一种柳氮磺吡啶的纯化工艺的制作方法
本发明涉及柳氮磺吡啶制备技术领域,尤其涉及一种柳氮磺吡啶的纯化工艺。
背景技术:
柳氮磺吡啶别名为5-[对-(2-吡啶胺磺酰基)苯]偶氮水杨酸,具有如下结构:
其是用于治疗炎症性肠病的磺胺类抗菌药。口服后被肠内细菌裂解成5-氨基水杨酸和磺胺吡啶。5-氨基水杨酸与肠壁结缔组织络合后较长时间停留在肠壁组织中起到抗菌消炎和免疫抑制作用。
目前柳氮磺吡啶的合成主要以磺胺吡啶(I)为原料,
亚硝酸钠为重氮化试剂,在水相中经重氮化反应后再与水杨酸耦合而得,例如可以参见WO2002018330中提供的制备方法;当经过上述反应,在成品粗品中带有的杂质很多,中国发明专利ZL03129670.X报道了已知结构的杂质共有九个:
接着其提供了一种柳氮磺吡啶的纯化方法,以DMF为溶剂,加入氢氧化钠类无机碱,然后再加入酸,充分酸析后离心过滤即得半精制品,再将半精制品在水溶液中加碱溶解,加热脱水,再酸析即可得到精制的柳氮磺吡啶。
虽然其提供的精制工艺能降低杂质的含量,将产品纯度由95.377%提高至96.567%,如果想在此基础上进一步进行提高产品的纯度,还是有很大的难度,并且必然会损失更多的柳氮磺吡啶粗品;而且由于其提供的纯化工艺需要进行两次加碱酸析步骤,操作较为繁琐,并且其所使用的粗品已经是经过预先纯化的,母液中柳氮磺吡啶无法得到回收套用,因此有必要开发新的纯化工艺,以解决现有技术中存在的母液中柳氮磺吡啶无法回收利用这个问题。
技术实现要素:
为了解决现有技术中存在的问题,本发明采用如下技术方案:
一种具有如下式A结构的化合物:
其中R1,R2和R3各自独立的为异丙基,乙基,甲基,氢。
优选的所述的式A化合物具有如下结构:
利用式a化合物制备得到的柳氮磺吡啶较其他胺盐收率更高,更容易从溶液中析出,且经游离后得到的柳氮磺吡啶的纯度也更高。
另一方面,本发明提供了一种具有式A结构化合物的制备方法:柳氮磺吡啶粗品与下列的式B化合物进行反应得到所述的式A化合物,
NR1R2R3
B
其中R1,R2,R3的定义与前文定义相同。
优选的本发明采用如下制备方法,柳氮磺吡啶粗品与式B化合物在溶剂中进行反应,制备得到所述的式A化合物。
所述溶剂优选为醇类溶剂,如甲醇,乙醇,异丙醇等;或水;其中更为优选的反应溶剂为水,利用水为反应溶剂,反应效率更高。
优选的,所述反应温度为40℃~回流。
更进一步的,本发明采用如下制备方法,柳氮磺吡啶粗品与式B化合物在水中进行反应,反应完成后,缓慢降温析晶,制备得到式A化合物。
所述的析晶温度为-10至60℃。
所述柳氮磺吡啶与所述式B化合物的摩尔用量比1:(1~10),优选为1:(1.5~4);
所述柳氮磺吡啶与水的重量比为1:(2~10),优选为1:(3-6);
另一方面,本发明提供了一种式A化合物的用途,用于制备柳氮磺吡啶精品,利用本发明得到的式A化合物制备得到的柳氮磺吡啶纯度高。
具体的本发明采用如下反应步骤:
所述式A化合物在酸作用下游离得到柳氮磺吡啶精品。
所述酸优选为盐酸,醋酸。
所述游离反应温度为40℃至回流。
所述的柳氮磺吡啶与所述酸的摩尔用量比为1:(1~12),优选为1:(3~8)。
所述酸优选为盐酸水溶液,优选的浓度为10~30%。
利用本发明提供的式A化合物制备得到的柳氮磺吡啶质量好,无需精制就可以从柳氮磺吡啶回收液中得到高纯度的柳氮磺纯品。并且经常规操作可以将游离后的胺进行回收,有利于工业化生产。
附图说明
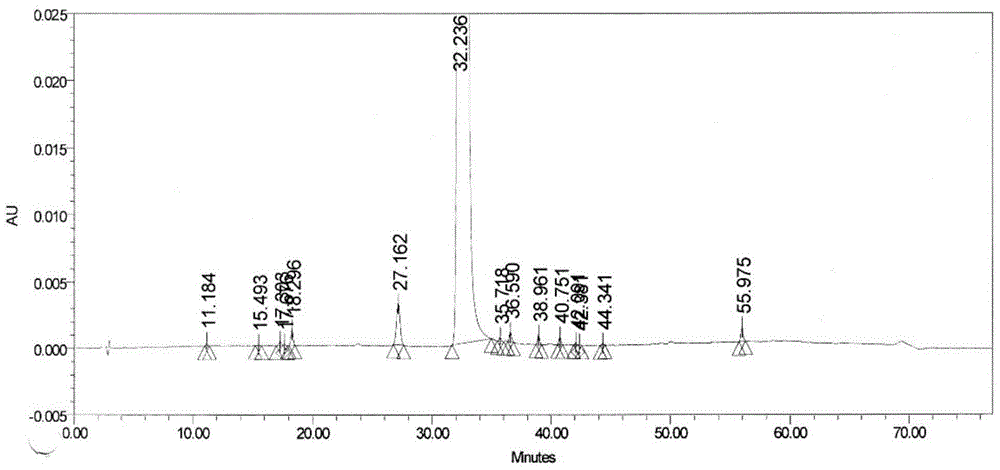
图1为柳氮磺吡啶粗品的HPLC谱图;
图2为按照实施例1制备得到的柳氮磺吡啶二异丙铵盐TGA谱图;
图3为按照实施例2制备得到柳氮磺吡啶的HPLC谱图;
图4为按照实施例4制备得到的柳氮磺吡啶的HPLC谱图;
图5为按照实施例6制备得到的柳氮磺吡啶的HPLC谱图;
图6为按照实施例8制备得到的柳氮磺吡啶的HPLC谱图;
图7为按照实施例10制备得到的柳氮磺吡啶的HPLC谱图。
具体实施方式
为了更好的理解本发明的内容,下面结合具体实施例来做进一步的说明,但具体的实施方式并不是对本发明的内容所做的限制。
实施例1:柳氮磺吡啶二异丙铵盐的制备
在500ml四口烧瓶中,投入柳氮磺吡啶粗品50g(HPCL纯度78.76%)、200g水、搅拌,再慢慢加入38g二异丙胺。投料毕,升温至75-80℃。在75-80℃保温反应0.5-1h。(料全溶解)反应毕,缓慢冷却至50-60℃,有晶体析出,并在50-60℃保温搅拌30min,再缓慢降温至0-5℃,抽滤,滤干。再用20g 0-5℃冰水淋洗滤饼。滤饼在75℃热风干燥12小时以上,得干品约47.75g。收率95.3%。粗品HPLC检测结果如表1。
TAG检测结果:失重:-18.8074%,-2.0086mg;残留物:81.1798%,8.6700mg;起始失重温度:32.45℃;结束失重温度:138.70℃;弯曲点:126.17℃;终点:124.98℃。
HPLC分离条件:
仪器:Agilent 1200
色谱柱:Reprosil pur C18-AQ,4.6mm×250mm×5μm
柱温:35℃
流动相A:称取1.0g磷的二氢钠和2.5g乙酸钠,加900ml水溶解,用冰醋酸调pH=4.8,用水稀释成1000ml
流动相B:流动相A/甲醇=10:40(V/V)混合
流速:1.0mL/min
波长:320nm
进样量:20μl
表1
实施例2:柳氮磺吡啶的制备
在另一500ml四口瓶中加入二异丙胺盐的干品47.75g,再加入160ml 15%稀盐酸,搅拌升温至80-85℃搅拌1h。抽滤,用200ml 80-85℃热水淋洗。在75℃热风烘箱中烘干,得到精品约35.2g收率92.8%,HPLC纯度99.60%,柳氮磺吡啶HPLC检测结果如表2。
表2
实施例3:柳氮磺吡啶三乙胺盐的制备
在500ml四口烧瓶中,投入柳氮磺吡啶粗品50g(HPCL纯度78.76%)、200g水、搅拌,再慢慢加入40g三乙胺。投料毕,升温至75-80℃。在75-80℃保温反应4小时。反应毕,缓慢冷却至30-40℃,有晶体析出,并在30-40℃保温搅拌30min,再缓慢降温至0-5℃,抽滤,滤干。再用20g 0-5℃冰水淋洗滤饼。滤饼在75℃热风干燥12小时以上,得干品约45.06g。收率90.3%。粗品HPLC检测结果如表1。
实施例4:柳氮磺吡啶的制备
在另一500ml四口瓶中加入三乙胺盐的干品40g,再加入160ml 15%稀盐酸,搅拌升温至80-85℃搅拌4h。抽滤,用200ml 80-85℃热水淋洗。在75℃热风烘箱中烘干,得到精品约28.7g收率90.0%,HPLC纯度99.42%,柳氮磺吡啶HPLC检测结果如表3。
表3
实施例5:柳氮磺吡啶胺盐的制备
在500ml四口烧瓶中,投入柳氮磺吡啶粗品50g(HPCL纯度78.76%)、200g水、搅拌,再慢慢加入40ml氨水(28%)。投料毕,升温至75-80℃。在75-80℃保温反应6小时。反应毕,缓慢冷却至10-20℃,有晶体析出,并在10-20℃保温搅拌1小时,再缓慢降温至0-5℃,抽滤,滤干。再用20g 0-5℃冰水淋洗滤饼。滤饼在75℃热风干燥12小时以上,得干品约35.36g。 收率85.2%。粗品HPLC检测结果如表1。
实施例6:柳氮磺吡啶的制备
在另一500ml四口瓶中加入实施例5制备得到的胺盐的干品30g,再加入160ml 15%稀盐酸,搅拌升温至80-85℃搅拌3h。抽滤,用200ml 80-85℃热水淋洗。在75℃热风烘箱中烘干,得到精品约25.64g收率89.1%,HPLC纯度99.26%,柳氮磺吡啶HPLC检测结果如表4。
表4
实施例7:柳氮磺吡啶二甲胺盐的制备
在500ml四口烧瓶中,投入柳氮磺吡啶粗品50g(HPCL纯度78.76%)、200g水、搅拌,再慢慢加入50ml二甲胺水溶液(40%)。投料毕,升温至75-80℃。在75-80℃保温反应10小时。反应毕,缓慢冷却至-10-0℃,有晶体析出,并在-10-0℃保温搅拌4小时,抽滤,滤干。再用20g 0-5℃冰水淋洗滤饼。滤饼在75℃热风干燥12小时以上,得干品约36.37g。收率82.1%。粗品HPLC检测结果如表1。
实施例8:柳氮磺吡啶的制备
在另一500ml四口瓶中加入实施例7制备得到的二甲胺盐的干品30g,再加入200ml 15%稀盐酸,搅拌升温至80-85℃搅拌3h。抽滤,用200ml 80-85℃热水淋洗。在75℃热风烘箱中烘干,得到精品约24.31g收率90.2%,HPLC纯度92.01%,柳氮磺吡啶HPLC检测结果如表5。
表5
实施例9:柳氮磺吡啶二乙胺盐的制备
在500ml四口烧瓶中,投入柳氮磺吡啶粗品50g(HPCL纯度78.76%)、200g乙醇、搅拌,再慢慢加入20g二乙胺。投料毕,升温至75-80℃。在75-80℃保温反应5小时。反应毕,缓慢冷却至0-10℃,有晶体析出,并在1-10℃保温搅拌1小时,抽滤,滤干。再用20g 0-5℃冰水淋洗滤饼。滤饼在75℃热风干燥12小时以上,得干品约43g。收率91.3%。粗品HPLC检测结果如表1。
实施例10:柳氮磺吡啶的制备
在另一500ml四口瓶中加入实施例9制备得到的胺盐的干品40g,再加入130ml醋酸,搅拌升温至80-85℃搅拌3h。抽滤,用200ml 80-85℃热水淋洗。在75℃热风烘箱中烘干,得到精品约30.32g收率89.7%,HPLC纯度98.42%,柳氮磺吡啶HPLC检测结果如表6。
表6
尽管已经结合了具体实施方式对本发明进行了充分的描述,应当注意的是对于本领域技术人员来说其各种变化和修改是显而易见的。这样的变化和修改将可以理解为包括在由所附权利要求所定义的本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!