高分散性碱土金属化合物微粉末、光学膜、图像显示装置和高分散性碱土金属化合物微粉末的制造方法以及微粉末分散性评价方法和微粉末分散性评价装置与流程
本发明涉及高分散性碱土金属化合物微粉末、光学膜、图像显示装置和高分散性碱土金属化合物微粉末的制造方法以及微粉末分散性评价方法和微粉末分散性评价装置,特别涉及表面附着有表面活性剂的高分散性碱土金属化合物微粉末、光学膜、图像显示装置和高分散性碱土金属化合物微粉末的制造方法以及微粉末分散性评价方法和微粉末分散性评价装置。
背景技术:
金属微粒被用于各种用途,可根据需要使其分散在溶剂中进行使用。例如针状的碳酸锶颗粒具有负的双折射性,作为光学用树脂填料,起到对具有正的双折射性的树脂的双折射进行控制的作用。因此,需要使碳酸锶颗粒高分散混配在树脂中,但为了提高树脂的透明性而使碳酸锶颗粒微细时,表面能增加,微粒之间容易发生凝聚,有损树脂的透明性,双折射的控制效果有可能降低。因此,需要利用表面活性剂对碳酸锶微粒的表面进行分散/被覆处理。
专利文献1中记载了一种有机无机复合颗粒,其在溶剂和/或树脂中能够以一次颗粒的形式分散,在无机颗粒的表面具有相互不同的两个以上有机基团。此外记载了:作为碳酸锶颗粒,平均粒径为200μm以下,优选为3nm~10μm。进一步,在实施例中记载了在分散液(溶剂为环己烷等)的状态下使用动态光散射光度计对碳酸锶颗粒的分散性进行测定;利用x射线衍射法(xrd)确认了颗粒为碳酸锶。
专利文献2中记载了一种用下述工序进行表面处理的碳酸盐微粒的表面处理方法,所述工序为:表面改性工序,用具有羧酸基的表面改性剂对碳酸盐微粒进行湿式处理;和分散工序,在分散剂存在下,利用分散机对改性后的微粒进行分散。对于碳酸盐微粒的尺寸,记载了由透射电子显微镜(tem)图像的投影面积估算的平均圆当量直径(heywood径)为80nm。此外记载了使用分散液(溶剂为乙醇),利用动态光散射法对碳酸锶微粒的分散性进行测定。
专利文献3中记载了一种碳酸锶微粒的制造方法,将氢氧化锶化合物的水溶液和氢氧化碱混合,向该混合液中导入二氧化碳。此外,对于碳酸锶,记载了用扫描型电子显微镜(sem)观测的长径为15~2000nm,在实施例中为50~500nm的范围内。
专利文献4中记载了一种方法,在碳酸锶微粒的表面预先用表面活性剂进行处理,所述表面活性剂包含亲水性基团和疏水性基团,进一步具有在水中形成阴离子的基团。该文献的实施例中记载了由电子显微镜图像测定的平均纵横比为2.70、长径的平均长度为110nm的针状碳酸锶粉末。并且记载了将该针状碳酸锶粉末分散在二氯甲烷中而得到的分散液的平均粒径为170nm。
现有技术文献
专利文献
专利文献1:日本特开2011-236111号公报(权利要求1、第0118段、实施例等)
专利文献2:日本特开2008-101051号公报(权利要求1、第0071、0098段等)
专利文献3:日本特开2007-001796号公报(权利要求1、6、第0029、0030段)
专利文献4:国际公开第2012/111692号(权利要求1、第0036、0037段)
技术实现要素:
发明所要解决的课题
为了评价碳酸锶微粒的分散性,需要利用将碳酸锶微粒制作时的干燥粉添加至有机溶剂中而得到的分散液对其粒度分布进行测定,或者利用电子显微镜(sem)照片进行观测,在评价上耗费工时。此外,在干燥粉末状态下没有方法判断表面活性剂是否完全被覆在各个一次颗粒的周围、是否可靠地进行分散处理、所添加的表面活性剂的量是否最合适。
此外,用表面活性剂进行表面处理后的现有的碳酸锶微粒分散在溶剂中时的分散性尚存在改善的余地,要求更高分散的碳酸锶微粒。
本发明的目的在于提供分散在溶剂中时具有高分散性的高分散性碱土金属化合物微粉末、光学膜、图像显示装置和高分散性碱土金属化合物微粉末的制造方法。
此外,本发明的目的在于提供即使不使碱土金属化合物微粉末实际分散在溶剂中也能够在粉末状态下对使碱土金属化合物微粉末分散在溶剂中时的分散性进行评价的碱土金属化合物微粉末分散性评价方法和碱土金属化合物微粉末分散性评价装置。
用于解决课题的手段
为了实现上述目的,本发明人进行了深入研究,结果发现,根据在粉末状态下利用小角x射线散射法对碱土金属化合物微粉末进行测定的结果,能够推测将该微粉末分散在溶剂中时的分散性。进一步利用该小角x射线散射法进行评价,结果确认到能够制作与以往相比为高分散的微粉末,从而完成了本发明。此外,本发明人发现,根据在粉末状态下利用小角x射线散射法对碱土金属化合物微粉末进行测定的结果,能够推测将该微粉末分散在溶剂中时的分散性,从而完成了本发明。
即,本发明为一种高分散性碱土金属化合物微粉末,该高分散性碱土金属化合物微粉末以碱土金属作为主要成分,在平均长径为10~100nm、平均纵横比为1.0~5.0的范围内的碱土金属化合物微粒的表面附着有表面活性剂,其特征在于,利用小角x射线散射法照射波长0.154nm的x射线进行测定,在散射角2θ为0.2~1.0°的范围内,在散射强度中具有散射峰。
此外,优选的是,上述散射角2θ为0.4~0.7°的范围内,且根据该散射角2θ,由布拉格公式求出的颗粒间距离d的计算值为12.6~22.1nm的范围内。
此外,在上述中,优选的是,上述碱土金属化合物为碱土金属碳酸盐。进一步,该情况下,优选的是,上述碱土金属碳酸盐为碳酸锶。
进一步,优选的是,上述表面活性剂包含亲水性基团和疏水性基团,进一步具有在水中形成阴离子的基团。
这些情况下,优选的是,在上述碱土金属化合物微粒的一次颗粒的周围均匀涂布上述表面活性剂,上述碱土金属化合物微粒等间隔地排列。
此外,本发明为一种光学膜,其特征在于,上述中任一项所述的高分散性碱土金属化合物微粉末分散在树脂中。
该情况下,优选的是,上述树脂为选自由聚碳酸酯、聚甲基丙烯酸甲酯、纤维素酯、聚苯乙烯、苯乙烯丙烯腈共聚物、聚富马酸二酯、聚芳酯、聚醚砜、聚烯烃、马来酰亚胺系共聚物、聚对苯二甲酸乙二醇酯、聚萘二甲酸乙二醇酯、聚酰亚胺、聚酰胺、聚氨酯组成的组中的一种以上。
进一步,本发明为一种图像显示装置,其特征在于,具备上述中任一项所述的光学膜。
此外,本发明为一种高分散性碱土金属化合物微粉末的制造方法,其为上述高分散性碱土金属化合物微粉末的制造方法,其特征在于,该制造方法包括:分散工序,在表面活性剂的存在下,对平均长径为10~100nm的范围内的碱土金属化合物微粒分散在水性溶剂中而成的第一分散液提供剪切力,由此使上述碱土金属化合物微粒的一次颗粒分散在上述水性溶剂中,同时使该一次颗粒与上述表面活性剂接触,从而得到第二分散液;和干燥工序,使上述第二分散液在100~300℃的温度进行加热干燥而制成粉末状。
即,本发明为一种微粉末分散性评价方法,该微粉末分散性评价方法是在粉末状态下对使碱土金属化合物微粉末分散在溶剂中时的分散性进行评价,其特征在于,该评价方法具有:x射线照射工序,利用小角x射线散射法对碱土金属化合物微粉末照射x射线,得到规定范围的散射角下的散射强度的光谱;散射强度分析工序,根据上述光谱,分析在散射角2θ为0.2~1.0°的范围内是否具有散射强度的散射峰;和分散性推定工序,基于上述散射峰的检测结果,推定上述溶剂中的上述碱土金属化合物微粉末的分散性。
此外,优选的是,上述分散性推定工序在检测出上述散射峰的情况下推定为上述溶剂中的上述碱土金属化合物微粉末的分散性相对较高。
进一步,优选的是,上述散射角2θ为0.4~0.7°的范围内。
此外,本发明为一种微粉末分散性评价装置,该微粉末分散性评价装置在粉末状态下对碱土金属化合物微粉末分散在溶剂中时的分散性进行评价,其特征在于,该评价装置具有:x射线照射单元,利用小角x射线散射法对碱土金属化合物微粉末照射x射线,得到规定范围的散射角下的散射强度的光谱;散射强度分析单元,根据上述光谱,分析散射角2θ为0.2~1.0°的范围内是否具有散射强度的散射峰;和分散性推定单元,基于上述散射峰的检测结果,推定上述溶剂中的上述碱土金属化合物微粉末的分散性。
此外,优选的是,上述分散性推定单元在检测出上述散射峰的情况下推定为上述溶剂中的上述碱土金属化合物微粉末的分散性相对较高。
进一步,优选的是,上述散射角2θ为0.4~0.7°的范围内。
发明效果
根据本发明,能够提供分散在溶剂中时具有高分散性的高分散性碱土金属化合物微粉末及其制造方法。
此外,根据发明,能够提供即使不使碱土金属化合物微粉末实际分散在溶剂中,也能够在粉末状态下对碱土金属化合物微粉末分散在溶剂中时的分散性进行评价的碱土金属化合物微粉末分散性评价方法和碱土金属化合物微粉末分散性评价装置。
附图说明
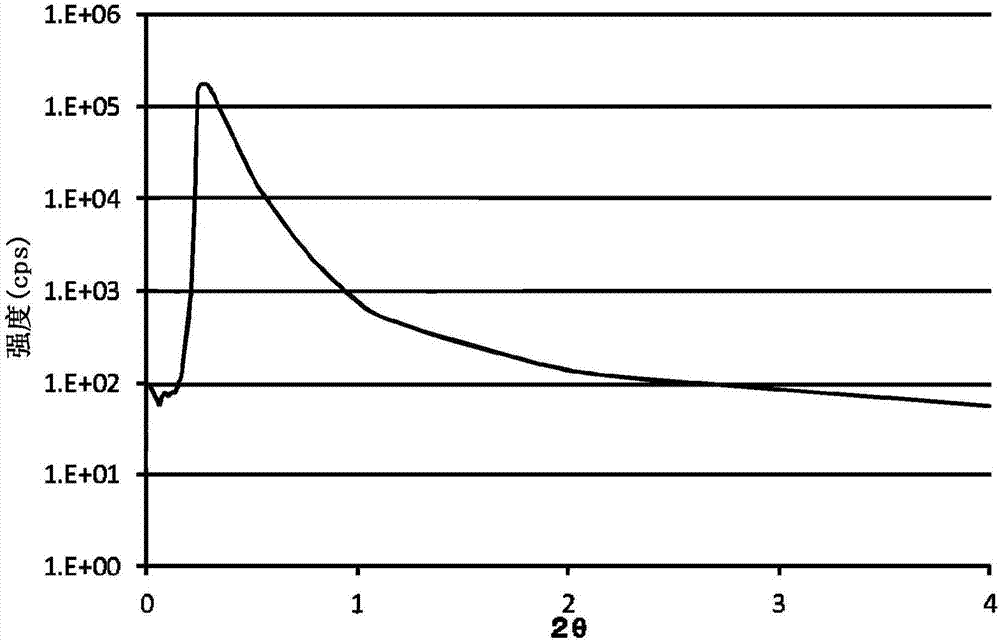
图1为示出利用小角x射线散射法测定的高分散性碳酸锶微粉末的光谱的示意图。
图2为示出分散性低的碳酸锶微粉末的光谱的示意图。
图3为示出微粉末分散性评价方法的流程的流程图。
图4为微粉末分散性评价装置的示意性构成图。
具体实施方式
1.高分散性碱土金属化合物微粉末
本发明的高分散性碱土金属化合物微粉末以碱土金属化合物作为主要成分,用表面活性剂对平均长径为10~50nm、平均纵横比为1.0~5.0的范围内的碱土金属化合物微粒进行表面处理而得到。即为在碱土金属化合物微粒的表面附着有表面活性剂,分散在溶剂时具有高分散性的高分散性碱土金属化合物微粉末。
(1)碱土金属化合物微粒(表面处理前)
用表面活性剂进行表面处理之前的碱土金属化合物微粒的平均长径为10~100nm的范围内,优选为15~40nm的范围内,更优选为20~30nm的范围内。平均长径低于10nm时,颗粒过小而容易发生凝聚,分散性容易劣化。另一方面,平均长径超过50nm时,颗粒过大,混合在树脂中时,透明性容易劣化。
此处,平均长径可以利用对碱土金属化合物微粒的扫描型电子显微镜(sem)照片进行目视或自动进行图像处理的方法进行测定。碱土金属化合物微粒的长径可以如下测定,将碳酸锶颗粒等碱土金属化合物颗粒视为长方形时,以长度方向的长度(长边的长度)计进行测定。此外,碱土金属化合物颗粒的短径可以如下测定,将碱土金属化合物颗粒视为长方形时,以宽度方向的长度(短边的长度)计进行测定。具体而言,对与图像的碱土金属化合物颗粒外接的面积最少的长方形进行计算,由该长边和短边的长度,求出长径和短径。进一步,“平均”是指,对统计学上有可靠性的个数(n个)的碱土金属化合物进行测定而得到的平均值,作为该个数,通常为300个以上,优选为500个以上,更优选为1000个以上。
碱土金属化合物微粒的平均纵横比为1.0~5.0的范围内,优选为2.0~4.5的范围内,特别优选为2.5~4.0的范围内。平均纵横比超过5.0时,微粒过于细长,容易弯折,从而容易导致粒径分布的劣化等。此外,纵横比过小的情况下,在双折射的控制上有时无法发挥效果。
需要说明的是,此处所说的纵横比是指颗粒的“长径/短径”。此外,平均纵横比是指纵横比的平均值,对一个颗粒的纵横比进行测定,计算出两个以上颗粒的平均值。
作为碱土金属化合物微粒的示例,可以举出碱土金属的碳酸盐、硫酸盐、硝酸盐、氧化物、氯化物、氢氧化物等。此外,作为碱土金属,可以举出钙、锶、钡、镭等。因此,作为碱土金属化合物微粒的示例,可以举出碳酸钙微粒、碳酸锶微粒、碳酸钡微粒等。这些之中,从在光学用树脂填料的用途等中控制双折射的观点出发,优选碳酸锶微粒。
(2)表面活性剂
本发明中使用的表面活性剂附着在碱土金属化合物微粒的表面,具有提高其在溶剂中的分散性的功能。作为表面活性剂的种类,没有特别限定,优选阴离子型表面活性剂。其中,特别优选包含亲水性基团和疏水性基团,进一步具有在水中形成阴离子的基团的化合物。亲水性基团优选包含碳原子数为1~8的氧化烯基的聚氧亚烷基。疏水性基团优选烷基或者芳基。烷基和芳基可以具有取代基。烷基的碳原子数通常为3~30的范围内,优选为10~18的范围内。芳基的碳原子数通常为6~30的范围内。在水中形成阴离子的基团优选为选自由羧酸基(-cooh)、硫酸基(-oso3h)、磷酸基(-opo(oh)2、-opo(oh)o-)组成的组中的酸基。这些酸基的氢原子可以用钠、钾等碱金属离子或铵离子取代。
这些之中,从碱土金属化合物微粒在溶剂中的分散性良好的方面出发,优选多元羧酸系的阴离子型表面活性剂、多磷酸系的阴离子型表面活性剂、非离子型表面活性剂的任一种以上。
作为多元羧酸系的阴离子型表面活性剂,可以举出下式(i)所表示的化合物。
[化1]
(其中,r1表示取代或未取代的烷基或者取代或未取代的芳基,e1表示碳原子数为1~8的范围内的亚烷基,a表示1~20的范围内、优选为2~6的范围内的数。需要说明的是,优选的是,r1为碳原子数为10以上、优选为10~18的范围内的烷基)
作为多磷酸系的阴离子型表面活性剂,可以举出下式(ii)所表示的化合物(单体)、或者下式(iii)所表示的化合物(双体)、或者式(ii)与式(iii)的混合物。
[化2]
(其中,r2表示取代或未取代的烷基或者取代或未取代的芳基,e2表示碳原子数为1~8的范围内的亚烷基,b表示1~20的范围内、优选为2~6的范围内的数。需要说明的是,优选的是,r2为碳原子数为10以上、优选为10~18的范围内的烷基)
[化3]
(其中,r3与r4可以相同或者也可以不同,表示取代或未取代的烷基或者取代或未取代的芳基,e3与e4可以相同或者也可以不同,表示碳原子数为1~8的范围内的亚烷基,c和d分别表示1~20的范围内、优选为2~6的范围内的数。需要说明的是,优选的是,r3与r4均为碳原子数为10以上、优选为10~18的范围内的烷基)
作为非离子型表面活性剂,可以举出下式(iv)所表示的化合物。
[化4]
(其中,r5表示取代或未取代的烷基或者取代或未取代的芳基,e5、e6均表示碳原子数为1~8的范围内的亚烷基,e、f均表示1~20的范围内、优选为2~6的范围内的数。需要说明的是,优选的是,r5为碳原子数为10以上、优选为10~18的范围内的烷基)
相对于碱土金属化合物微粉末,表面活性剂可以单独使用,也可以混合两种以上使用。此外,表面活性剂在碱土金属化合物微粉末的表面上可以仅附着一层,也可以附着两层以上。附着两层以上的情况下,可以在各层使用相同的表面活性剂,也可以在各层使用不同的表面活性剂。需要说明的是,使用傅里叶变换红外分光测定装置(ft-ir)对颗粒表面的红外吸收光谱进行测定,由此能够确认碱土金属化合物微粉末的表面是否附着有表面活性剂。
(3)小角x射线散射测定法的散射峰
本发明的高分散性碱土金属化合物微粉末的特征在于,利用小角x射线散射(saxs)法照射波长0.154nm的x射线进行测定的情况下,进行峰值搜索,结果在散射角2θ为0.2~1.0°的范围内具有散射峰。
此处,小角x射线散射法为下述方法:向物质照射x射线,对散射的x射线中出现在2θ=10°以下的低角区域的光谱进行测定,从而对物质的结构进行评价。小角x射线散射法通常用于评价1~100nm左右的分子水平的周期性、取向性。
基于saxs法的测定可以使用从物质的分子水平的结构(1~100nm的宏观结构)至原子水平的结构(0.2~1nm的微观结构)均能够评价的x射线结构评价装置(小角x射线散射测定装置)来进行。通过进行小角x射线散射测定,能够对下述(1)~(3)的项目等进行解析、分析。
(1)形状、尺寸(粒径)的测定“漫散射”:分子的形状、尺寸、粒径、空隙、
(2)结晶性周期结构的测定“干涉性散射、基于有无散射强度的峰”:层状(片状)结构的周期性、
(3)物质的不均匀结构的测定“干涉性散射”:相变化、固溶体-金属的不均匀结构。
图1为示出用小角x射线散射法测定的高分散性碳酸锶微粉末的光谱的示意图,图2为示出分散性低的碳酸锶微粉末的光谱的示意图。任一图中均是纵轴表示散射强度,横轴表示散射角2θ。如图1所示,可知:在本发明的高分散性碱土金属化合物微粉末中,在散射角2θ为0.2~1.0°的范围内,在散射强度中具有散射峰(极大)(图中的虚线圈围出的部分)。另一方面,如图2所示,可知在分散性低的碳酸锶微粉末中,在散射角2θ为0.2~1.0°的范围内,在散射强度中不具有散射峰。
对于小角x射线散射测定法的散射峰,在后述的“3.微粉末分散性评价方法和微粉末分散性评价装置”中进行详细说明。
(4)颗粒间距离d
高分散性碱土金属化合物微粉末优选根据上述散射角2θ由布拉格公式求出的颗粒间距离d的计算值为14~20nm的范围内。
此处,布拉格公式是指下式(1)。
2dsinθ=nλ…式(1)
(其中,d表示颗粒间距离(也称为“晶面间距”),θ表示上述散射角,n表示整数,λ表示x射线的波长)
据认为颗粒间距离d低于14nm的情况下,颗粒之间的凝聚力增高。或者表面活性剂的添加量过少,颗粒之间过于接近,使其分散在溶剂中时不易分散。另一方面,颗粒间距离d超过21nm的情况下,表面活性剂的添加量过多,或者颗粒之间过于分离,使其分散在溶剂中时,分散性有时会变差。
目前,根据有机溶剂分散液的动态光散射来测定粒度分布的情况下,颗粒越小,则布朗运动越大,从而发生多重散射而难以测定精确的粒度。但是,在本发明中,在散射角2θ为0.2~1.0°的范围内存在散射峰,因此在微粉末的状态下,颗粒之间保持某一恒定周期的间隔进行排列。即,利用表面活性剂对一次颗粒实施完全的被覆处理。并且,将这样状态的微粉末添加至溶剂中时,一次颗粒完全进行表面处理,因此颗粒容易分散在溶剂中。对于有无在微粉末的状态下测定的上述散射峰进行评价,由此能够预测使其分散在溶剂中时的分散性。因此,能够在干燥微粉末的状态下评价分散性,而无需如以往那样将微粉末实际分散在溶剂中,在分散液的状态下测定粒度分布,从而能够降低评价工时。
如上所述,本发明的高分散性碱土金属化合物微粉末具有散射峰,但为了满足这样的条件,除了表面活性剂的种类、浓度之外,高分散性碱土金属化合物微粉末的制造条件等也会有影响。以下对高分散性碱土金属化合物微粉末的制造方法进行说明。
2.高分散性碱土金属化合物微粉末的制造方法
本发明的高分散性碱土金属化合物微粉末的制造方法的特征在于,具备:分散工序,获得将平均长径为10~100nm的范围内的碱土金属化合物颗粒分散在水性溶剂中而成的分散液(第一分散液),在表面活性剂的存在下,提供剪切力,由此使上述碱土金属化合物颗粒的一次颗粒分散在水性溶剂中,同时使该一次颗粒与表面活性剂接触,由此得到分散液(第二分散液);和干燥工序,使该第二分散液在100~200℃的温度进行加热干燥而制成粉末状。对于进行表面处理之前的碱土金属化合物微粉末的制造方法没有特别限定,可以举出使成为原料的碱土金属化合物反应而生成水性浆料,并使其熟化的方法。
以下,作为高分散性碱土金属化合物微粉末的一例,对制造高分散性碳酸锶微粉末的工序进行说明。
(1)反应工序
该工序如下进行,一边对成为原料的氢氧化锶的水溶液或者水性悬浮液(以下称为水性浆料)进行搅拌,一边在晶体生长抑制剂的存在下导入二氧化碳气体,使氢氧化锶发生碳氧化,由此制造纵横比低的球状碳酸锶微粒。对于水性浆料所含的氢氧化锶的浓度没有特别限制,通常为1~20质量%的范围,优选为2~18质量%的范围,更优选为3~15质量%的范围。
晶体生长抑制剂优选羧基数量为两个且羟基与这些羧基的总量为3~6个的有机酸。作为晶体生长抑制剂的优选实例,可以举出酒石酸、苹果酸和丙醇二酸。作为晶体生长抑制剂,可以使用具有羟基和两个羧基且以总量计具有至少3个基团的有机酸,但从附着在制造的颗粒的表面,控制粒成长,保持微细的状态提高分散性的方面出发,更优选上述分子内含有一个以上羟基的二羧酸或其酸酐,特别优选dl-酒石酸。相对于氢氧化锶100质量份,晶体生长抑制剂的用量通常为0.1~20质量份的范围,优选为1~10质量份的范围。
相对于氢氧化锶1g,二氧化碳气体的流量通常为0.5~200ml/分钟的范围,优选为0.5~100ml/分钟的范围。通过反应工序,能够得到例如平均纵横比小于1.5、近似球状的微细的球状碳酸锶微粒。需要说明的是,球状碳酸锶微粒的制造方法记载于国际公开第2011/052680号中。
(2)熟化工序
熟化工序是使含有由反应工序得到的球状碳酸锶微粒的水性浆料在规定的温度、时间熟化,使其晶粒生长为针状的碳酸锶微粒的工序。熟化工序可以在温水中进行。熟化温度为75~115℃的范围内,优选为80~110℃的范围内,特别优选为85~105℃的范围内。熟化温度低于75℃时,球状碳酸锶微粒的晶体生长不充分,存在平均纵横比过低的倾向,超过115℃时,球状碳酸锶微粒的短径的晶体生长被促进,存在纵横比变低的倾向。此外,对于熟化时间没有特别限定,通常为1~100小时的范围内,优选为5~50小时的范围内,特别优选为10~30小时的范围内。
需要说明的是,上述“(1)反应工序”和“(2)熟化工序”是从作为原料的氢氧化锶得到针状的碳酸锶微粒的工序,能够获得作为市售品的碳酸锶微粒的情况下,可以利用市售品。
(3)表面处理工序
表面处理工序为下述工序:对平均长径为10~100nm的范围内的碳酸锶微粒分散在水性溶剂中而得到的分散液提供剪切力,由此使一次颗粒分散,同时使其与表面活性剂接触而得到高分散性碳酸锶。作为表面活性剂,可以使用上述表面活性剂。
对于表面处理工序中使用的分散液,进行熟化工序的情况下,可以使用熟化工序后的水性浆料,使用市售的碳酸锶微粒的情况下,可以使用将其分散在水溶液中而得到的分散液。表面处理工序可以一边施加剪切力一边向分散液中添加表面活性剂来进行。水性浆料中的碳酸锶颗粒的含量优选为1~30质量%的范围。对于水性浆料中的表面活性剂的投入量,表面活性剂的添加总量通常为1~60质量%的范围内,优选为10~50质量%的范围内,更优选为20~40质量%的范围内。剪切力的提供可以使用搅拌桨叶混合机、均混器、磁力搅拌器、空气搅拌器、超声波均化器、clearmix、filmix等公知的搅拌装置来进行。
使用两种以上表面活性剂进行表面处理的情况下,相对于水性浆料中的碳酸锶颗粒100质量份,水性浆料中的各表面活性剂的投入量通常为1~40质量份的范围,优选为3~30质量份的范围。表面活性剂可以同时投入或逐次投入。
(4)干燥工序
干燥工序是在100~300℃的范围内的温度对上述“(3)表面处理工序”中得到的水性浆料进行加热干燥而得到高分散性碳酸锶微粉末的干燥物的工序。干燥温度低于100℃时,干燥容易不充分,干燥温度超过200℃时,容易产生碳酸锶微粉末的破损等。干燥温度优选为110~180℃的范围内,更优选为120~160℃的范围内。干燥工序可以利用使用喷雾干燥机、鼓式干燥机和盘式干燥机等热干燥机的公知的干燥方法来进行。
本发明的高分散性碱土金属化合物微粉末分散在溶剂中时的分散性优异,因此可以用于各种用途。例如高分散性的碳酸锶微粉末可以出于抑制双折射或者反之出于有意地表现双折射的目的而进行使用。作为这样的用途,可以举出液晶显示装置的光学膜。作为光学膜,可以举出保护膜、防反射膜、亮度提高膜、棱镜膜、视角改善膜、相位差膜等。作为膜的树脂材料,可以举出三乙酰纤维素、聚对苯二甲酸乙二醇酯、聚环状烯烃、聚碳酸酯、聚甲基丙烯酸甲酯等。光学膜可以通过将碳酸锶微粉末作为填充剂混合在树脂材料中,利用溶液流延制膜法、熔融挤出法等公知的方法进行制膜,进一步可以根据需要通过进行拉伸处理来制造。
作为使本发明的高分散性碱土金属化合物微粉末分散的溶剂,没有特别限定,可以根据用途等适当选择。作为这样的溶剂的种类,没有特别限定,可以根据树脂的性质等适当选择。作为溶剂的示例,优选有机溶剂,作为有机溶剂的示例,可以举出醇(例如乙醇、1-丙醇、2-丙醇、1-丁醇、乙二醇)、二氯甲烷、nmp、四氢呋喃、mek、乙酸乙酯、环己烷、甲苯等。这些有机溶剂中,特别优选二氯甲烷。除了仅使用上述一种以外,还可以组合两种以上使用。
3.微粉末分散性评价方法和微粉末分散性评价装置
以下,参照图3、图4对本发明的微粉末分散性评价方法和微粉末分散性评价装置进行说明。图3为示出本发明的微粉末分散性评价方法的流程的流程图。如该图所示,本发明的微粉末分散性评价方法是在粉末状态下对碱土金属化合物微粉末分散在溶剂中时的分散性进行评价的微粉末分散性评价方法,具有:x射线照射工序(s1),利用小角x射线散射法对碱土金属化合物微粉末照射x射线,得到规定范围的散射角下的散射强度的光谱;散射强度分析工序(s2),根据该光谱,对在散射角2θ为0.2~1.0°的范围内是否具有散射强度的散射峰进行分析;和分散性推定工序(s3),基于散射峰的检测结果,推定溶剂中的碱土金属化合物微粉末的分散性。
图4为示出本发明的微粉末分散性评价装置的一实施方式的框图。如该图所示,微粉末分散性评价装置1主要的构成要素有:光源控制部11、x射线照射部12、x射线检测部13、光谱生成部14、散射峰检测部15、分散性推定部16。
光源控制部11、x射线照射部12、x射线检测部13、光谱生成部14是实施本发明的x射线照射工序(s1)的单元,散射峰检测部15是实施本发明的散射强度分析工序(s2)的单元,分散性推定部16是实施本发明的分散性推定工序(s3)的单元。以下对各构成进行详细说明。
光源控制部11是在x射线照射部12中用于控制x射线的照射量等的单元。x射线照射部12是用于对试样s照射x射线的单元。x射线照射部12具有用于照射x射线的x射线管球(未图示)。x射线管球是生成例如cu的kα射线的管球,接收来自光源控制部11的控制信号,照射波长1nm以下的x射线。
试样s载置于未图示的工作台上,按照被来自x射线照射部12的x射线照射的方式进行配置。照射至试样s的x射线的一部分发生散射。x射线检测部13是用于对从试样s散射的x射线中的小角度x射线(小角x射线)的强度(散射强度)进行检测的单元。对于x射线检测部13检测的小角x射线没有特别限定,通常2θ为10°以下。载置试样s的工作台可以变更倾斜角度,通过一边变更工作台的倾斜角度一边照射x射线,能够变更散射角2θ来测定散射强度。x射线检测部13检测的小角x射线在光谱生成部14中,以散射强度与2θ的光谱的形式进行记录。
散射峰检测部15是根据在光谱生成部14生成的光谱的轮廓来分析散射角2θ为0.2~1.0°的范围内是否具有散射强度的散射峰的单元。分散性推定部16为下述单元:在散射峰检测部15检测出散射峰的情况下,判断试样s的分散性高,在未检测出峰的情况下,判断试样s的分散性低。
光谱生成部14、散射峰检测部15、分散性推定部16可以使用例如中央运算装置(cpu)、存储装置(硬盘、存储器)等公知的硬件和存储在存储装置中的软件而构成。
光谱生成部14可以由例如将从x射线检测部13发送的x射线强度的信号以散射强度和2θ的轮廓的形式存储在存储装置中的软件和实行该过程的中央运算装置来实现。由此,从x射线检测部13逐次发送的散射强度的数据与由工作台的角度计算出的散射角2θ的信息一起以光谱数据形式存储在存储装置中,同时可以显示在未图示的显示装置等。
散射峰检测部15可以由根据光谱生成部14中生成的光谱判别2θ为0.2~1.0°的范围内是否存在散射强度的极大值的软件和实行该过程的中央运算装置来实现。作为软件,可以利用通过微分法计算出极大值的公知的算法等。由此,能够自动检测出散射角2θ为0.2~1.0°的范围内是否存在散射强度的散射峰。
分散性推定部16可以由报知检测出散射峰的情况下分散性高、未检测出散射峰的情况下分散性低的至少任一方的软件和实行该过程的中央运算装置来实现。例如检测出散射峰的情况下,可以在画面上显示分散性高,反之未检测出散射峰的情况下,可以在画面上显示散射性低。
以下,对光谱的散射峰与分散性的关系进行说明。如上述图1所示,可知:在分散性高的碳酸锶微粉末中,在散射角2θ为0.2~1.0°的范围内,在散射强度中具有散射峰(极大)(图中的虚线圈围出的部分)。另一方面,如图2所示,可知在分散性低的碳酸锶微粉末中,在散射角2θ为0.2~1.0°的范围内,在散射强度中不具有散射峰。
即,进行散射角2θ下的峰值搜索,结果在0.2~1.0°的范围内检测出散射峰,这是指微粒之间保持某一恒定的距离、以恒定周期规则准确地进行排列。
目前,根据有机溶剂分散液的动态光散射测定粒度分布的情况下,颗粒越小,则布朗运动越大,从而发生多重散射而难以测定精确的粒度。但是,在本发明中,判别在散射角2θ为0.2~1.0°的范围内是否存在散射峰,由此在微粉末的状态下颗粒之间保持某一恒定周期的间隔的状态进行排列。并且,将这样状态的微粉末添加至溶剂中时,在溶剂中,颗粒容易分散。通过评价有无在微粉末的状态下测定的上述散射峰,能够预测分散在溶剂中时的分散性。因此,能够在干燥微粉末的状态下评价分散性,而无需如以往那样将微粉末实际分散在溶剂中,在分散液的状态下测定粒度分布,从而能够降低评价的工时。
需要说明的是,散射峰的高度越高,则可以评价分散性越高。此处,作为散射峰的高度,可以为分散强度的值(绝对值),也可以为散射峰的散射强度相对于规定的2θ(例如θ=0°)的散射强度的比例(相对值)。通过如此进行,不仅能够推测分散性的好坏,分散性好的情况下,还能够推测其分散度。
此外,可以预先测定各种碱土金属化合物微粉末、将它们实际分散在各种溶剂中而测定的分散性的指标(例如粉末的润湿性等)和散射峰的散射强度,将其结果(碱土金属化合物微粉末的种类、溶剂的种类、分散性的指标、散射强度)注册在数据库中。并且,可以从数据库中选择成为测定对象的碱土金属化合物微粉末和溶剂,根据测定得到的光谱的分散峰的分散强度的值,自动提取分散性的指标的数值。通过如此进行,根据碱土金属化合物微粉末的种类和溶剂的种类,以分散性作为具体的指标,能够更精确地进行评价。
如使用图4所示的微粉末分散性评价装置的情况那样,本发明的微粉末分散性评价方法也可以用一个装置进行x射线照射工序(s1)、散射强度分析工序(s2)、分散性推定工序(s3),但可以人工进行一部分工序,或者可以用其它装置进行一部分工序。
在人工进行一部分工序的情况下,例如对于x射线照射工序(s1)和散射强度分析工序(s2),也可以使用通常市售的小角x射线散射装置自动检测出散射峰,对于分散性推定工序(s3),也可以根据有无散射峰的结果,人工进行判断。
此外,用其它装置进行一部分工序的情况下,例如对于x射线照射工序(s1),也可以使用通常市售的小角x射线散射装置取得光谱数据,对于散射强度分析工序(s2)和分散性推定工序(s3),也可以根据用小角x射线散射装置得到的光谱数据,使用通常的个人计算机等,实行散射峰的检测和分散性的推定。
进一步,本发明优选根据散射角2θ,由布拉格公式计算出颗粒间距离d。在此,布拉格公式是指下式(1)。
2dsinθ=nλ…式(1)
(其中,d表示颗粒间距离(也称为“晶面间距”),θ表示上述散射角,n表示整数,λ表示x射线的波长)
据认为若颗粒间距离d小,则颗粒之间的凝聚力高。即,颗粒之间过于接近,或者附着在颗粒表面的表面活性剂的附着量过少,则使其分散在溶剂中时难以分散。另一方面,若颗粒间距离d大,则颗粒之间过于分离,规则性变低,或者颗粒表面的表面活性剂的附着量过多,使其分散在溶剂中时分散性容易变差。如此,通过测定颗粒间距离d,能够评价分散性的好坏。分散性也根据碱土金属化合物微粒的种类而不同,但在颗粒的长径为50nm以下的纳米颗粒中,优选的颗粒间距离d通常为10nm~30nm的范围内。需要说明的是,在未完全实施表面处理的纳米颗粒中,在本发明的测定中,未观测到散射峰,无法求出d值。
4.光学膜
本发明的高分散性碱土金属化合物微粉末与树脂混合而形成树脂组合物,通过将其成膜,能够制成光学膜。以下对光学膜进行说明。
作为树脂组合物所含的树脂,只要为通常光学膜中所使用的树脂,则没有特别限定,可以根据目的选择各种树脂。作为这样的树脂,可以举出选自由聚碳酸酯、聚甲基丙烯酸甲酯、三乙酰纤维素等纤维素酯、聚苯乙烯、苯乙烯丙烯腈共聚物、聚富马酸二酯、聚芳酯、聚醚砜、聚环状烯烃等聚烯烃、马来酰亚胺系共聚物、聚对苯二甲酸乙二醇酯、聚萘二甲酸乙二醇酯、聚酰亚胺、聚酰胺、聚氨酯组成的组中的一种以上。
碱土金属化合物微粉末相对于树脂组合物整体的含量优选为0.1~50质量%的范围内。碱土金属化合物微粉末的含量低于0.1质量%时,基于碱土金属化合物微粉末的双折射控制效果变得过小。反之,碱土金属化合物微粉末的含量超过50质量%时,碱土金属化合物微粉末相对于树脂的比例变得相对过大,因此成膜出的膜的透明性变差。碱土金属化合物微粉末相对于树脂组合物整体的含量更优选为0.5~40质量%的范围内,特别优选为1~35质量%的范围内。
通过将上述树脂和碱土金属化合物微粉末混合,能够制成树脂组合物。碱土金属化合物微粉末与树脂的混合可以利用使用超声波均化器、搅拌叶片、液体喷射式粉碎机的方法等公知的方法进行。
此外,可以调整将树脂组合物和适当的溶剂混合而得到的掺杂溶液来进行光学膜的成膜。作为这样的溶剂的种类,没有特别限定,可以根据树脂的性质等适当选择使用。作为溶剂的示例,优选有机溶剂,作为有机溶剂的示例,可以举出醇(例如乙醇、1-丙醇、2-丙醇、1-丁醇、乙二醇)、二氯甲烷、nmp、四氢呋喃、mek、乙酸乙酯、环己烷、甲苯等。这些有机溶剂中,特别优选二氯甲烷。不仅可以使用上述的一种,也可以组合两种以上使用。
树脂相对于溶剂的比例以质量比计优选为1:10~10:1的范围内。对于掺杂溶液,可以将树脂和溶剂混合制成树脂混合溶液,向其中添加碱土金属化合物微粉末并混合,或者可以将碱土金属化合物微粉末和溶剂混合制成粉末混合溶液,向其中添加树脂并混合。进一步,可以分别准备上述的树脂混合溶液和粉末混合溶液,将两者混合而制成掺杂溶液。碱土金属化合物微粉末、树脂和溶剂可以用使用超声波均化器、搅拌叶片、液体喷射式粉碎机的方法等公知的方法进行混合。
树脂组合物、掺杂溶液可以用公知的方法进行成膜而制成光学膜。作为成膜方法,可以举出上述的熔融挤出成膜法、溶液流延成膜法等公知的成膜法。熔融挤出成膜法是将树脂组合物加热熔融而制成熔融物,将其在支撑体上呈膜状流延而进行冷却固化的方法。此外,溶液流延成膜法是将掺杂溶液在支撑体上流延而使溶剂蒸发来进行膜化的方法。
根据树脂的种类,有时在成膜时在树脂溶液中产生对流而形成贝纳德漩涡结构。贝纳德漩涡结构形成时,碱土金属化合物微粉末凝聚,光学膜的透明性劣化。此外,由于该凝聚,基于碱土金属化合物微粉末的双折射调整作用降低。因此,出于提高与支撑体的润湿性,抑制贝纳德漩涡的形成的目的,优选在树脂组合物、掺杂溶液中添加表面改性剂。使用聚碳酸酯作为树脂的情况下,容易形成贝纳德漩涡,因此添加表面改性剂所带来的透明性提高等效果显著。作为表面改性剂,可以举出乙烯基系表面活性剂、氟系表面活性剂、硅油等。
成膜后的膜可以根据用途等进行适当拉伸。作为拉伸方法,可以举出单轴拉伸、双轴拉伸等。双轴拉伸可以为逐次拉伸或同时拉伸。拉伸可以使用拉幅机等公知的拉伸装置进行。
如此得到的光学膜含有微细且高分散的碱土金属化合物微粉末,因此透明性优异,且通过调整碱土金属化合物微粉末相对于光学膜整体的含量,能够调整光学膜本身的双折射。碱土金属化合物微粉末本身示出负的双折射,因此可以根据目标光学膜的用途等来调整光学膜的双折射。
例如通过向聚碳酸酯、聚环状烯烃那样的示出正的固有双折射的树脂中添加碱土金属化合物微粉末,能够抵消树脂的固有双折射而制成双折射接近零的光学膜。作为这样的光学膜,可以举出例如保护膜。作为保护膜,除了层积在偏振片表面等的通常的保护膜之外,也包含直接层积在偏振元件表面而保护偏振元件的偏振元件保护膜。
或者通过向聚碳酸酯、聚环状烯烃那样示出正的双折射的树脂中添加少量的碱土金属化合物微粉末,可以制成具有正的双折射的光学膜。进一步,通过向这些示出正的双折射的树脂中添加大量的碱土金属化合物微粉末,可以制成具有负的双折射的光学膜。此处所说的“双折射”是指上述的面内双折射率(δnxy)的值。作为这样的示出正或负的面内双折射率的光学膜,可以举出相位差膜。作为相位差膜,可以举出1/4波片、1/2波片等。
反之,通过用于聚甲基丙烯酸甲酯、聚苯乙烯等示出负的双折射的树脂、双折射小的树脂,也能够制成表现出负的双折射的光学膜。作为这样的光学膜,可以举出相位差膜。作为相位差膜,可以举出1/4波片、1/2波片等。
作为本发明的光学膜,除了相位差膜、保护膜之外,还可以举出防反射膜、防眩膜、亮度提高膜、棱镜膜、视角改善膜等。
光学膜的雾度可以为10%以下,可以优选为5%以下,更优选为1%以下。需要说明的是,也可以根据光学膜的用途而有意使雾度劣化。例如通过向树脂组合物中添加玻璃珠等光散射性微粒,可以使雾度劣化而制成防眩膜。此外,光学膜的光线透过率可以为85%以上,可以优选为88%以上,更优选为90%以上。
本发明的光学膜也可以与其它光学膜层积而制成光学层积体。作为其它光学膜,可以举出例如偏振膜(也称为偏振元件)、基材膜等。作为光学层积体,可以举出将作为本发明的光学膜的保护膜和偏振膜层积而得到的偏振片、将作为本发明的光学膜的相位差膜和偏振膜层积而得到的椭圆偏振片、将作为本发明的光学膜的相位差膜和基材膜层积而得到的相位差板等。
5.图像显示装置
本发明的图像显示装置的特征在于,具备本发明的光学膜。作为图像显示装置的种类,可以举出液晶显示装置(lcd)、有机电致发光显示装置等。此外,作为图像显示装置的用途,可以举出电视机、计算机用监视器、移动电话、智能手机、pda等便携式信息终端等。
实施例
以下基于实施例对本发明进行具体的说明,但这些实施例并不限定本发明的目的。
(1)sem观察
(1-1)表面未处理品
关于表面未处理的碳酸锶微粒,向纯水中添加碳酸锶微粒,用超声波均化器使其分散。将该分散液滴加在试样工作台上,使其自然干燥后,对碳酸锶微粒进行锇涂布(オスミウムコーティング)而制作样品。利用电解放射型扫描型电子显微镜(fe-sem)对制作的样品进照片拍摄。根据所得到的fe-sem照片,测定各颗粒的长度方向长度(最大长度)和宽度方向长度(最小长度),将长度方向长度的平均值作为颗粒的平均长径。此外,根据所得到的长径、短径的值,计算出纵横比。
(1-2)表面处理品
关于表面处理的碳酸锶微粒,将制作的微粒0.01g溶解于二氯甲烷9.99g中,在超声波浴中分散1分钟,使所得到的分散液真空干燥。然后,进行锇涂布而制作样品。利用电解放射型扫描型电子显微镜(fe-sem)对制作的样品进行照片拍摄。根据所得到的fe-sem照片,测定各颗粒的长度方向长度(长径)和宽度方向长度(短径),将长度方向长度的平均值作为颗粒的平均长径。此外,根据所得到的长径、短径的值,计算出纵横比。
(2)小角x射线散射测定
分别将实施表面处理的碳酸锶微粒和未处理的微粒填充至毛细管柱中,利用小角散射测定装置nano-viewer(株式会社rigaku制)进行测定。曝光时间分成上、中、下3份分别进行5分钟曝光。装置的详细参数如下,
管球:cukα
输出功率:40kv-30ma
狭缝:1stslit=0.40mm
2ndslit=0.20mm
3rdslit=0.45mm
测定法:透过法
计数器:pilatus100k
相机长度:570mm
(3)粒度分布测定
使用激光衍射式粒度分布测定装置(microtrac9320hra、日机装株式会社制造)或者动态光散射式粒度分布测定装置(nanotracupa-150、日机装株式会社制造)进行测定。平均粒径测定用的试样使用下述试样,将高分散性碳酸锶微粉末添加至有机溶剂(二氯甲烷:wako特级)中,制作浓度1%的分散液,用1μm玻璃过滤器过滤而得到的试样。测定将5次连续测定的平均值作为测定值。
(4)表面活性剂
在下述示例中使用的表面活性剂如下。
·表面活性剂a:多元羧酸阴离子型表面活性剂(聚马来酸酐-聚氧乙烯酯共聚物)
·表面活性剂b:多元羧酸阴离子型表面活性剂(在上述化学式(i)中,r1为碳原子数为13的烷基、e1为c2h4、a平均为9.3的聚合物)
·表面活性剂c:多元羧酸阴离子型表面活性剂(在上述化学式(i)中,r1为碳原子数为12的烷基、e1为c2h4、a平均为2.3的聚合物)
·表面活性剂d:多元羧酸阴离子型表面活性剂(在上述化学式(i)中,r1为碳原子数为18的烷基、e1为c2h4、a平均为2的聚合物)
·表面活性剂e:多磷酸阴离子型表面活性剂(上述化学式(ii)与式(iii)的混合物,r2、r3、r4均为碳原子数为13的烷基、e2、e3、e4均为c2h4、b、c、d的合计为6.0的聚合物)
·表面活性剂f:聚乙二醇胺型非离子表面活性剂(在上述化学式(iv)中,r5为碳原子数为18的烷基、e5、e6均为c2h4、e和f的合计为4.0的聚合物)
1.实施例1
(1)碳酸锶微粒的制造
(a)反应工序
向水温10℃的纯水3l中加入dl-酒石酸(特级试剂、纯度:99%以上)并搅拌,使其溶解于水性悬浮液中。投入氢氧化锶八水合物(特级试剂、纯度:96%以上)366g并混合,制备浓度5.6质量%的氢氧化锶水性悬浮液。将该氢氧化锶水性悬浮液维持在10℃并持续搅拌,同时以0.5l/分钟的流量(相对于氢氧化锶1g为22ml/分钟的流量)向水性悬浮液中吹入二氧化碳气体直至水性悬浮液的ph为7,从而生成碳酸锶颗粒。然后,进一步持续30分钟的搅拌,得到碳酸锶颗粒水性悬浮液。
(b)熟化工序
将所得到的碳酸锶颗粒水性悬浮液投入不锈钢罐中,在95℃的温度进行12小时的加热处理,使碳酸锶颗粒成长为针状。然后,自然冷却至室温,制造碳酸锶微粒的水性浆料。将该浆料用鼓式干燥机进行干燥,得到碳酸锶微粒。利用上述的“(1)sem观察”的“(1-1)表面未处理品”中所记载的方法观察碳酸锶微粒,结果平均长径为35nm。将该结果示于表1。
(2)高分散性碳酸锶微粉末的制造
(a)表面处理工序
向上述制作的碳酸锶微粒的水性浆料(固体浓度:6质量%)中添加表面活性剂a并使其溶解,用搅拌器搅拌5分钟。接着,将表面活性剂b按照成为28质量%的方式添加至水性浆料中并使其溶解,用搅拌器搅拌5分钟后,利用clearmix施加剪切力,进行分散处理。由此,得到高分散性碳酸锶颗粒的水浆料。在表面上按照两种表面活性剂成为两层的方式进行层积。
(b)干燥工序(鼓式干燥机)
将搅拌混合后的水性浆料吹入加热为110~120℃的旋转式鼓式干燥机中,得到高分散性碳酸锶微粉末。用电子显微镜观察所得到的高分散性碳酸锶微粉末,结果确认到针状颗粒的微粉末。利用上述的“(1)sem观察”的“(1-2)表面处理品”中所记载的方法观察高分散性碳酸锶微粉末,结果平均长径为35nm。将表面活性剂的种类等条件示于表1。
(3)高分散性碳酸锶微粉末的小角x射线散射测定
使用上述得到的高分散性碳酸锶微粉末,利用“(2)小角x射线散射测定”和“(3)粒度分布测定”中所记载的方法进行测定。进行峰值搜索,结果在2θ=0.46处观察到散射峰,d值为19.2nm。此外,在使用该粉末的有机溶剂分散液下的粒度分布测定中的d50为50nm以下。将该结果示于表2。
2.实施例2
在实施例1的“(a)表面处理工序”中,将表面活性剂c按照成为30质量%的方式进行添加并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定基于小角x射线测定和粒度分布的平均长径等。将该结果示于表1、表2。
3.实施例3
在实施例1的“(a)表面处理工序”中,将表面活性剂e按照成为30质量%的方式进行添加并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定平均长径等。将该结果示于表1、表2。
4.实施例4
在实施例1的“(a)表面处理工序”中,将表面活性剂d按照成为30质量%的方式进行添加并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定平均长径等。将该结果示于表1、表2。
5.实施例5
在实施例1的“(1)碳酸锶微粒的制造”中,使用平均长径为20nm的碳酸锶微粒,在“(a)表面处理工序”中,将表面活性剂d按照成为47质量%的方式进行添加并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定平均长径等。将该结果示于表1、表2。
6.实施例6
在实施例1的“(1)碳酸锶微粒的制造”中,使用平均长径为20nm的碳酸锶微粒,在“(a)表面处理工序”中,将表面活性剂c按照成为47质量%的方式进行添加并溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定平均长径等。将该结果示于表1、表2。
7.比较例1
在实施例1的“(a)表面处理工序”中,将表面活性剂a按照成为8质量%的方式进行添加并使其溶解,用搅拌器搅拌5分钟。接着,将表面活性剂f按照成为23质量%的方式添加至水性浆料中并使其溶解,用搅拌器搅拌5分钟。与实施例1同样地测定平均长径等。
8.比较例2
在实施例1的“(a)表面处理工序”中,将表面活性剂c按照成为10质量%的方式添加至水性浆料中并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定平均长径等,同时与比较例1同样地计算出分布。将该结果示于表1、表2。
9.比较例3
在实施例1的“(a)表面处理工序”中,将表面活性剂c按照成为20质量%的方式添加至水性浆料中并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。与实施例1同样地测定平均长径等,同时与比较例1同样地计算出分布。将该结果示于表1、表2。
10.比较例4
在实施例1的“(2)高分散性碳酸锶微粉末的制造”中,不利用“(a)表面处理工序”进行表面处理,与实施例1同样地测定平均长径等。在小角x射线散射中,未观测到散射峰。将该结果示于表1、表2。
11.比较例5
在实施例1的“(a)表面处理工序”中,将表面活性剂c按照成为30质量%的方式进行添加并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。在小角x射线散射中,不是以干燥粉进行测定,而是使其按照固体浓度成为1质量%的方式分散于环己烷中进行测定。其结果未观测到散射峰。将该结果示于表1、表2。
12.比较例6
在实施例1的“(a)表面处理工序”中,将表面活性剂c按照成为30质量%的方式进行添加并使其溶解,在碳酸锶颗粒的表面将一种表面活性剂层积一层。在小角x射线散射中,不是以干燥粉进行测定,而是使其按照固体浓度成为1质量%的方式分散于水中进行测定。其结果未观测到散射峰。将该结果示于表1、表2。
根据以上结果可知,在实施例1~6中,在高分散性碱土金属化合物微粉末的状态下,在2θ为0.2~1.0°的范围内,在散射强度中均具有散射峰。此外,根据粒度分布测定的结果可知,分散性良好。可知在这些实施例中,表面活性剂添加最适量,对一次颗粒完全实施被覆处理、分散处理。
[表1]
[表2]
<聚碳酸酯(pc)膜>
以下对使用上述实施例4和比较例4中制造的高分散性碳酸锶微粉末制造聚碳酸酯膜的示例进行说明。
13.实施例7
(1)添加srco3的掺杂液制作方法
对二氯甲烷25g添加聚碳酸酯(以下称为“pc”)6g,搅拌6小时,制作pc-二氯甲烷溶液。接着,对二氯甲烷10g添加0.48g实施例4的表面处理粉末1,放入超声波浴中30秒,直接用孔径1μm的膜过滤器进行过滤而无需加压,制作分散液1。将pc-二氯甲烷分散液和分散液1混合,同时添加0.026g乙烯基系表面改性剂,用超声波均化器进行分散处理,得到添加srco3的掺杂液a-1。
(2)pc膜成膜方法
使用baker式涂布器,以湿式厚度11密尔将添加srco3的掺杂液a-1涂布在聚对苯二甲酸乙二醇酯(以下称为“pet”)膜上。将其在40℃干燥2分钟、在80℃干燥4分钟、在120℃干燥30分钟。从pet膜上剥离pc膜,得到pc膜a-1。利用膜拉伸装置(井元制作所制造、imc-1a8d型)在160℃对pc膜a-1进行自由端单轴拉伸至2.0倍,得到pc拉伸膜a-1。
(3)透过率和雾度测定
使用分光光度计(日本分光公司制造),进行pc拉伸膜a-1的可见光透过率和雾度测定。
(4)膜的相位差评价
用千分尺测定pc拉伸膜a-1的膜厚。然后,使用相位测定装置(王子计测设备株式会社制造,kobra-wr)测定拉伸的膜的相位差(δnxy)。将该结果示于表3。
14.比较例7
使用比较例4的粉末(未进行表面处理的srco3)代替实施例4的粉末,除此以外,使用与实施例7同样的方法。由此,得到pc拉伸膜h-1。与实施例7同样地对所得到的膜的特性进行评价。将该结果示于表3。
[表3]
比较实施例7和比较例7可知,与未进行表面处理的比较例7相比,进行表面处理的实施例7的雾度低、且双折射的双折射(δnxy×10-3)的值低。由此可知,从透明性和双折射表现的观点出发,优选进行了表面处理的碳酸锶微粒。
<聚甲基丙烯酸甲酯(pmma)膜>
以下对使用上述实施例4和比较例4中制造的高分散性碳酸锶微粉末,制造聚甲基丙烯酸甲酯膜的示例进行说明。
15.实施例8
(1)添加srco3的掺杂液制作方法
对二氯甲烷25g添加聚甲基丙烯酸甲酯(以下称为“pmma”)6g,搅拌3小时,制作pmma-二氯甲烷溶液。接着,对二氯甲烷10g添加0.48g实施例4的表面处理粉末1,放入超声波浴中30秒,直接用孔径1μm的膜过滤器进行过滤而无需加压,制作分散液1。将pmma-二氯甲烷分散液和分散液1混合,用超声波均化器进行分散处理,得到添加srco3的掺杂液i-1。
(2)pmma膜成膜方法
使用baker式涂布器,以湿式膜厚11密尔将添加srco3的掺杂液i-1涂布在pet膜上。将其在40℃干燥2分钟、在80℃干燥15分钟、在85℃干燥30分钟。从pet膜上剥离pmma膜,得到pmma膜i-1。使用膜拉伸装置(井元制作所制造、imc-1a8d型)在90℃对pmma膜i-1进行自由端单轴拉伸至2.0倍,得到pmma拉伸膜i-1。与实施例7同样地对所得到的膜的特性进行评价。将该结果示于表4。
16.比较例8
使用比较例4的粉末(未进行表面处理的srco3)代替实施例4的粉末,除此以外,使用与实施例8同样的方法。由此,得到pmma拉伸膜n-1。与实施例7同样地对所得到的膜的特性进行评价。将该结果示于表4。
[表4]
比较实施例8和比较例8可知,与未进行表面处理的比较例8相比,进行了表面处理的实施例8的雾度低、且双折射的双折射(δnxy×10-3)的值低。因此可知,从透明性和双折射表现的观点出发,优选进行了表面处理的碳酸锶微粒。综上可知,本发明的碱土金属化合物微粉末特别适合例如偏振片的相位差膜等透明性高且要求高双折射性的光学膜。
- 还没有人留言评论。精彩留言会获得点赞!