用于丙烯酸类粘合剂的胺共促进剂的制作方法
根据35U.S.C.§119(e),本申请要求于2014年3月19日提交的题为“用于丙烯酸类粘合剂的胺共促进剂(AMINECO-ACCELERATORFORACRYLICADHESIVES)”的美国临时专利申请No.61/955,251的优先权,其公开内容以引用的方式并入本文。
技术领域:
本发明涉及用于自由基固化的丙烯酸类粘合剂的共促进剂,该共促进剂提高了还原剂的活性。这些共促进剂使粘合剂的开放时间具有令人惊讶的时长、并使其具有快速固化速度,使粘合剂固化速率对酸性粘合促进剂不敏感,并且使丙烯酸类粘合剂牢固地粘结到各种金属(包括铝)上。
背景技术:
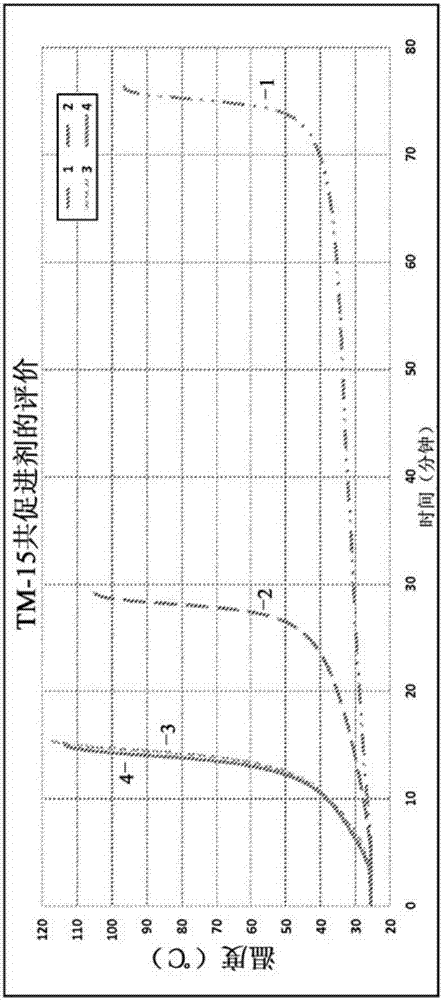
:丙烯酸类结构粘合剂组合物是公知的商业制品,其在工业中被广泛用于粘结金属和塑料材料。结构性粘合剂的承重性能和应力释放性能、以及其粘结强度(其粘结强度能够超过被粘结材料的强度)使得这些粘合剂成为了接合材料的机械方法(如铆接或点焊)的有吸引力的替代或部分替代方法,特别是优选在使负荷应力分布于较大区域而不是使应力集中于若干点的情况中更是如此。这种粘合剂的使用能够减少或消除机械接合方法所必需的高成本精加工操作,呈现出更悦目的外观,并且至少减少了含有一种或多种金属成分的组件的腐蚀可能性。此外,这种粘合剂能够用来粘结各种金属而不用进行大面积的表面处理。丙烯酸类结构性粘合剂被广泛用来向被接合的金属和或聚合物材料提供能够赋予结构性强度的粘结。丙烯酸类结构性粘合剂可代替焊接和机械紧固技术而被用来接合金属部件。其结构性要求包括高接合强度以及良好的时效模式。测量粘结强度的典型方法为搭接剪切试验、高速冲击试验和T-剥离试验。丙烯酸类结构性粘合剂的一种普遍用途是在汽车车身板和门中形成边缘法兰(hemflange)。常规的丙烯酸类结构性粘合剂通常包含一种或多种烯烃类反应性单体(例如甲基丙烯酸甲酯和甲基丙烯酸)、增韧剂和氧化还原引发剂体系的混合物。增韧剂可与或可不与反应性单体反应或聚合。在聚合过程中,可以使用诸如不饱和聚酯和丙烯酸氨基甲酸酯预聚物之类的反应性聚合物接枝到被引发单体上、或使被引发单体交联。此外,全配方的丙烯酸类结构性粘合剂通常包含用于改善对基材的粘合性、耐环境性、冲击强度、柔韧性、耐热性等的其它添加剂。环氧树脂赋予更高的耐热性。技术实现要素:在本发明的一个实施方案中,提供了一种固化性组合物,该固化性组合物包含自由基聚合性丙烯酸类单体、氧化剂、芳族叔胺还原剂、以及含有双环二氮杂化合物的叔胺共促进剂,该双环二氮杂化合物增加了所述氧化剂和所述还原剂之间的反应速率。在本发明优选的实施方案中,所述还原剂包括N,N-二异丙醇对氯苯胺、N,N-二异丙醇对溴苯胺、N,N-二异丙醇-对溴间甲基苯胺、N,N-二甲基对氯苯胺、N,N-二甲基对溴苯胺、N,N-二乙基对氯苯胺、N,N-二乙基对溴苯胺、N,N-二甲基对苯胺、或N,N-二异丙醇对甲苯胺中的至少一种,并且最优选为N,N-二异丙醇对甲苯胺。在本发明的另一个实施方案中,所述共促进剂包括1,4-二氮杂双环[2.2.2]辛烷。在本发明另一优选的实施方案中,所述叔胺还原剂包括N,N-二异丙醇对甲苯胺,并且所述共促进剂包括1,4-二氮杂双环[2.2.2]辛烷,或者所述组合物不含胺化合物。在本发明的一个实施方案中,所述组合物不含异氰酸酯化合物。在本发明的另一个实施方案中,所述自由基聚合性单体包括甲基丙烯酸甲酯、或甲基丙烯酸四氢糠酯中的至少一种。在本发明的另一个实施方案中,所述组合物还包含增韧剂,优选甲基丙烯酸缩水甘油酯/羧基封端的丁二烯(GMA/CTB)加合物、核-壳冲击改性剂、或嵌段共聚物弹性体中的至少一种。在本发明的一个实施方案中,所述组合物还包含粘合促进剂,优选甲基丙烯酸羟乙酯磷酸酯或甲基丙烯酸中的至少一种。在本发明的另一个优选实施方案中,所述氧化剂包括过氧化苯甲酰。在本发明的另外的实施方案中,所述组合物以两部份提供:在部分A中:(a)所述至少一种自由基聚合性单体、和(b)所述还原剂;在部分B中:氧化剂;其中所述共促进剂存在于部分A或部分B中的至少一者中;优选地,所述部分A与部分B的重量比为约1:1至约15:1。在本发明的另一个实施方案中,所述组合物置于两个基材之间并固化,从而在其间提供粘结,通过搭接剪切强度测量,所述粘结具有至少1000psi的粘结强度。在本发明的一个实施方案中,所述两个基材中的至少一个包括铝。附图说明图1示出了理想的丙烯酸类粘合剂的固化曲线,该固化曲线证明了“瞬时固化”温度曲线的反应进展。图2示出了现有技术的固化组合物的温度曲线与本发明实施方案间的比较。图3示出了现有技术的固化组合物的温度曲线与本发明实施方案间的比较。图4示出了现有技术的固化组合物的ROBDS和DMOT数据与本发明实施方案间的比较。具体实施方式在本发明的第一方面中,提供了一种丙烯酸类粘合剂组合物,该丙烯酸类粘合剂组合物包含自由基聚合性单体、引发体系和共促进剂,该共促进剂通过提高氧化剂和还原剂之间的反应速率来提高引发体系的反应性。在本发明优选的实施方案中,自由基聚合性单体包括甲基丙烯酸甲酯(MMA),该引发剂体系包括N,N-二异丙醇对甲苯胺(DIIPT)和过氧化苯甲酰(BPO),并且共促进剂包括1,4-二氮杂双环[2.2.2]辛烷(或三亚乙基二胺(TDA))。在本发明的一个实施方案中,丙烯酸类粘合剂以主要的量包含10重量%至90重量%的至少一种自由基聚合性单体(主要单体)。代表性的单体包括(甲基)丙烯酸的酯,如甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸丁酯、丙烯酸甲酯、丙烯酸丁酯、丙烯酸环己酯、丙烯酸己酯、丙烯酸-2-乙基己酯、丙烯酸月桂酯、丙烯酸乙酯、二甲基丙烯酸二乙二醇酯、甲基丙烯酸二环戊二烯氧乙酯、甲基丙烯酸环己酯、甲基丙烯酸-2-乙基己酯、甲基丙烯酸月桂酯、甲基丙烯酸缩水甘油酯和甲基丙烯酸四氢糠酯(THFMA)。优选的单体有助于提高已固化聚合物的刚性,并且其选自这样的甲基丙烯酸酯,该甲基丙烯酸酯的均聚物的Tg为至少50℃、优选60℃、并且有些高达105℃。在本发明另一实施方案中,主要单体可以与诸如甲基丙烯酸、丙烯酸、取代的(甲基)丙烯酸之类的烯烃不饱和羧酸单体或者诸如衣康酸、马来酸和富马酸之类的二元酸组合。本发明可包括的其他任选共单体为:丙烯腈、甲基丙烯腈、丙烯酰胺和甲基丙烯酰胺;乙酸乙烯酯、偏二氯乙烯;和丁二烯,例如2,3-二氯-1,3-丁二烯和2-氯-1,3-丁二烯。其它可使用的单体包括马来酸酯和富马酸酯。在一个实施方案中,可使用单体甲基丙烯酸四氢糠酯、甲基丙烯酸和甲基丙烯酸甲酯的混合物。在另一实施方案中,任选地优选包括含有主要单体的反应性稀释剂。主要单体中可任选地包含的共单体包括:具有OH官能团的单烯烃不饱和单体,如(甲基)丙烯酸-2-羟乙酯、(甲基)丙烯酸-3-羟丙酯、(甲基)丙烯酸-4-羟丁酯、(甲基)丙烯酸-4-羟基环己酯、单(甲基)丙烯酸-1,6-己二醇酯、单(甲基)丙烯酸新戊二醇酯、二甲基丙烯酸-1,6-己二醇酯和二甲基丙烯酸-1,4-丁二醇酯。优选包含0.0重量%至10重量%(基于A部分的重量)的多官能交联共单体,例如三羟甲基丙烷二(甲基)丙烯酸酯(trimethylohpropanedi(meth)acrylate)、三羟甲基乙烷二(甲基)丙烯酸酯、季戊四醇三(甲基)丙烯酸酯、二季戊四醇五(甲基)丙烯酸酯和环氧基二丙烯酸酯,例如,乙氧基化的双酚A二甲基丙烯酸酯。本发明包含环境温度引发体系。可以在优选粘合剂体系中使用的环境温度引发体系为熟知的氧化还原对体系,本文中无需详细地讨论。基本上,这样的体系包含在室温下共反应的至少一种氧化剂和至少一种还原剂,以产生能够有效引发加成聚合反应的自由基,并固化该粘合剂。基本上能够使用任何已知的可共反应的氧化剂和还原剂。代表性的氧化剂包括但不限于:有机过氧化物,例如过氧化苯甲酰和其它二酰基过氧化物;氢过氧化物,例如氢过氧化枯烯;过酸酯,例如β-丁基过氧化苯甲酸酯;酮过氧化物,例如甲基乙基酮过氧化氢;过渡金属的有机盐,例如环烷酸钴;和含有不稳定氯的化合物,例如磺酰氯。在本发明的一个优选实施方案中,氧化剂包括有机过氧化物,最优选为过氧化苯甲酰(BPO)。在本发明的另一优选实施方案中,还原剂包括芳族叔胺。代表性的还原剂包括N,N-二异丙醇对氯苯胺、N,N-二异丙醇对溴苯胺、N,N-二异丙醇-对溴间甲基苯胺、N,N-二甲基对氯苯胺、N,N-二甲基对溴苯胺、N,N-二乙基对氯苯胺、N,N-二乙基对溴苯胺、N,N-二甲基对苯胺、N,N-二甲基对甲苯胺、N,N-二乙基对甲苯胺、N,N-二异丙醇对甲苯胺(DIIPT)或其它对卤化苯胺衍生物中的至少一种。在本发明的一个优选实施方案中,还原剂的含量为聚合性粘合剂组合物的约0.05重量%至约10重量%,优选约0.1重量%至约6.0重量%,并且氧化剂的含量为还原剂的约0.5重量%至约50重量%。在本发明最优选的实施方案中,还原剂包括DIIPT并且氧化剂包括过氧化苯甲酰。在本发明的另一实施方案中,丙烯酸类粘合剂组合物包含共促进剂。该共促进剂与促进剂/还原剂不同,这是因为该共促进剂不会促进氧化剂的分解,因此不会单独促进组合物的固化。然而,当将共促进剂与上述还原剂一同使用时,共促进剂会提高体系的反应性,从而比单独使用还原剂时提供更多的“瞬时固化”。在本发明优选的实施方案中,共促进剂包括环状叔胺、并且优选的叔胺包括双环二氮杂化合物,其中氮原子位于环中的环接合处。在本发明最优选的实施方案中,共促进剂包括1,4-二氮杂双环[2.2.2]辛烷(TDA)。在本发明的实施方案中,基于组合物的总重量,共促进剂的含量为0.10重量%至3.0重量%。虽然希望不受理论的限制,但是认为本发明的共促进剂参与反应,以提高还原剂的活性,而不直接促进氧化剂的分解。其机理被认为是由环状叔胺的较高碱性来驱动的,该环状叔胺与组合物中的酸性组分络合,以提高还原剂和氧化剂的反应性。此外,本发明的共促进剂并不会与氧化剂反应并且通过它们自身来固化组合物,并且必须与如上所述的主要还原剂一起使用。在本发明的一个优选实施方案中,组合物优选不含异氰酸酯官能度,即反应性NCO基团。有时,将异氰酸酯和/或聚氨酯加入到丙烯酸类结构性粘合剂中,然而在本发明的这个实施方案中,基本上不含异氰酸酯官能度的组合物是优选的。在本发明最优选的实施方案中,组合物完全不含异氰酸酯反应性组分。在本发明的另外实施方案中,丙烯酸类组合物还包含增韧剂。合适的增韧剂的例子包括各种固体和液体弹性聚合物材料,并且特别是如美国专利No.4,223,115、No.4,452,944、No.4,769,419、No.5,641,834和No.5,710,235中描述的烯烃封端的液体弹性体,和如美国专利No.4,223,115、No.4,452,944、No.4,467,071和No.4,769,419中描述的具有异氰酸酯官能团的预聚物和具有羟基官能团的单体的烯烃氨基甲酸乙酯反应产物。优选的氨基甲酸乙酯改性的烯烃封端的液体弹性体包括美国专利No.4,769,419中公开的那些,包括烯烃单环氧化合物与共轭二烯烃的聚羧酸均聚物的反应产物,并且最优选为美国专利No.4,769,419的实施例1中公开的甲基丙烯酸缩水甘油酯/羧基封端的丁二烯(GMA/CTB)加合物。A-B-A三嵌段的嵌段共聚物是可使用的增韧剂。在一个例子中,嵌段A为聚苯乙烯、α-甲基苯乙烯、叔丁基苯乙烯、或其它环烷基化苯乙烯、以及上述这些物质的部分或全部的混合物,并且嵌段B为Tg为0℃或以下的弹性链段,例如来自共轭二烯(如丁二烯)、异丁烯或其它烯烃(如乙烯-丙烯单体)的弹性链段。市售可得的嵌段共聚物增韧剂包括得自EnichemElastomersAmericas,Inc的在本发明的一个优选实施方案中,增韧剂为基于苯乙烯-[异戊二烯]-苯乙烯(25重量份-[50重量份]-25重量份)的三嵌段聚合物。其他市售嵌段共聚物包括:得自KratonPolymers,Inc的系列,例如KratonSBS和SIS系列的共聚物。其它高分子量的增韧剂包括(例如)嵌段共聚物和无规共聚物,包括但不限于聚乙烯、聚丙烯、苯乙烯-丁二烯、聚氯丁二烯、EPDM、氯化橡胶、丁基橡胶、苯乙烯/丁二烯/丙烯腈橡胶和氯磺化聚乙烯。在本发明另一优选的实施方案中,增韧剂包括苯乙烯-丁二烯-苯乙烯嵌段共聚物。其它增韧剂包括烯烃封端的液体弹性体,其中弹性体部分基于丁二烯的均聚物、丁二烯和至少一种可与其共聚的单体(例如,苯乙烯、丙烯腈、甲基丙烯腈(例如聚(丁二烯-(甲基)丙烯腈)或聚(丁二烯-(甲基)丙烯腈-苯乙烯)及其混合物)的共聚物;以及改性的弹性体聚合物材料,例如丁二烯均聚物、以及通过痕量(最多5重量%)的具有至少一种官能单体(例如丙烯酸、甲基丙烯酸、马来酸酐、富马酸、苯乙烯和甲基丙烯酸甲酯)的弹性体材料与丁二烯共聚改性的共聚物,以产生(例如)甲基丙烯酸封端的聚丁二烯均聚物和/或共聚物。增韧剂包括具有羧基酯连接基团和至少一个初生仲羟基的烯烃封端的聚链二烯,例如美国专利No.5,587,433(其内容通过引用并入本文中)中公开的那些。如共同拥有的美国专利No.5,641,834(其内容通过引用并入本文中)中公开的那样,可利用异氰酸酯任选地将仲OH基团带帽(caped)。加合的羟基封端的聚丁二烯的具体例子包括:使酸酐改性的OH封端PBD与二元酸酐(邻苯二甲酸酐)反应,然后与环氧化物(例如缩水甘油基取代物)反应而得到的反应产物。正如美国专利No.6,225,408所教导的,进一步的增韧剂体系利用两种具有不同分子量的聚合物的组合。其中教导的具体例子为主要量的主增韧剂(重均分子量(MW)小于约18,000)与次要量的辅增韧剂(MW大于约18,000)的组合。具体例子为甲基丙烯酸缩水甘油酯封端的CTBN橡胶和苯乙烯-[异戊二烯]-苯乙烯的三嵌段共聚物的60:40混合物。在本发明的另外实施方案中,将粘合促进剂加入到丙烯酸类组合物中。本文中可使用的粘合促进剂为已知的含磷化合物,该含磷化合物为次膦酸的单酯、具有一单元的乙烯基或烯丙基不饱和基团的膦酸和磷酸的单酯和二酯。乙烯基不饱和基团是优选的。含磷粘合促进剂的代表实例为(但不限于):磷酸、2-甲基丙烯酰氧乙基磷酸酯、双-(2-甲基丙烯酰氧乙基)磷酸酯、2-丙烯酰氧乙基磷酸酯、双-(2-丙烯酰氧乙基)磷酸酯、甲基-(2-甲基丙烯酰氧乙基)磷酸酯、甲基丙烯酰氧乙基磷酸乙酯、丙烯酰氧乙基磷酸甲酯、丙烯酰氧乙基磷酸乙酯、丙烯酰氧乙基磷酸丙酯、丙烯酰氧乙基磷酸异丁酯、丙烯酰氧乙基磷酸乙基己酯、丙烯酰氧乙基磷酸卤代丙酯、丙烯酰氧乙基磷酸卤代异丁酯、或丙烯酰氧乙基磷酸卤代乙基己酯、乙烯基膦酸、环己烯-3-膦酸、α-羟基丁烯-2-膦酸、1-羟基-1-苯基甲烷-1,1-二膦酸、1-羟基-1-甲基-1,1-二膦酸、1-氨基-1-苯基-1,1-二膦酸、3-氨基-3-羟基丙烷-1,1-二膦酸、氨基-三(亚甲基膦酸)、γ-氨基-丙基膦酸、γ-缩水甘油醚氧基丙基膦酸、磷酸-单-2-氨基乙基酯、烯丙基膦酸、烯丙基次膦酸、β-甲基丙烯酰氧乙基次膦酸、二烯丙基次膦酸、(β-甲基丙烯酰氧乙基)次膦酸和烯丙基甲基丙烯酰氧乙基次膦酸。优选的粘合促进剂为甲基丙烯酸-2-羟乙酯-磷酸酯(HEMA-磷酸酯)。在本发明的另一实施方案中,粘合促进剂包括具有丙烯酸酯官能度的酸,该酸包括丙烯酸、甲基丙烯酸、巴豆酸、异巴豆酸、富马酸、马来酸、肉桂酸、2-甲基马来酸、衣康酸、2-甲基衣康酸、山梨酸和α,β-亚甲基戊二酸。粘合促进剂的其它优选种类包括二甲基丙烯酸金属盐。一种特别优选的粘合促进剂包括二甲基丙烯酸锌。这些粘合促进剂起到双重作用,与金属表面进行金属相互作用以及交联以增强聚合物网络。在本发明的一个实施方案中,二甲基丙烯酸金属盐的含量为0.05重量%至4.0重量%。在本发明优选的实施方案中,二甲基丙烯酸金属盐的含量为约0.5重量%至约2.0重量%。全配方的粘合剂中所通常考虑的其它任选的添加剂包括抗氧化剂、抑制剂、抗流挂添加剂、触变剂、加工助剂、蜡、UV稳定剂、圆弧抑制剂和滴落抑制剂。典型的添加剂的例子为热解法二氧化硅、氧化铝、受阻酚、取代的氢醌、硅烷处理的滑石、云母、长石和硅灰石。虽然本发明的添加剂可以呈现许多形式,但是最优选的粘合剂体系是以多包装或两部分粘合剂体系提供的,其中一个包装或部分包含聚合性或反应性组分和还原剂,第二包装或部分包含氧化剂。在使用时将两部分混合在一起以引发反应性固化。用于分配粘合剂的优选装置为两腔室的盒,该盒在喷嘴中配备有静态搅拌器,并且对于较大规模的应用,优选装置为计量混合配料设备。在混合各个包装后,将混合后的粘合剂体系施加至待接合的一个或两个表面,然后将这两个表面以彼此接触的方式放置。A:B的优选混合比通常包括1:1至15:1,并且更优选为4:1至10:1的。本发明的粘合剂体系可以用来将诸如钢、铝和铜之类的金属表面粘结到各种基材上,所述基材包括金属、塑料和其它聚合物、增强塑料、纤维、玻璃、陶瓷、木材等。本发明的特征是:本文描述的粘合剂组合物能够用来粘结金属基材(例如钢、铝和铜),而仅需在粘合剂施加之前对金属表面进行稍许预处理(如果要进行预处理的话)。因此,甚至能够有效粘结油性金属表面,否则在使用当前大多数可得的底漆和粘合剂时,通常需要对金属表面进行深入的预处理来清洁。另外,本发明的粘合剂体系在室温下提供了有效的粘结强度,因而对于将粘合剂体系施加至基材或对于形成处理强度和尺寸稳定性而言,都不需要加热。虽然本发明的粘合剂优选用来粘结金属表面,但是本发明粘合剂组合物可以作为粘合剂、底漆或涂层施加至能够接受粘合剂的任何表面或基材。优选被本发明粘合剂粘结的金属包括锌、铜、镉、铁、锡、铝、银、铬、这些金属的合金、和这些金属的金属涂层或镀层,例如包括热浸镀、电镀锌钢和合金化热浸镀锌钢的镀锌钢。粘合剂可以刷涂、辊涂、喷涂、点缀、刀涂、滤筒施用,特别是可以从双盒中施加到一个基材上,但是优选施用到两个基材上至所期望的厚度,优选不超过60密耳。在可能预期两个基板的相对运动的安装中,在固化过程中可以将基材夹牢。例如,为了粘结金属表面,将粘附量的粘合剂组合物施加至一个表面上,优选施加至两个表面上,则两个表面隔着粘合剂组合物而彼此相对。为了最佳的结果,粘合剂应该具有小于60密耳的厚度。表面的光滑性和它们之间的间隙(例如,在存在螺母和螺栓的情况下)将决定最佳粘合所需的膜厚度。将两个金属表面和介于其间的粘合剂组合物维持接合,直到粘合剂组合物已经充分固化并粘结两个表面。在室温下的初始固化时间之后,可通过后序烘烤接合部分来促进固化的进行。另外,正如美国专利No.5,487,803和5,470,416中教导的,特别是在折边操作中,优选掺入玻璃珠来控制粘合层厚度。虽然本发明已经结合具体的实施方案进行了描述,但应认识到,这些实施方案仅用于说明本发明的原理。本领域的普通技术人员将认识到,本发明的组合物、装置和方法可以按照其他方式和实施方案构造并实施。因此,本文的描述不应被理解为限制本发明,其它实施方式也落入由所附权利要求书所限定的本发明的范围内。实施例实施例1-固化速率通过测试方法15(TM-15)来测量丙烯酸粘合剂的固化速率。在此测试中,监测经过在25℃下预处理的大约20g的混合粘合剂达到由反应放热产生的最大温度所需的时间(混合后的时间)。还记录了所达到的最大温度。这使得能够相对简单地比较不同粘合剂配方之间的反应速率(引发和进展)。通过仔细地观察温度随时间的变化能够获得额外的信息。在观察温度随固化过程的变化速率时,特别地,在观察最小温度上升所花费的时间量相对于通过固化峰的温度上升速率时,可以估计开放时间与处理强度之比(以下将更详细地描述)。在理想的反应情形下,由于抑制剂阻断了聚合反应的进行,所以在一段时间内温度未升高,然后由于粘合剂固化非常迅速,因此立即升温至最大温度并产生高粘结强度(如下所示)。这通常被称为“瞬时”固化,能够在图1中看到。进行最初的实验以比较各种胺促进剂对所选择的丙烯酸粘合剂体系模型的TM-15固化速度的影响。根据表1中的配方制备了无胺母料配方。表1-母料试剂量丙烯酸酯单体60.2g橡胶增韧剂28g抑制剂80ppm蜡1g甲基丙烯酸1gHEMA-磷酸酯3.5g粘合促进剂0.5g气相二氧化硅4g然后以下表2中所示的量向98.2g的母料中加入还原剂和共促进剂以形成比较样品,能够使用LordAccelerator17(购自LORDCorporation,采用BPO系氧化剂)将该比较样品固化。表2-还原剂&共促进剂试剂12342ADIIPT1.80g0.80g0.80g0.80g1.50gDMA1.00g2.50gDMP1.00gTDA1.00g样品1包含还原剂N,N-二异丙醇对甲苯胺(DIIPT)而不包含共促进剂。样品2包含DIIPT和其它还原剂N,N-二甲基对苯胺(DMA)。样品3包含还原剂DIIPT和常见的叔胺添加剂二甲基哌嗪(DMP)。样品4包含还原剂DIIPT和本发明的共促进剂1,4-二氮杂双环[2.2.2]辛烷(TDA)。对于样品1-4,对相等重量的胺化合物(即还原剂和促进剂的组合)作了直接比较。图2显示了TM-15曲线。在此数据组中并且如图2中所示出,可以看出:在相同的重量负载(weightloading)下,与包含单独的DIIPT、或者同时包含DIIPT和DMA的情况相比,含有TDA的配制物表现出更高的固化速度,并且稍微好于含有DMP的配制物。为了进一步实验,样品2A尝试调节DIIPT/DMA促进剂体系的量以与DIIPT/TDA的固化速度相匹配,这是因为为了进行公平的比较,重要的是评估具有相似固化速度的丙烯酸粘合剂的性质。注意的是,不可能调节仅含有DIIPT配制物的固化速度,使得固化速度接近15分钟。图3显示了速率调节的样品的TM-15曲线。从图3的数据组中可以看出:即使DMA负载显著增加(高于TDA的摩尔当量)并且同时增加DIIPT的负载以提高固化速度,但是升温速率仍不具有与含有TDA的样品相同的“瞬时”固化特性。样品2A的早期温度上升和显著降低放热峰表明反应动力学是显著不同的。如上所述,胺促进剂包装必须克服加入的抑制剂和酸性粘合促进剂的固化抑制。据认为,由TDA产生的更强的“瞬时”固化是由于更强的碱性,该碱性与酸形成强的络合并消除对固化速率的抑制。在含有DMP的配制物中也观察到了较轻程度的这种现象,然而,认为DMP的单环结构阻碍了TDA显示出优异性能。将看到在以下讨论的开放时间和处理强度的时间中反映了这些固化特性。实施例2-开放时间vs处理强度的时间开放时间被定义为在施加有粘合剂珠之后、直至将基材配合在一起以提供与将基材在初始施加粘合剂时即配合在一起时的粘结强度基本上相当的粘结强度这一段时间。“基本上相当”的粘结强度通常理解为立即配合基材所达到的粘结强度的约10%以内。处理强度的时间被定义为在施加粘合剂珠并配合之后使粘结强度超过100psi所需要的时间。对于丙烯酸类粘合剂,较长的开放时间通常导致较长的处理强度的时间。通常最理想的是使处理强度的时间稍微长于开放时间,这被称为“瞬时”固化或有时称为“指令时固化”。通常,丙烯酸类粘合剂呈现出在2,000psi至3,000psi之间的最终粘结强度。通过被称为延迟配合开放时间(DMOT)的测试来测量开放时间。在此测试中,将粘合剂迅速地施加至一系列1”x4”的铝试件上,然后在依次增加的时间间隔的延迟下将试件配合,使得两个试件之间存在0.5”的重叠,并且粘结线厚度(bondlinethickness)为10密耳。然后使粘合剂完全固化(通常一整夜),并将试件剪切拉开。比较了各个试件与匹配延迟时间实际上为零的试件的搭接剪切强度(LSS)和失效模式(粘合与凝聚),然后通过强度的降低>10%的原始强度和/或凝聚失效的降低<80%(>20%粘结失效)来判断开放时间的终点。通过被称为粘结强度发展速率(ROBSD)的测试来测量处理强度的时间。在此测试中,将粘合剂施加至一系列1”x4”的铝试件上,然后将该试件立即配合第二试件上,使得两个试件之间存在0.5”的重叠,并且粘结线厚度为10密耳。然后在依次增加的时间间隔下将试件剪切拉开,评价随着粘合剂的固化和粘结强度的增加,搭接剪切强度随时间的变化。通过粘结强度达到100psi所花费的时间来判断处理强度的时间。下表和图4显示了ROBSD和DMOT测试的数据。正如前面提到的,最重要的是比较具有大致相同的TM-15固化速率的丙烯酸类粘合剂体系。表中的ROBSD数据直接显示了使用TDA作为共促进剂获得了较快的反应动力学。注意的是,ROBSD时间显著长于TM-15时间,这是因为薄粘合线和金属试件大大减少了在反应过程中产生的热。因此与TM-15测试中相比,这些固化反应发生的温度更加接近于恒温(或低温)。ROBSD与DMOT之比清楚地显示了使用TDA获得了更加理想的“瞬时”固化,特别是与含有DMA的样品(其显示出最低的ROBSD/DMOT比值)相比,并且凸显了相对于当前反应促进剂DMP的改善。实施例3-固化速率对酸性化合物的灵敏性用于丙烯酸类粘合剂的粘合促进剂通常包括已知用来促进与金属间的粘结的酸性化合物。一个优选的粘合促进剂为甲基丙烯酸-2-羟基乙酯-磷酸酯(HEMA-磷酸酯)。然而,已知HEMA-磷酸酯会减慢DIIPT固化的粘合剂的固化速度,并且因此在粘合力和固化速率之间存在取舍关系。例如,在市售可得的Lord410/19(使用DIIPT和N,N-二甲基苯胺的组合作为还原剂、并使用BPO作为氧化剂)中,加入约万分之五的HEMA-磷酸酯,将会使20g配制物的放热峰的时间减慢1分钟。令人惊奇的是,DIIPT和TDA的组合使得配制物对HEMA-磷酸酯的负载量的甚至较大的变化不敏感,这使得可使用较大量的粘合促进剂而不改变固化速率。例如,在含有DIIPT和TDA的实验配制物中,添加3%的HEMA-磷酸酯仅使20g配制物的放热峰时间减慢12秒(基本上在测试的误差范围内)。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1