一种基于氢键络合的多肽或蛋白纳米粒及其制备方法和应用与流程
本发明属于生物医药技术领域,更具体地,涉及一种基于氢键络合的多肽或蛋白类药物纳米制剂及其制备方法和应用。
背景技术:
多肽或蛋白类药物具有特异性强和功效高等优点,因而广泛地应用于调控生理过程以及疾病治疗。多肽或蛋白类分子由于其亲水性及高分子量特征而难以跨越体内多种生物屏障,诸如粘液屏障,胃肠道屏障等,从而到达靶向部位发挥其生理功能;多肽或蛋白类药物容易被体内的酶降解,其体内血液循环时间也短,因此需要通过反复给药治疗,这造成病人的顺应性较差。通过对多肽或蛋白类药物的纳米制剂化,可以延长其体内循环时间,提高靶向输送效率,改善病人给药条件和途径,改善药物的贮藏条件等。
多肽或蛋白类纳米制剂的制备目前主要通过以下方式:多肽和蛋白类药物在偏离其等电点ph条件下通常带有电荷性,可以利用带相反电荷的载体依靠电荷作用对其进行包裹。然而当多肽和蛋白带有微弱电荷性时,却无法进行有效包裹;另一方面,多肽和蛋白分子可能含有疏水性区域,也可以利用载体的疏水性通过疏水作用进行包裹,然而纳米制剂过程中需要有机溶剂参与,因此会影响多肽和蛋白类药物的生物活性,且载药效率通常较低。
因此,研发一种新的多肽或蛋白类药物的纳米制剂化方法和载药纳米颗粒就显得尤为重要。
技术实现要素:
本发明所要解决的技术问题是克服现有技术中多肽或蛋白类药物在制备纳米制剂过程中存在的缺陷和不足,提供一种具有普适性的基于氢键相互作用高效地负载多肽或蛋白类药物的纳米输送体系,具有极高的包封效率,并可进一步通过静电/疏水相互作用,对载药体系表面进行功能化修饰。
本发明的第一个目的是提供一种基于氢键络合的载药纳米粒。
本发明的第二个目的是提供所述载药纳米粒的制备方法。
本发明的第三个目的是提供所述载药纳米粒的应用。
本发明的上述目的是通过以下技术方案给予实现的:
一种基于氢键络合的载药纳米粒,所述载药纳米粒包含多酚类化合物以及多肽或蛋白类药物;具体地,为包含作为氢键供体的多酚类化合物载体以及作为氢键受体的多肽或蛋白类药物。
由于多酚类化合物含有许多酚羟基官能团,其可作为氢键供体与一些含氢键受体的大分子组装形成多功能的膜、微胶囊或颗粒等,而多肽和蛋白类药物上含有丰富的肽键,可以作为氢键受体,因此通过多酚类化合物的氢键络合作用可以对多肽和蛋白类药物进行纳米制剂化;现有技术中报道蛋白质与多酚相互作用会引起凝聚而沉淀,而发明人研究发现,当多肽和蛋白类药物的分子量小于50kda时,多酚类化合物可与多肽和蛋白类药物通过氢键络合作用形成纳米颗粒,从而高效地负载蛋白以及多肽,且不会对蛋白以及多肽的活性产生显著影响,并且可以通过其他相互作用,如静电、疏水相互作用等,对纳米颗粒表面进行功能化的修饰;得到的纳米载体,可以通过多种给药途径有效地输送蛋白及多肽药物进入体内发挥药效。
优选地,所述多肽或蛋白类药物的分子量为0.5~50kda,具体包括0.5~1kda,1~5kda,5~10kda,10~50kda。
优选地,所述载药纳米粒还含有稳定剂和/或功能分子。
优选地,所述载药纳米粒的粒径为40~500nm,具体包括40~100nm,10~200nm,200~500nm。
优选地,所述载药纳米粒为核壳结构,包含核心和包被在核心上的稳定剂和/或功能分子,所述核心包含多酚类化合物以及多肽或蛋白类药物。
优选地,所述载药纳米粒为复合纳米颗粒。
更优选地,所述稳定剂和/或功能分子为壳聚糖、聚乙烯吡咯烷酮或透明质酸。
优选地,所述多肽或蛋白类药物为胰高血糖素样肽-1类似物(如艾塞那肽)、鲑鱼降钙素或胰岛素等。
优选地,所述多酚类化合物为单宁酸。
本发明基于氢键络合的载药纳米粒可进一步制备成不同的制剂类型,通过多种给药途径,有效的输送蛋白及多肽进入体内,发挥药效。因此,本发明还请求保护所述基于氢键络合的载药纳米粒在制备载药纳米制剂中的应用。
本发明基于氢键络合的载药纳米粒可利用逐步滴加法、倾倒法或快速纳米复合(fnc)技术来制备。
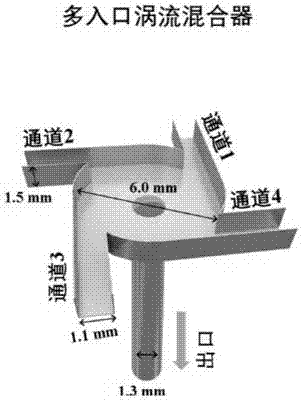
优选地,本发明采用fnc技术在多入口涡流混合器中来制备负载多肽和蛋白类药物的纳米粒,所述多入口涡流混合器包括位于上部的第一部件、位于中部的第二部件和位于下部的第三部件,所述第一部件、第二部件和第三部件为具有相同直径的圆柱体;第一部件设置有多个通道(优选为4个,分别为通道1,通道2,通道3,通道4),第二部件设置涡流混合区域和多个导流区域,第三部件设置通道;第一部件的通道与第二部件的导流区域流体连通;第二部件的导流区域均与涡流混合区域流体连通;第二部件的涡流混合区域与第三部件的通道流体连通;fnc技术可以在纯水相中,利用静电相互作用或其他相互作用,对药物进行负载,制备装置为多入口涡流混合器,高通量,可控性强,制得的纳米制剂分布均匀且粒径较小,批次间差异小。整个过程中均无有机溶剂参与,特别适合于蛋白、核酸等生物大分子的制剂化;上述技术及装置记载在本发明人前期申请号为pct/us2017/014080的专利中。
本发明所述载药纳米颗粒的制备方法,为采用fnc技术,将多酚类化合物的溶液通过通道1和2,多肽或蛋白类药物溶液通过通道3和4,到达涡流混合区域中混合;或将多酚类化合物的溶液通过通道1,多肽或蛋白类药物溶液通过通道2,稳定剂通过通道3,水通过通道4,到达涡流混合区域中混合;四个通道液体匀速流动,得到包含多酚类化合物与多肽或蛋白类药物的纳米颗粒溶液。
优选地,采用fnc技术,将得到的纳米颗粒溶液通过通道1和2,功能分子通过通道3和4,到达涡流混合区域中混合,四个通道液体匀速流动,得到表面涂覆功能分子的载药纳米粒。
具体地,本发明利用多酚类物质-单宁酸(ta),通过单宁酸上的酚羟基基团与蛋白及多肽中肽键的氢键相互作用,对多种蛋白及多肽类药物进行负载。利用fnc技术成功将电性较强的等电点较低(~4.5)的胰岛素、带电性较弱的艾塞那肽以及等电点较高(~9.3)的降钙素进行纳米制剂化,具有普适性以及极高的负载效率(包封率>90%)。并进一步利用fnc技术,通过静电/疏水相互作用,对载药纳米粒表面进行功能化修饰以及肠溶微胶囊制剂化,其中负载艾塞那肽的纳米粒及微胶囊制剂可以有效通过口服途径被小肠吸收。
作为一种优选的实施方式,所述载药纳米粒为负载艾塞那肽纳米颗粒,其粒径为40~500nm,多分散性指数(pdi)为0.1~0.4,包封率为70~99%,载药量为10~60%,其制备方法包括如下步骤:
s1.将单宁酸(ta)溶液通过通道1和2,艾塞那肽(exe)溶液通过3和4,到达涡流混合区域中混合,得到ta与exe组成的纳米复合核心;四个通道液体匀速流动,流速为5ml/min~50ml/min(优选为30ml/min);
s2.将s1所得混合液通过通道1和2,壳聚糖溶液通过通道3和4,到达涡流混合区域中混合,得到表面涂覆壳聚糖的纳米颗粒;四个通道液体匀速流动,流速为5ml/min~50ml/min(优选为30ml/min);
s3.将s2所得混合液通过通道1和2,尤特奇溶液通过通道3,醋酸(hac)水溶液通过通道4到达涡流混合区域中混合,得到肠溶微胶囊制剂;四个通道液体匀速流动,流速为30ml/min;
优选地,s1所述的艾塞那肽溶液的ph为5.5~6.8(优选6.2);具体为将艾塞那肽溶解于0.2%的醋酸(hac)溶液中,后用naoh溶液将ph值调至5.5~6.8(优选6.2)。
优选地,s1所述单宁酸(ta)溶液浓度为0.1~0.9mg/ml(优选为0.3mg/ml)。
优选地,s1所述艾塞那肽(exe)浓度为0.1~0.9mg/ml(优选为0.3mg/ml)。
优选地,s2所述壳聚糖溶液ph值为5.0~6.4(优选为6.2)。
优选地,s2所述壳聚糖溶液浓度为0.1~1mg/ml(优选为0.5mg/ml)。
优选地,s3所述尤特奇浓度为0.3~1.5mg/ml(优选为1.5mg/ml)。
优选地,s3反应体系ph值为3.5~5.2(优选ph=5.2)。
优选地,所述单宁酸与艾塞那肽的质量比为0.3~3:1。
作为一种优选的实施方式,所述载药纳米粒为负载降钙素纳米颗粒,其粒径为60~900nm,多分散性指数(pdi)为0.1~0.4,包封率为70~98%,载药量为20~60%,其制备方法包括如下步骤:
s1.将单宁酸(ta)溶液通过通道1,降钙素(sa)溶液通过通道2,壳聚糖(cs)溶液通过通道3,双蒸水通过通道4到达涡流混合区域中混合,得到负载sa的纳米颗粒;四个通道液体匀速流动,流速为30ml/min;
s2.将s1所得混合液通过通道1和2,透明质酸溶液通过通道3和4,到达涡流混合区域中混合,得到表面涂覆透明质酸的纳米颗粒;四个通道液体匀速流动,流速为30ml/min。
优选地,所述的降钙素(sa)溶液的浓度为0.5mg/ml。
优选地,所述单宁酸(ta)溶液浓度为0.1~1.0mg/ml(优选为0.4mg/ml)。
优选地,所述壳聚糖(cs)溶液ph值为5.0~6.4(优选为6.2)。
优选地,所述壳聚糖溶液浓度为0.2~0.5mg/ml(优选为0.5mg/ml)。
优选地,所述透明质酸溶液ph为6.2。
优选地,所述透明质酸溶液浓度为0.1~1.0mg/ml(优选为0.25mg/ml)。
作为一种优选的实施方式,所述载药纳米粒为负载胰岛素纳米颗粒,其粒径为50~500nm,多分散性指数(pdi)为0.1~0.4,包封率为70~98%,载药量为10~60%,其制备负方法包括如下步骤:
s1.将单宁酸(ta)溶液通过通道1,胰岛素(ins)溶液通过通道2,pvp溶液(hepes,25mm)通过通道3,水通过通道4到达涡流混合区域中混合,得到负载胰岛素的纳米颗粒;四个通道液体匀速流动,流速为5ml/min~50ml/min(优选为40ml/min);
优选地,所述胰岛素溶液的ph为3.8~5.0(优选4.0);具体为将胰岛素溶解于ph=2的盐酸(hcl)溶液中,后用naoh溶液将ph值调至3.8~5.0(优选4.0)。
优选地,所述单宁酸(ta)溶液浓度为0.3~0.5mg/ml(优选为0.5mg/ml)。
优选地,所述胰岛素溶液的浓度为0.5mg/ml。
优选地,所述pvp溶液浓度为0.8~1.2mg/ml(优选为1mg/ml)。
优选地,所述pvp分子量为3.5~360kda(优选为10kda)。
优选地,所述pvp溶液ph为5.0~6.0(优选为5.8),具体为将pvp溶解于25mmhepes缓冲液中,利用naoh或hcl溶液将ph调节至5.0~6.0(优选为5.8)。
优选地,所述纳米颗粒混合溶液的最终ph值为4.5~6.0(优选5.2)
与现有技术相比,本发明具有以下有益效果:
本发明所述基于氢键络合的药物负载体系,具有极高的包封效率,同时可以对多种不同结构与性质的蛋白及多肽类药物进行高效负载(包封率>90%),相比于基于静电/疏水作用力的制剂体系具有更强的普适性,并且制备过程中无有机溶剂参与。利用这种氢键络合方式几乎可以应用于所有的水溶性多肽和蛋白类药物的纳米制剂化,后续加工修饰更方便,可以通过静电/疏水或氢键相互作用进一步对颗粒表面性质以及形态进行调控,纳米颗粒加工过程在纯水相中进行,可以高效保持药物活性功能。
附图说明
图1所示为多入口涡流混合器的结构示意图。
图2所示为实施例1中流速对nc颗粒粒径、pdi的影响。
图3所示为实施例1中ph值对nc颗粒粒径pdi的影响。
图4所示为实施例1中ta浓度对nc颗粒粒径、pdi和表面电位的影响。
图5所示为实施例1中ta浓度对nc颗粒包封率和载药量的影响。
图6所示为实施例1中cs浓度对np颗粒粒径、pdi和表面电位的影响。
图7所示为实施例1中ph对np颗粒粒径、pdi和表面电位的影响。
图8所示为实施例1中ph对e-np包封率的影响。
图9所示为实施例1中尤特奇浓度对e-np包封率的影响。
图10所示为实施例2中e-np在ph=2.5环境中的释放行为。
图11所示为实施例2中最优条件下nc、np和e-np在生理条件下的释放行为。
图12所示为实施例3中载药纳米颗粒的小肠上皮渗透行为。
图13所示为实施例4中口服np和e-np的艾塞那肽血清药代动力学曲线。
图14示实为实施例5中ta浓度对nsc粒径、pdi和表面电位的影响。
图15所示为实施例5中壳聚糖浓度对nsc粒径、pdi和表面电位的影响。
图16所示为实施例5ha浓度为nsp粒径、pdi和表面电位的影响。
图17所示为实施例6中流速对nip粒径、pdi的影响。
图18所示为实施例6中ta浓度对nip粒径、pdi和包封率的影响。
图19所示为实施例6中pvp分子量对nip粒径、pdi和包封率的影响。
图20所示为实施例6中pvp浓度对nip粒径、pdi和包封率的影响。
图21所示为实施例6中ph对nip粒径、pdi和包封率的影响。
图22所示为实施例7中nip纳米颗粒糖尿病大鼠皮下注射降血糖效果。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
实施例1利用fnc技术制备负载艾塞那肽的纳米颗粒及肠溶微胶囊制剂
1、制备单宁酸(ta)和艾塞那肽(exe)纳米复合核心
ta溶解于去离子水中,浓度为0.1~0.9mg/ml。exe溶解于0.2%的醋酸溶液(hac)中,后用naoh溶液将ph值调至不同ph值(5.5~6.8),exe溶液浓度为0.3mg/ml。ta溶液分布于第一以及第二通道,exe溶液分布于第三及第四通道,如图1所示,四个通道流速一致,调节通道流速,5ml/min,10ml/min,20ml/min,30ml/min,40ml/min,50ml/min,使两种溶液通过四个通道到达涡流混合区进行混合,得到ta与exe的纳米复合核心(nc)。
图2所示为流速对nc粒径以及多分散性指数(pdi)的影响。ta浓度为0.3mg/ml,exe浓度为0.3mg/ml。当流速为5~30ml/min时,颗粒粒径随流速升高而减小,当流速位于30~50ml/min之间时,颗粒粒径随流速升高而增大。在实验流速范围内,pdi随流速增大而减小。当流速为30ml/min时,得到的颗粒最小且较为均一。选择流速30ml/min这一优选条件。
图3所示为ph对nc粒径以及多分散性指数的影响。随ph降低,颗粒粒径逐渐增大,当ph为5.5时,未形成稳定的悬浮颗粒,而是发生快速沉淀。在ph=6.2时,具有较小的粒径以及最小的pdi,因此选择ph=6.2这一优选条件。
图4所示为ta/exe比例对nc粒径、pdi以及表面电位的影响。exe浓度保持为0.3mg/ml,流速为30ml/min。当ta浓度为0.3~0.9mg/ml时,颗粒粒径及pdi基本不变,分别为~45nm和~0.25;电位随ta浓度增加略有增大,由25mv升高至30mv。
图5所示为不同比例下nc的包封率及载药量。在实验范围内,ta浓度对pdi,粒径以及表面电位基本无影响,均能得到粒径较小,分布均一且表面带负电的纳米颗粒。图5所示为不同ta浓度下,nc的艾塞那肽包封率和载药量,随ta浓度由0.9mg/ml降低至0.3mg/ml,nc的包封率基本不变,均高于95%,载药量由24%升高至48%,为了得到较高的载药量,exe的浓度选择0.3mg/ml这一优选条件。
2、利用fnc技术在nc表面涂覆一层壳聚糖(cs)
cs溶解于0.2%的醋酸(hac)水溶液中,浓度为0.1mg/ml,0.2mg/ml,0.3mg/ml,0.4mg/ml,0.5mg/ml。第一步制得的nc混悬液分布于第一及第二通道,cs水溶液分布于第三及第四通道,如图1所示,四个通道流速一致,调节通道流速为30ml/min,使两种液体通过四个通道到达涡流混合区域进行混合,得到表面涂敷cs的纳米颗粒(np)。
图6所示为不同cs浓度下制得的np的粒径,多分散性指数以及表面电位。随cs浓度由0.5mg/ml降低至0.2mg/ml,颗粒粒径由101nm升高至140nm,电位由18mv降低至12mv,当cs浓度降低至0.1mg/ml时,发生沉淀。当cs浓度为0.5mg/ml时,具有最小的粒径以及较小的pdi,最终选择cs浓度为0.5mg/ml这一优选条件。
研究了体系ph值对np的影响。图7所示为不同ph值下制备得到的np。随ph由6.2降低至5.0,粒径由103nm增加至131nm,pdi由0.27增加至0.526,这是由于随ph值降低,ta与exe的结合能力增强,导致在涂敷cs过程中发生聚集,粒径增大,分散均匀度降低;表面点位由18.3mv升高至26.4mv,这是由于ph降低,cs的解离程度增加,正电性增强,导致颗粒的表面点位增大。随ph由6.2升高至6.7,粒径降低至83nm,pdi增大至0.38;这是由于ph升高,ta与exe相互作用力减弱,ta/exe核心在涂敷cs过程中发生部分解离,导致颗粒粒径减小,粒径及分散均匀度下降;表面电位降低至12mv,这是由于ph升高后,cs解离程度降低,正电性减少,导致np表面电位减小。涂敷过程中ph值发生改变会导致ta/exe核心性质发生改变,并进一步影响涂敷后np的粒径及pdi,因此选择ph=6.2(制备nc时的ph值)作为涂敷过程的优选ph值。
3、肠溶微胶囊制剂制备
eugraditl100-55溶解于ph=11的naoh水溶液中,浓度为0.3~1.5mg/ml,待完全溶解后利用hcl溶液将ph值调整至7.4。np颗粒混悬液置于通道1和通道3,eugraditl100-55溶液置于通道2,醋酸(hac)水溶液置于通道4用以调控体系的ph值,设定通道流速为30ml/min,使三种液体通过四个通道到达涡流混合区进行混合,得到eudragitl100-55包裹的肠溶微胶囊(e-np),最终体系ph值为3.5~5.2。
ph值高于5.5时,eudragitl100-55处于溶解状态,与np复合后,无法形成微封装的颗粒。当ph小于5.5时,可以得到包裹np的微米级颗粒或沉淀。图8所示为体系不同ph值得到的e-np对np的包封率。ph值由5.2降低至3.5,e-np的包封率由97.7%降低至57.3%。这是由于随ph降低,eudragitl100-55电离程度降低,其带有的负电荷随之降低,对正电性np的结合程度下降,导致沉淀过程中未与eudragitl100-55结合的np增加,包封率下降。当ph=5.2时,具有最高的包封率,因此选择体系ph=5.2作为制备e-np的优选条件。
图9为不同eudragitl100-55浓度下e-np对np的包封率。当eudragitl100-55浓度高于0.9mg/ml时,e-np具有较高的包封率(~97%);当eudragitl100-55浓度低于0.9mg/ml时,随eudragitl100-55浓度降低至0.3mg/ml,e-np包封率由97%降低至75%。这是由于eudragitl100-55浓度较高时,相对于np过量,可以高效包裹np;eudragitl100-55浓度较低时,相对于np不足,不足以结合所有的np,导致包封率降低。因此制备过程中eudragitl100-55浓度应大于0.9mg/ml。
实施例2体外药物释放实验
利用ritc标记的exe在优选条件下制备nc、np及e-np三种制剂待用。将e-np制剂置于ph=2.5的hcl溶液中,在37℃,150rpm的摇床中孵育,每隔2h,10000rpm离心10min,取上清,测量其中exe浓度。图10为ph=2.5时,不同浓度尤特奇制备得到的e-np的exe释放曲线,由图可知,尤特奇浓度为0.9mg/ml,1.2mg/ml,1.5mg/ml时,2h内分别有50%、18%和7%的exe释放出来,由此说明当尤特奇浓度较低时,大量exe释放出来,无法有效保护exe通过胃部强酸环境,当尤特奇浓度升高至1.5mg/ml时,可以有效保护exe避免尾部酸性环境快速释放,最终选择尤特奇浓度1.5mg/ml作为优选条件。
取1ml的nc、np、e-np颗粒悬浮液,置于1ml的透析管(mw=50kda)中,封口。将透析管置于20ml10mm的pbs缓冲液(ph=7.4)中,在37℃,150rpm的摇床中孵育,每隔特定时间取出1ml缓冲液(随后用1ml的空白缓冲液补充,保持体系体积不变),通过荧光强度测量释放出的exe的浓度。图11所示为三种不同制剂在生理条件下的释放曲线。nc具有最快的释放速率,np由于有cs涂覆层的保护作用,exe释放速率小于nc,e-np表现出与np相近的释放速率,且24h后,有80%以上的exe释放量,由此说明,通过尤特奇涂覆得到的肠溶微胶囊制剂e-np可以在小肠环境中迅速崩解,快速释放出np。
实施例3载艾塞那肽纳米颗粒的小肠渗透性试验
利用ritc标记的exe在优选条件下制备nc、np两种制剂待用。对sd大鼠禁食12h,麻醉后,通过手术取出大鼠小肠的十二指肠、空肠、回肠部分各5cm长度,后将大鼠处死。将不同部位的小肠两端结扎,并注入1ml的exe溶液、nc颗粒悬浮液、np颗粒悬浮液,后将小肠置于10ml的krebsringer缓冲液中,37℃下,150rpm的摇床内透析,每隔0.5h,取出1ml的透析液,测量其中exe的浓度,并利用公式(3-3)计算得到不同exe制剂的表观渗透系数(pappvalue)。
其中,
如图12所示为np,nc和exe溶液在小肠二指肠、空肠、回肠的表观渗透系数(papp)值。np在十二指肠、空肠、回肠处均具有远远高于nc和exe溶液的表观渗透系数。np具有最高的小肠穿透效率,这是由于np表面涂敷了一层cs,正电性的cs增强了颗粒与粘液层的相互作用,提高了颗粒与小肠上皮细胞的亲和性,促进exe被小肠上皮细胞吸收;同时这层cs还具有可逆的打开小肠上皮紧密连接的能力,通过细胞旁路途径进一步促进exe运输穿过小肠。比较相同载药纳米颗粒在小肠不同部位的表观渗透系数可以发现,exe的穿透效率也依赖于小肠的部位。相同载药颗粒在十二指肠及空肠处,相比于回肠处具有更高的穿透效率,这可能是因为十二指肠及空肠具有较薄的粘液层(<300μm),颗粒可以更容易到达小肠上皮被吸收,而回肠部位粘液层较厚(~500nm),约为十二指肠及空肠部位的2倍,载药颗粒较难到达小肠上皮。
实施例4载艾塞那肽纳米颗粒的口服生物利用度
将200~250g的sd鼠分为4组,实验前所有实验组禁食10~12h。组1皮下注射exe溶液(50μg/kg),组2灌胃np颗粒悬浮液(500μg/kg),组3灌胃e-np颗粒悬浮液(500μg/kg),组4灌胃exe溶液(500μg/kg)。每隔特定时间,眼眶取血,利用艾塞那肽elisa试剂盒测量血液中exe的浓度,得到exe血药浓度与时间的曲线,如图13所示。皮下注射exe溶液组作为100%对照,通过比较口服颗粒组和皮下注射exe溶液组曲线下面积可以得到生物利用度。np的口服生物利用为:13.8%,e-np的口服生物利用为:15.1%。口服exe溶液组作为阴性对照,exe血药浓度接近于零。
实施例5利用fnc技术制备负载鲑鱼降钙素(sa)的纳米颗粒。
1、负载sa纳米核心(nsc)的制备
ta溶解于水中,浓度为0.1mg/ml,0.2mg/ml,0.3mg/ml,0.4mg/ml,0.5mg/ml,0.75mg/ml,1.0mg/ml;sa溶解于0.2%的hac溶液中,浓度为0.5mg/ml;壳聚糖(cs)溶解于0.2%的hac溶液中,浓度为0.2mg/m,0.3mg/ml,0.4mg/ml,0.5mg/ml,后用naoh溶液将ph值调至6.2;ta溶液分布于第一通道,sa溶液分布于第二通道,cs溶液分布于第三通道,第四通道为水,四个通道流速一致,调节通道流速为30ml/min,到达涡流混合区进行混合,得到ta/sa/cs的纳米颗粒(nsc)。
图14为不同ta浓度得到的nsc颗粒,由图可知,当ta浓度逐渐升高时,颗粒粒径逐渐增大。当ta浓度由0.1升高至0.75时,pdi逐渐减小(由0.6降低至0.2),表面电位为13~15mv之间,当ta浓度为1mg/ml时,电位降低至10mv左右,同时形成的颗粒为微米级颗粒。当ta浓度为0.4mg/ml时,颗粒具有较小的粒径和pdi,因此选择ta浓度为0.4mg/ml。
图15为改变壳聚糖浓度得到的nsc。由图可知,当cs浓度为0.2mg/ml时形成微米级颗粒。当cs浓度由0.3mg/ml升高至0.5mg/ml时,颗粒粒径逐渐减小,pdi处于0.2~0.25范围内,表面电位位于13~18mv范围内。当cs浓度为0.5mg/ml时,得到最小的粒径以及较小的pdi,因此选择cs浓度为0.5mg/ml。2、nsc表面涂覆透明质酸(ha)
ha(分子量为35kda)溶解于水中,浓度为0.1mg/ml,0.25mg/ml,0.5mg/ml,0.75mg/ml,1.0mg/ml;nsc颗粒悬浮液分布于第一,二通道,ha溶液分布于第三、四通道,四个通道流速一致,调节通道流速为30ml/min,到达涡流混合区进行混合,得到表面涂覆ha的颗粒(nsp)。
图16所示为不同ha浓度下得到的nsp,随ha浓度由1.0mg/ml降低至0.25mg/ml,nsca的粒径由116nm升高至205nm,pdi由0.13降低至0.06,表面电位由-25mv降低至-16mv,当ha浓度降低至0.1mg/ml时,粒径急剧增大至500nm以上,pdi也升高至约0.4,因此选择ha浓度为0.25mg/ml。
实施例6利用fnc技术制备载胰岛素(ins)的纳米颗粒
ta溶解于水中,浓度为0.3mg/ml,0.4mg/ml,0.5mg/ml;ins溶解于ph=2的hcl溶液中,后用naoh溶液调节ph值至4.0,浓度为0.5mg/ml;pvp溶解于25mmhepes缓冲溶液中,浓度为0.8mg/m、1.0mg/ml、1.2mg/ml,后用naoh溶液将ph值调至5.0~6.0;pvp分子量为3.5kda、10kda、40kda、360kda。ta溶液分布于第一通道,ins溶液分布于第二通道,pvp溶液分布于第三通道,第四通道为水,四个通道流速一致,调节通道流速为5ml/min,10ml/min,20ml/min,30ml/min,40ml/min,50ml/min,到达涡流混合区进行混合,得到ta/ins/pvp的纳米颗粒(nip),最终纳米颗粒的ph值为4.5~6.0。
图17所示为流速对nip粒径以及多分散性指数(pdi)的影响。ta浓度为0.5mg/ml,ins浓度为0.5mg/ml,pvp浓度为1mg/ml,最终颗粒溶液的ph值为5.2。当流速为5~20ml/min时,随流速升高,颗粒粒径逐渐减小,pdi逐渐减小。当流速位于20~50ml/min之间时,颗粒粒径及pdi随流速增大基本保持不变。当流速为40ml/min时,得到的颗粒最小且较为均一。选择流速40ml/min这一优选条件。
图18所示为ta浓度对nip的粒径、pdi、包封率影响,其中ins浓度固定为0.5mg/ml。随ta浓度由0.3mg/ml升高至0.5mg/ml,nip的粒径由111nm降低至100nm,pdi基本保持不变(0.15~0.16),包封率均大于95%。当ta浓度为0.5mg/ml时,粒径较小,且分布较为均一,选择ta浓度为0.5mg/ml作为优选条件。
图19所示为pvp分子量对nip粒径、多分散性指数(pdi)及包封率的影响。ta浓度为0.5mg/ml,ins浓度为0.5mg/ml,pvp浓度为1mg/ml,流速为30ml/min,ph值为5.2。当分子量由3.5kda增大为10kda时,nc粒径及pdi分别由164nm减小至100nm,0.21降低至0.16。当分子量由10kda增大至360kda,粒径及pdi分别由100nm增大至123nm,0.21增大至0.41。pvp分子量对nc包封率基本无影响,在实验范围内,包封率均高于90%。当pvp分子量为10kda时,得到的颗粒最小且较为均一。选择pvp分子量为10kda这一优选条件。
图20所示为pvp浓度对nip粒径、多分散性指数(pdi)及包封率的影响。ta浓度为0.5mg/ml,ins浓度为0.5mg/ml,流速为30ml/min,ph值为5.2,pvp分子量为10kda。随pvp浓度由0.8mg/ml升高至1.2mg/ml,nc粒径由129nm降低至97nm,pdi基本保持不变,包封率均高于90%。当pvp浓度为1.0mg/ml时,得到的颗粒最小且较为均一。选择pvp浓度为1.0mg/ml这一优选条件。
图21所示为最终所获纳米颗粒溶液的ph值对nip粒径、多分散性指数(pdi)及包封率的影响。ta浓度为0.5mg/ml,ins浓度为0.5mg/ml,pvp浓度为1mg/ml流速为30ml/min,pvp分子量为10kda。当ph值为4.8时,nc粒径约为500nm,pdi约为0.47。随ph由4.8升高至5.2,nc粒径由498nm降低至93nm,pdi由0.64降低至0.15,包封率基本保持不变,均高于90%。当ph为5.2时,得到的颗粒较小且较为均一。选择ph为5.2这一优选条件。
实施例7负载胰岛素纳米颗粒(nip)的体内降血糖效果评价
将链脲佐菌素(stz)诱导的糖尿病大鼠(sd鼠,200~220g)随机分为4组每组5只,实验前所有实验组禁食10~12h。组1皮下注射生理盐水,组2皮下注射胰岛素溶液(2iu/kg),组3皮下注射胰岛素纳米粒nip(2iu/kg),组4皮下注射皮下注射胰岛素纳米粒nip(4iu/kg)。每隔特定时间段,从尾静脉取血,利用血糖仪测量血糖水平,如图22所示。皮下注射生理盐水,糖尿病大鼠几乎无血糖波动。然后,皮下注射胰岛素溶液以及nip颗粒后,糖尿病大鼠具有显著降血糖现象,而且相比于皮下注射胰岛素溶液组,同等剂量(2iu/kg)的nip颗粒表现出更显著降糖效果,延长了对应的降血糖时间。此外我们发现进一步增加nip颗粒胰岛素注射剂量(4iu/kg)时,差异性更为明显。该实验结果表明胰岛素与单宁酸(ta)、pvp基于氢键络合作用形成的纳米粒可以保留胰岛素活性,同时具有更缓慢胰岛素释放及更高效降血糖效果。
- 还没有人留言评论。精彩留言会获得点赞!