d,2H).MASS=312.28合成例28(LDD-2198):3-(Ν'-羟基-N-(4-甲氧基苯基)碳亚氨基)-1H-吲唑-5-羧酸(化合物28)将3-(氯(肟基)甲基)-1H-吲唑-5-羧酸(30mg,0.13mmol)溶解于乙醇(1.5mL)中。接着,添加4-甲氧基苯胺(41mg,47mmol),然后在80℃下搅拌混合物2小时。利用乙醇将所获得的残余物浓缩而除去,然后通过硅胶层析进行精制,从而获得目标化合物(收率:31%)。1HNMR(400MHz,DMSO-d6)δ10.60(s,1H),8.59(s,1H),8.11(s,1H),7.89(dd,1H),7.54(dd,1H),6.61(s,4H),3.58(s,3H).MASS=326.31步骤(e-5)的一般过程将在合成例25获得的化合物(10mg,0.02mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,在0℃下逐渐滴加四甲基硅烷(TMS)-重氮甲烷(100μL),然后在0℃下搅拌30分钟。在真空下进行浓缩后,通过硅胶层析进行精制,从而获得目标化合物。合成例29(LDD-2196):甲基-3-(Ν'-羟基-N-苯基碳亚氨基)-1H-吲唑-5-羧酸(化合物29)将在合成例25获得的化合物(10mg,0.02mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,在0℃下逐渐滴加四甲基硅烷-重氮甲烷(100μL),然后在0℃下搅拌30分钟。在真空下进行浓缩后,通过硅胶层析进行精制,从而获得目标化合物(收率:83%)。1HNMR(400MHz,DMSO-d6)δ12.27(s,1H),9.56(dd,1H),9.20(s,1H),8.65(dd,1H),8.25(dd,1H),7.54(m,2H),7.19(t,1H),7.06(dd,2H),3.55(s,3H).MASS=310.31步骤(f-6)的一般过程合成示意图3,将化学结构式14的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(HOBt,0.04mmol)、哌啶(0.09mmol)、TEA(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物。合成例30(LDD-1821):Ν'-羟基-N-苯基-5-(哌啶-1-羰基)-1H-吲唑-3-羧基酰亚胺酰胺(化合物30)将在合成例25获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(0.04mmol)、哌啶(0.09mmol)、TEA(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:65%)。1HNMR(400MHz,DMSO-d6)δ10.67(s,1H),8.36(s,1H),7.83(s,1H),7.55(d,1H),7.34(d,1H),7.02(t,2H),6.73(t,1H),6.64(d,2H),3.56(m,4H),1.58(m,6H).MASS=363.42合成例31(LDD-2199):N-(4-氟苯基)-Ν'-羟基-5-(哌啶-1-羰基)-1H-吲唑-3-羧基酰亚胺酰胺(化合物31)将在合成例26获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(0.04mmol)、哌啶(0.09mmol)、TEA(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:72%)。1HNMR(400MHz,DMSO-d6)δ10.67(s,1H),8.41(s,1H),7.88(s,1H),7.58(dd,1H),7.38(dd,1H),6.915(m,2H),6.71(m,2H),3.33(m,4H),1.62(m,6H).MASS=381.41合成例32(LDD-2350):Ν'-羟基-N-(4-羟基苯基)-5-(哌啶-1-羰基)-1H-吲唑-3-羧基酰亚胺酰胺(化合物32)将在合成例27获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(0.04mmol)、哌啶(0.09mmol)、TEA(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:65%)。1HNMR(400MHz,DMSO-d6)δ13.25(s,1H),10.34(s,1H),8.84(s,1H),7.96(s,1H),7.74(s,1H),7.51(d,1H),7.31(dd,1H),6.53(m,2H),6.44(m,2H),3.39(m,4H),1.60(m,6H).MASS=379.42合成例33(LDD-2200):Ν'-羟基-N-(4-甲氧基苯基)-5-(哌啶-1-羰基)-1H-吲唑-3-羧基酰亚胺酰胺(化合物33)将在合成例28获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(0.04mmol)、哌啶(0.09mmol)、TEA(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:41%)。1HNMR(400MHz,DMSO-d6)δ10.48(s,1H),8.16(s,1H),7.82(s,1H),7.56(dd,1H),7.36(dd,1H),3.55(s,3H),3.46(m,4H),1.62(m,6H).MASS=393.45合成例34(LDD-1987):Ν'-羟基-N-苯基-5-(4-丙基哌嗪-1-羰基)-1H-吲唑-3-羧基酰亚胺酰胺(化合物34)将在合成例25获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(0.04mmol)、1-丙基哌嗪(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:41%)。1HNMR(400MHz,DMSO-d6)δ10.69(s,1H),8.37(s,1H),7.87(s,1H),7.59(d,1H),7.39(dd,1H),7.06(t,2H),6.77(t,1H),6.69(d,2H),3.45(m'4H),2.37(m,4H),2.29(t,2H),1.47(q,2H),0.88(m,3H).MASS=406.49合成例35(LDD-1988):Ν'-羟基-5-(4-(2-甲氧基乙基)哌嗪-1-羰基)-N-苯基-1H-吲唑-3-碳酰亚胺酰胺(化合物35)将在合成例25获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,向反应混合物添加羟基苯并三唑(0.04mmol)、1-(2-甲氧基乙基)哌嗪(0.09mmol)及二氯化乙烯(0.09mmol),在常温下搅拌混合物3小时。然后用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:36%)。1HNMR(400MHz,DMSO-d6)δ10.70(s,1H),8.39(s,1H),7.87(s,1H),7.59(d,1H),7.39(dd,1H),7.06(t,2H),6.77(t,1H),6.69(d,2H),3.46(m,6H),3.24(s,1H),2.52(m,2H),2.40(m,4H).MASS=422.49合成例36(LDD-2351):5-(4-(2-(1H-咪唑-1-基)乙基)哌嗪-1-羰基)-N'-羟基-N-苯基-1H-吲唑-3-碳酰亚胺酰胺(化合物36)将在合成例25获得的化合物(20mg,0.06mmol)溶解于二甲基甲酰胺(0.6mL)中。接着,向反应混合物添加羟基苯并三唑(0.08mmol)、1-(2-(1H-咪唑-1-基)乙基)哌嗪(0.19mmol)及二氯化乙烯(0.19mmol),在常温下搅拌混合物1小时。通过MC:MeOH=10:1混合溶剂和饱和的NaHCO3水溶液对所获得的残余物进行提取。通过利用以氨饱和的氯仿和甲醇的硅胶层析进行精制,从而获得目标化合物(收率:16%)。1HNMR(400MHz,DMSO-d6)δ10.74(s,1H),8.37(s,1H),7.870(s,1H),7.65(s,1H),7.58(dd,1H),7.38(dd,1H),7.20(m,1H),7.05(t,2H),6.87(m,1H),6.77(t,1H),6.68(d,2H),4.10(t,2H),3.46(m,4H),2.68(t,2H),2.42(m,4H).MASS=458.53合成例37(LDD-2352):5-(4-(2-(1H-咪唑-1-基)乙基)哌嗪-1-羰基)-N-(4-氟苯基)-N'-羟基-1H-吲唑-3-碳酰亚胺酰胺(化合物37)将在合成例25获得的化合物(20mg,0.06mmol)溶解于二甲基甲酰胺(0.6mL)中。接着,向反应混合物添加羟基苯并三唑(0.08mmol)、1-(2-(1H-咪唑-1-基)乙基)哌嗪(0.19mmol)及二氯化乙烯(0.19mmol),在常温下搅拌混合物1小时。通过MC:MeOH=10:1混合溶剂和饱和的NaHCO3水溶液对所获得的残余物进行提取。通过利用以氨饱和的氯仿和甲醇的硅胶层析进行精制,从而获得目标化合物(收率:29%)。1HNMR(400MHz,DMSO-d6)δ10.74(s,1H),8.42(s,1H),7.90(s,1H),7.65(s,1H),7.58(d,1H),7.39(dd,1H),7.20(t,1H),6.92(m,3H),6.70(m,2H),4.10(t,2H),3.50(m,4H),2.68(t,2H),2.40(m,4H).MASS=476.52合成例38(LDD-2354):5-(4-(2-(1H-咪唑-1-基)乙基)哌嗪-1-羰基)-N'-羟基-N-(4-羟基苯基)-1H-吲唑-3-碳酰亚胺酰胺(化合物38)将在合成例25获得的化合物(20mg,0.06mmol)溶解于二甲基甲酰胺(0.6mL)中。接着,向反应混合物添加羟基苯并三唑(0.08mmol)、1-(2-(1H-咪唑-1-基)乙基)哌嗪(0.19mmol)及二氯化乙烯(0.19mmol),在常温下搅拌混合物1小时。通过MC:MeOH=10:1混合溶剂和饱和的NaHCO3水溶液对所获得的残余物进行提取。通过利用以氨饱和的氯仿和甲醇的硅胶层析进行精制,从而获得目标化合物(收率:14%)。1HNMR(400MHz,DMSC-d6)δ13.29(s,1H),10.41(s,1H),8.88(s,1H),7.99(s,1H),7.79(s,1H),7.71(s,1H),7.55(d,1H),7.36(dd,1H),7.22(m.1H),6.90(m.1H),6.55(m,2H),6.49(m,2H),4.10(t,2H),3.50(m,4H),2.67(t,2H),2.42(m,4H).MASS=474.52合成例39(LDD-2353):5-(4-(2-(1H-咪唑-1-基)乙基)哌嗪-1-羰基)-N'-羟基-N-(4-甲氧基苯基)-1H-吲唑-3-碳酰亚胺酰胺(化合物39)将在合成例25获得的化合物(20mg,0.06mmol)溶解于二甲基甲酰胺(0.6mL)中。接着,向反应混合物添加羟基苯并三唑(0.08mmol)、1-(2-(1H-咪唑-1-基)乙基)哌嗪(0.19mmol)及二氯化乙烯(0.19mmol),在常温下搅拌混合物1小时。通过MC:MeOH=10:1混合溶剂和饱和的NaHCO3水溶液对所获得的残余物进行提取。通过利用以氨饱和的氯仿和MeOH的硅胶层析进行精制,从而获得目标化合物(收率:31%)。1HNMR(400MHz,DMSO-d6)δ13.29(s,1H),10.41(s,1H),8.88(s,1H),7.99(s,1H),7.79(s,1H),7.71(s,1H),7.55(d,1H),7.36(dd,1H),7.22(m.1H),6.90(m.1H),6.55(m,2H),6.49(m,2H),4.10(t,2H),3.50(m,4H),2.67(t,2H),2.42(m,4H).MASS=488.55合成例40(LDD-2319):叔丁基(2-(2-(2-(4-(3-(Ν'-羟基-N-苯基碳亚氨基)-1H-吲唑-5-羰基)哌嗪-1-基)乙氧基)乙氧基)乙基)氨基甲酸酯(化合物40)将在合成例25获得的化合物(20mg,0.07mmol)溶解于二甲基甲酰胺(0.6mL)中。接着,向反应混合物添加羟基苯并三唑(0.11mmol)、叔丁基(2-(2-(2-(哌嗪-1-基)乙氧基)乙氧基)乙基)氨基甲酸酯(0.14mmol)及二氯化乙烯(0.14mmol),在常温下搅拌混合物2小时。用乙酸乙酯和水对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:90%)。1HNMR(400MHz,DMSO-d6)δ10.68(s,1H),8.36(s,1H),7.82(s,1H),7.53(d,1H),7.33(dd,1H),6.99(t,2H),6.71(m,2H),6.63(d,2H),3.49(m,8H),3.29(m,4H),3.01(m,2H),2.49(m,2H),2.37(m,4H),1.32(s,9H).MASS=595.7合成例41(LDEH2320):5-(4-(2-(2-(2-氨基乙氧基)乙氧基)乙基)哌嗪-1-羰基)-N'-羟基-N-苯基-1H-吲唑-3-碳酰亚胺酰胺(化合物41)将在合成例25获得的化合物(10mg,0.02mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,在0℃下添加三氟乙酸(0.15mL),在0℃下搅拌30分钟。在真空下进行浓缩后,通过利用以氨饱和的氯仿和甲醇的硅胶层析进行精制,从而获得目标化合物(收率:85%)。1HNMR(400MHz,DMSO-d6)δ10.75(s,1H),8.40(s,1H),7.86(s,1H),7.57(d,1H),7.38(dd,1H),7.03(t,2H),6.75(t,1H),6.68(d,2H),3.54(m,12H),2.86(m,2H),2.49(m,2H),2.42(m,4H).MASS=495.58合成示意图4下面,在合成例所记载的反应过程和取代基指代所述合成示意图中的反应过程和取代基。一般合成过程步骤(a)的一般过程合成示意图4,将化学结构式2的化合物(20mg,0.11mmol)溶解于1,4-二恶烷(1mL)中。接着,向溶液添加R2-N-羟基苯亚胺基氯化物(0.17mmol)和TEA(0.22mmol)。在常温下搅拌所述混合物18小时后,用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物。合成例42(LDD-2194):3-(Ν'-羟基苯酰亚胺酰胺)-1H-吲唑-5-羧酸(化合物42)将在合成例2获得的化合物(20mg,0.11mmol)溶解于1,4-二恶烷(1mL)中。接着,向溶液添加N-羟基苯亚胺基氯化物(0.17mmol)和TEA(0.22mmol)。在常温下搅拌所述混合物18小时后,用乙酸乙酯和饱和的NH4Cl水溶液对所获得的残余物进行提取。然后,通过硅胶层析进行精制,从而获得目标化合物(收率:23%)。1HNMR(400MHz,DMSO-d6)δ12.31(s,1H),8.62(m,1H),7.63(dd,1H),7.43(m,3H),7.33(m,2H),7.16(dd,1H),6.81(s,2H).MASS=296.29合成例45(LDD-1945):N'-羟基-N-(5-(哌啶-1-羰基)-1H-吲唑-3-基)苯酰亚胺酰氨(化合物45)将在合成例44获得的化合物(10mg,0.04mmol)溶解于1,4-二恶烷(0.4mL)中。接着,向溶液添加N-羟基苯亚胺基氯化物(0.04mL)和TEA(0.08mmol)。在常温下搅拌所述混合物18小时,对所产生的沉淀物进行过滤,然后用亚甲基氯清洗,从而获得目标化合物(收率:31%)。1HNMR(400MHz,DMSO-d6)δ12.28(s,1H),7.91(s,1H),7.43(m,3H),7.32(d,2H),7.18(m,2H),6.48(s,2H),3.57(m,4H),1.54(m,6H).MASS=363.42步骤(b-2)的一般过程合成示意图4,将化学结构式17的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,在0℃下逐渐滴加四甲基硅烷-重氮甲烷(100μL),然后在0℃下搅拌30分钟。在真空下进行浓缩后,通过硅胶层析进行精制,从而获得目标化合物。合成例43(LDD-2195):甲基-3-(Ν'-羟苯酰亚胺酰氨)-1H-吲唑-5-羧酸(化合物43)将在合成例42获得的化合物(10mg,0.03mmol)溶解于二甲基甲酰胺(0.3mL)中。接着,在0℃下逐渐滴加四甲基硅烷-重氮甲烷(100μL),然后在0℃下搅拌30分钟。在真空下进行浓缩后,通过硅胶层析进行精制,从而获得目标化合物(收率:85%)。1HNMR(400MHz,DMSO-d6)δ12.31(s,1H),8.66(m,1H),7.640(dd,1H),7.43(m,3H),7.33(m,2H),7.19(dd,1H),6.87(s,2H),3.84(s,3H).MASS=310.31步骤(c-3)的一般过程合成示意图4,将化学结构式2的化合物(100mg,0.56mmol)溶解于二甲基甲酰胺(2mL)中。接着,向反应混合物添加羟基苯并三唑(0.85mmol)、各种胺(0.68mmol)、TEA(1.7mmol)及二氯化乙烯(1.13mmol),在常温下搅拌混合物12小时。用饱和的NaHCO3水溶液和乙酸乙酯对所获得的残余物进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。利用二氯甲烷和核酸形成沉淀物,对此进行过滤,从而获得目标化合物。合成例44:(3-氨基-1H-吲唑-5-基)(哌啶-1-基)甲酮(化合物44)合成示意图4,将化学结构式2的化合物(100mg,0.56mmol)溶解于二甲基甲酰胺(2mL)中。接着,向反应混合物添加羟基苯并三唑(0.85mmol)、各种胺(0.68mmol)、TEA(1.7mmol)及二氯化乙烯(1.13mmol),在常温下搅拌混合物12小时。用饱和的NaHCO3水溶液和乙酸乙酯对所获得的残余物进行提取。通过盐水清洗结合的有机层,用无水Na2SO4进行干燥,在真空下浓缩。利用二氯甲烷和核酸形成沉淀物,对此进行过滤,从而获得目标化合物(收率:41%)。1HNMR(400MHz,DMSO-d6)δ11.58(s,1H),7.81(m,1H),7.24(s,2H),5.50(s,2H),3.40(m,4H),1.61(m,6H).MASS=244.30试验例试验例1:利用茜素红染色(Alizarinredstaining)的细胞筛选诱导直接细胞重编程(Directcellreprogramming)的低分子化合物的筛选用于寻找诱导细胞重编程的化合物的方法基于以往的诱导骨形成的肌肉细胞的骨细胞转换研究PMID:23044010及18974773和逆转素论文(ChenS2004)。将C2C12小鼠骨骼肌细胞以每孔1×104的密度分注于24-孔形式的细胞培养皿。在分注后经过24小时,向3个孔以1浓度添加感兴趣的化合物。作为阳性对照组(Positivecontrol),对细胞用100nM逆转素或2uMBIO进行处理。这两个化合物是被认为诱导肌细胞的骨细胞转换的化合物(ChenS2004andKimWHrecapitulatepapers)。作为阴性对照组(Negativecontrol),对细胞用DMSO进行处理,或没有进行处理。对细胞用化合物进行处理总共72小时,在48小时后,为了补充化合物而进一步进行处理。化合物的细胞毒性由细胞中消失的部分和当培养时浮漂的细胞碎片可以确定,而对引发毒性的化合物以250nM浓度重新进行筛选。72小时后,将细胞培养皿转换成骨形成分化诱导培养基(用添加有10%FBS,50μg/mL抗坏血酸-2-磷酸盐,0.1μΜ地塞米松,10mMβ-甘油磷酸酯,50unitsmL-1青霉素,50-1链霉素的DMEM进行处理14天)。骨形成诱导培养基基于在以往的直接细胞重编程(reversinepapaers,ChenS2004andChenS2007)中的描述。对从肌肉细胞直接细胞重编程成的骨细胞通过检测在骨系细胞的钙沉积(GregoryCA2004:PMID:15136169)是否存在的茜素红染色(alizarinredstaining)进行检测。茜素红染色(Alizarinredstaining)在通过PBS清洗细胞一次后,利用磷酸盐缓冲甲醛水进行固定20分钟。通过蒸馏水清洗所固定的细胞,然后将该细胞浸渍于溶解于水中的1%茜素红-S(Sigma-Aldrich)5分钟。利用蒸馏水清洗剩下的染色液。在干燥细胞后,利用光学显微镜(CKX41Olympus)拍摄染色的细胞照片。染色的细胞被视为红色部分,并将其选择为“命中(hit)”化合物来进行了更多分析。利用如上所述的试验方法对所合成的吲唑化合物的细胞重编程能力进行评价。当分化成成骨细胞的过程中,LDD-1664、LDD-1821、LDD-1945、LDD-2199、LDD-2200呈现与作为阳性对照组的逆转素的重编程能力相当的强大的重编程能力,且LDD-1986、LDD-1987、LDD-1988也呈现重编程能力。在可以确认分化成脂肪细胞的过程的油红0染色中,LDD-1821、LDD-1986、LDD-1987、LDD-1988物质呈现强大的重编程能力。尤其,在LDD-1821的情况下,分化成成骨细胞和脂肪细胞的过程中都呈现强大的重编程能力(参见图1、图2及图3)。【表1】细胞染色结果++(强阳性),+(弱阳性),-(阴性)试验例2:激酶(Kinase)试验吲唑衍生物的对于GSK-3β和AuroraA的抑制活性通过HTRF(homogeneoustime-sesolvedfluorescence)试验进行确认。HTRF试验是在ATP的存在下确认肽物质的磷酸化的试验方法。磷酸化的物质由TR-FRET(TimeResolved-FluorescenceResonance)信号探测。重组的GSK-3β和AuroraA激酶由Millipore(Billerica,MA)购买。使用HTRFKinEASE试剂盒(Cisbio)进行试验,用1μΜ的吲唑物质对GSK-3β和AuroraA分别进行处理,然后确认抑制的磷酸化程度。试验由溶解于激酶反应缓冲液(250mMHEPES(pH7.0)、0.5mM原钒酸盐(orthovanadate)、0.05%BSA、0.1%NaN3)中的物质-酶混合物和肽物质构成。在添加检测缓冲液后,TR-FRET信号由EnVisionmulti-labelreader检测。用1μΜ的LDD-1664、LDD-1667、LDD-1820及LDD-1821对GSK-3β和AuroraA分别进行处理,酶活性的抑制程度如下表所示。如表2的结果所示,4种衍生物都在1μΜ浓度下针对GSK-3β和AuroraA呈现小于50%的抑制活性。并且,呈现卓越的细胞重编程能力的LDD-1821在1μΜ浓度下针对两个酶完全未呈现抑制活性。由此可知,通过与作为GSK-3β或AuroraA的抑制剂的BIO和逆转素不同的机制诱导细胞重编程。[表2]激酶试验试验例3:MTT比色试验将C2C12成肌细胞(myoblast)以每孔3×103个细胞的密度分注于96-孔板。在培养细胞一天后,用物质进行处理。然后,培养细胞两天,对细胞用包含3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑溴化物(MTT)的无血清细胞培养基(Dulbecco'sModifiedEagleMedium,DMEM)进行处理3小时。在除去培养基后,在二甲基亚砜(dimethylsulfoxide,DMSO)中,在摇动的同时培养10分钟。在570nm用微读数器(VersaMax,MolecularDevices公司制)测量吸光度(参见图4)。通过MTT试验对整理在下表中的化合物的细胞毒性进行评价,结果,被评价的所有吲唑衍生物都呈现10μΜ以上的IC50。在这一点上,吲唑衍生物与呈现高细胞毒性的现有BIO和逆转素相比,具有明显的优点。[表3]MTT比色分析的结果化合物IC50(μΜ)LDD-1821>10LDD-1986>10LDD-1987>10LDD-1988>10LDD-1945>10LDD-2194>10LDD-2195>10LDD-2196>10LDD-2197>10LDD-2198>10LDD-2199>10LDD-2200>10以上对本
发明内容的特定部分进行了详细阐述,对具有相关行业普遍知识的人员来说,上述具体技术只是一种优选实施例,并非用来限制本发明的范围。因此,本发明的实质范围可以根据权利要求及其等同物进行定义。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1