一种吲哚美辛粉体的制作方法
本发明涉及药物制剂技术领域,特别涉及一种吲哚美辛粉体。
背景技术:
吲哚美辛化学名为:2-甲基-1-(4-氯苯甲酰基)-5-甲氧基-1h-吲哚-3-醋酸,分子式为c19h16clno4,分子量为357.79,结构式如下式:
吲哚美辛是白色或微黄色结晶性粉末,熔点是158~162℃,溶于丙酮,略溶于乙醇、氯仿、乙醚,几乎不溶于水,无味,几乎无臭。
吲哚美辛具有抗炎、解热及镇痛作用,其作用机理为通过对环氧化酶的抑制而减少前列腺素的合成,制止炎症组织痛觉神经冲动的形成,抑制炎性反应,包括抑制白细胞的趋化性及溶酶体酶的释放等。至于退热作用,是由于其作用于下视丘体温调节中枢,引起外周血管扩张及出汗,使散热增加,这种中枢性退热作用也可能与在下视丘的前列腺素合成受到抑制有关。急性毒性试验结果:大鼠经口ld50为12mg/kg;小鼠经口ld5为50mg/kg。
吲哚美吲几乎不溶于水,为难溶性药物,难溶性药物(poorlywater-solubledrug)在水中溶解度小,药物难以被机体吸收,体内消除速度较快,血药浓度容易出现峰谷现象,口服制剂生物利用度低,且难以实现剂型的多样化。难溶性药物的吸收速率通常取决于溶出速率,溶出速度随分散度的提高而提高。
技术实现要素:
本发明提供了一种吲哚美辛粉体,解决现有的吲哚美辛制剂溶出度较低的问题。
为解决上述技术问题,本发明的技术方案为:
一种吲哚美辛粉体,是由下述制备方法制成:取稳定剂水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加吲哚美辛丙酮溶液直至溶液体系变浑浊,静置,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
其中,优选地,所述稳定剂为聚乙二醇2000、聚乙二醇6000、聚乙二醇10000、聚乙烯吡咯烷酮、十二烷基苯磺酸钠、十二烷基硫酸钠、羟甲基纤维素纳和甘露醇中的任意一种。稳定剂用于抑制晶体生成并防止吲哚美辛小颗粒重新聚合。
其中,优选地,所述稳定剂的质量百分比浓度为0.5~1.5%。
其中,优选地,所述吲哚美辛丙酮溶液的浓度为0.04~0.06mol/l。
其中,优选地,所述稳定剂水溶液与所述吲哚美辛丙酮溶液的体积比为10~15:1。
其中,优选地,所述静置的时间为6~10h。
本发明有益效果:
本发明中的吲哚美辛粉体,通过粒径分析可知,粉体粒径可达d50=2.36μm。本发明可有效地提高吲哚美辛的溶解度和溶出速率,从而可以有效地提高吲哚美辛的生物利用度。
体外溶出试验表明,本发明制备的吲哚美辛粉体,与同晶态的吲哚美辛相比,在0.1n盐酸溶液、ph4.5、ph6.0和ph7.4的磷盐缓冲液中,吲哚美辛的水溶性均有提高,且均有较高的溶出速率。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例1溶出曲线图;
图2为本发明实施例2溶出曲线图;
图3为本发明实施例3溶出曲线图;
图4为本发明实施例4溶出曲线图;
图5为本发明实施例5溶出曲线图;
图6为本发明实施例6溶出曲线图;
图7为本发明实施例7溶出曲线图;
图8为本发明实施例8溶出曲线图。
具体实施方式
下面将结合本发明具体实施例,对本发明的的技术方案进行清楚、完整的描述,所描述的实例仅仅是本发明的部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员,在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明的保护范围。
实施例1
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取质量百分比浓度为1.0%的聚乙二醇2000水溶液33.5l,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加2.79l浓度为0.05mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置8h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
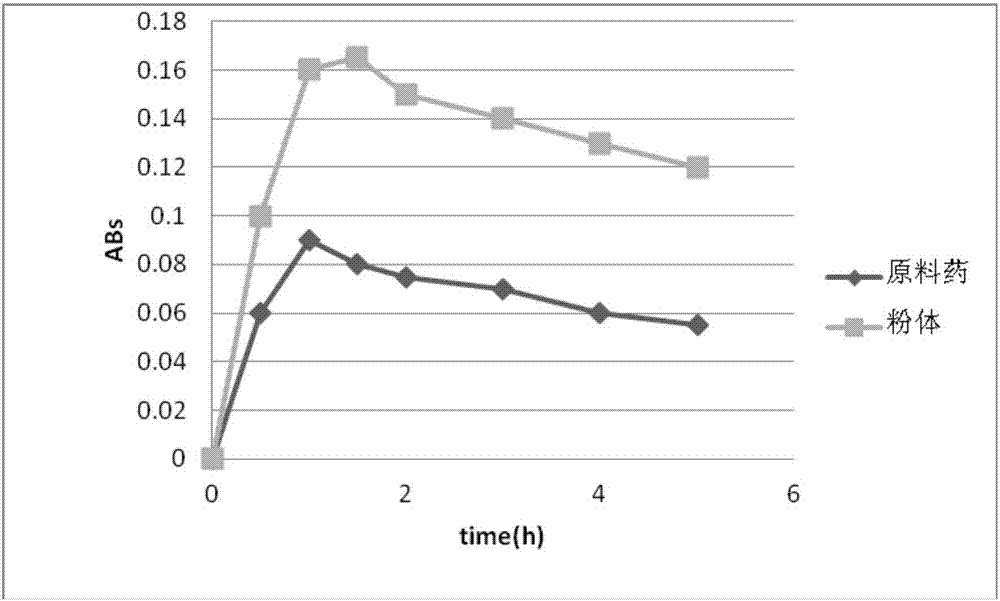
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图1所示。
由图1的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=2.36μm。
实施例2
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取34.9l质量百分比浓度为0.5%的聚乙二醇6000水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加3.49l浓度为0.04mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置8h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图2所示。
由图2的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=4.36μm。
实施例3
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取34.9l质量百分比浓度为1.5%的聚乙二醇10000水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加2.33l浓度为0.06mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置6h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图3所示。
由图3的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=5.36μm。
实施例4
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取34.2l质量百分比浓度为0.8%的聚乙烯吡咯烷酮水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加3.11l浓度为0.045mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置7h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图4所示。
由图4的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=3.76μm。
实施例5
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取18.17l质量百分比浓度为1.2%的十二烷基苯磺酸钠水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加1.40l浓度为0.05mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置9h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图5所示。
由图5的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=5.24μm。
实施例6
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取17.79l质量百分比浓度为1.3%的十二烷基硫酸钠水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加1.27l浓度为0.055mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置8h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图6所示。
由图6的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=5.44μm。
实施例7
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取15.37l质量百分比浓度为1.4%的羟甲基纤维素纳水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加1.40l浓度为0.05mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置7h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图7所示。
由图7的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=5.45μm。
实施例8
本实施例提供一种吲哚美辛粉体,是由下述制备方法制成:取15.53l质量百分比浓度为1.0%的甘露醇水溶液,将之置于0~5℃的冰水浴中,在超声的条件下,缓慢滴加1.55l浓度为0.045mol/l的吲哚美辛丙酮溶液直至溶液体系变浑浊,静置9h后,高速离心直至分离完全;将所得的固体样品洗涤离心,除去上层清液,得到的产物于50℃的真空干燥箱中干燥,过80目筛,得到吲哚美辛粉体。
取上述方法制备的样品及原料药,置于广口瓶中,再加入200mlph7.4的磷盐缓冲液作为溶出介质,于37℃摇床中振晃,在不同时间点取样,经微孔滤膜过滤,测其吸光度,比较样品的溶解度。溶出曲线图如图8所示。
由图8的溶出曲线可以看出,与原料药相比,固体分散体的溶出速率和溶出度均有明显的提高。本实施例吲哚美辛粉体的粒径d50=5.12μm。
上述实施例制得的固体分散体同样也在0.1n盐酸溶液、ph4.5、ph6.0磷酸盐缓冲液中作溶出度试验,结果显示,本发明制得的固体分散体与原料药相比,溶出速率和溶出度也是显著提高的。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!