一种用于制备均匀的高摩尔质量环亚氨醚聚合物的方法与流程
本发明涉及一种制备具有高摩尔质量和低(聚合)分散的环亚氨醚聚合物的方法,以及具有这种性质的聚噁唑啉聚合物。本发明还涉及含有该聚合物的药物组合物和医疗设备。
背景技术:
制药和生物技术工业面临着改进药物和候选药物(drug-candidate)的疗效的重大挑战。对于某些类型药物的给药,传统的剂型并非总是特别适合。例如,大多非常有效的药物和候选药物在水中溶解性低,因此人体吸收并不充分;小分子药物容易发生快速肾脏清除,以及生物药容易发生快速酶降解。
已经使用多种方法来改进这类药物的药代动力学特性和药效动力学特性,其中一种是将药物与天然或合成聚合物缀合,另一种是使用聚合物赋形剂形成固体分散体和固溶体。
聚(乙二醇)(peg)是最常用的药物缀合载体。与未改性的形式相比,通过聚乙二醇化(pegylation)增加药物的分子量产生了许多重要的药理学的优点,比如提高了药物的溶解性,延长了循环的半衰期,减少了免疫原性,增加了药物稳定性,还增加抗蛋白质降解的能力。peg通过增加药物的水力半径(hydrodynamicradius),从而(部分地)屏蔽其与人体的相互作用(包括免疫系统和蛋白酶),由此改进药物的疗效。特别地,与其他水溶性聚合物相比,后者的屏蔽特性是peg成功的主要驱动力,这被认为是源于peg的良好水合作用。
尽管聚乙二醇化普遍采用,但是其使用中仍具有许多缺点。有时,可以观察到超敏反应和peg抗体的形成。还可以观察到,当使用高分子量的peg时,其累积在肝脏中,导致所谓的大分子综合征。peg分子的链长在酶的作用下可减短,比如p450或乙醇脱氢酶(alcoholdehydrogenase),增加了有毒的副产物。
至于小治疗分子,通常观察得到,由于仅存在一个或两个可激活的端羟基,仅可与peg获得相对较低的载药量。此外,具有治疗基团、探测基团或靶向基团的peg或peg树枝(pegdendrons)的正交功能化并不容易实现。
此外,制备peg是相对困难和危险的,因为需要爆炸性的且剧毒的浓缩的环氧乙烷单体。而且,peg具有有限的存储性,即存储时需要抗氧化剂来避免过氧化物的形成。
因此,需要良好界定的、不产生免疫性的且无毒的聚合物载体以用于药物递送。
为满足该目的,聚噁唑啉(polyoxazolines,pox)被认为是潜在的备选物。其通常具有制备简单、稳定性好、低毒和低免疫性、高负载能力、良好的亲水性能和良好的抗蛋白质效果(goodproteinrepellenteffect)的优点。
然而,商业可获得的合成聚合材料如peg和pox有一个重要的缺点,事实上它们并非均匀的,它们是含有多种链长的聚合物的宽混合物。分子量的不均匀性(‘多分散性’)—或如今被iupac所推的‘分散性’)—是聚合物生产过程的固有结果。反应混合物中的小分子杂质引起聚合过程中的副反应,从而导致初步的链终止。
因此容易理解的是,在生物医药学应用中特别需要使用良好界定的、化学上均质的材料。就高摩尔质量聚合物而言,通常需要最小化低摩尔质量聚合物碎片的水平。换言之,在生物医药学应用中尽可能降低聚合物分散度。该目标特别难以达到,因为生物医药学应用经常需要高分子量聚合物而分散度随摩尔质量增加而增加,特别是对于环亚氨醚聚合物来说,由于在制备此聚合物过程中,会通过所谓的β-消除发生内部的链转移。
wo2008/106186公开了末端激活的聚噁唑啉(pox)化合物,其含有在末端具有单活性官能团的pox聚合物,所述官能团可与靶分子反应生成靶分子-pox缀合物(conjugate)。在该国际专利申请中,可发现的是,聚合物修饰药物的商业开发经常需要采用分子量(mws)高达40,000da或更高并且分子量分布或多分散度(pds)小于1.1的聚合物,但是已经有大量的工作表明,采用pox常规技术不能获得在上述范围内的mws和pds。wo2008/106186描述了用于获得低pd值的pox衍生物,涉及通过阴离子交换色谱法纯化反应混合物。实施例36描述了mw为15.200da且pd为1.09的聚乙基噁唑啉聚合物(polyethyloxazolinepolymer)。
us2009/156782描述了一种用于制备2-乙基-2-噁唑啉和2-异丙基-2-噁唑啉的单分散均相共聚物和无规共聚物的方法。us2009/156782中描述的该方法包括将反应混合物对水透析纯化。用作参考的实施例1描述了具有9700(dp=86)分子量(mn)和1.02分散度(mw/mn)的聚(2-异丙基-2-噁唑啉)聚合物的制备。
us3,326,929描述了使用酰基氯(acidchlorides)或酸酐处理噁唑啉单体,以充分纯化所述噁唑啉单体,从而通过阳离子活性催化剂便可在链不断裂下实现聚合。但是所得聚合物的准确的mn和分散度并未报道。
us4,281,137描述了从2-噁唑啉混合物中去除水和生色杂质(color-causingimpurities)残留,用于减少这些杂质对于噁唑啉聚合物的颜色的不良影响。这是通过将该单体与氯硅烷或亚磷酸盐反应来达到的。但是该us专利未包含所得聚合物mn和分散度的任何信息。
t.x.viegas等(2011)bioconjugatechem.22,976-986描述了5kda和10kda且分散度小于1.1的聚乙基噁唑啉聚合物的合成的具体过程。
技术实现要素:
本发明的发明人开发了一种通过环亚氨醚单体(cycliciminoethermonomer)的阳离子开环聚合制备均匀的高摩尔质量聚合物的方法。该方法具有的优势在于:反应混合物中的杂质基本完全被去除,从而在聚合过程中不会出现显著的副反应。通过在低温下进行聚合反应还可以进一步抑制副反应的发生。本方法因此可以方便地用于制备生物医药等级的具有高摩尔质量且窄分子量分布(低分散)的环亚氨醚聚合物。
根据本发明的方法包括以下步骤:
a)提供含有环亚氨醚单体和任选的溶剂的第一液体反应混合物;
b)通过阳离子开环聚合将在第一反应混合物中的环亚氨醚单体进行聚合以制得含有(i)聚合材料和(ii)溶剂和/或未反应的环亚氨醚单体的经聚合的第一反应混合物;
c)将溶剂和/或未反应的环亚氨醚单体与所述经聚合的第一反应混合物中含有的聚合材料分离;
d)将分离的未反应的环亚氨醚单体和/或分离的溶剂与其他组分结合,以提供含有环亚氨醚单体和溶剂的第二液体反应混合物;以及
e)通过阳离子开环聚合将在第二反应混合物中的环亚氨醚单体进行聚合以制得聚合度(dp)至少为100的均匀的聚合物。
尽管发明人不期望受到理论的束缚,应当理解的是在本方法的步骤a)到c)中,可有效地移除能够引起聚合反应初步终止(preliminarytermination)的污染物,从而提供的第二液体反应混合物基本不含有这样的污染物。因此,所述第二液体反应混合物能够聚合得到高摩尔质量且非常低分散的聚合物。
本发明还涉及具有高分子量且低分散的聚(2-噁唑啉)聚合物。具体地,本发明涉及的聚合物含有至少50wt.%的衍生自2-取代-2噁唑啉单体的重复单元,所述2-取代-2噁唑啉单体选自2-甲基-2-噁唑啉、2-乙基-2-噁唑啉、2-正丙基-2-噁唑啉、2-异丙基-2-噁唑啉及其组合,所述聚合物具有至少250的dp和小于θmax的分散度,其中θmax是通过下列方程计算得出:
当250≤dp<900时,θmax=1.2+(dp-250)/8000;
当900≤dp<1000时,θmax=1.28125+(dp-900)/500;
当1000≤dp时,θmax=1.48125+(dp-1000)/9770。
本发明还涉及含有此聚合物的药物组合物或医疗设备。
具体实施方式
相应地,本发明一方面涉及通过带有5元环或6元环的环亚氨醚单体的阳离子开环聚合制备均匀的高摩尔质量聚合物的方法,所述方法包括以下步骤:
a)提供含有环亚氨醚单体和任选的溶剂的第一液体反应混合物;
b)通过阳离子开环聚合将在第一反应混合物中的环亚氨醚单体进行聚合以制得含有(i)聚合材料和(ii)溶剂和/或未反应的环亚氨醚单体的经聚合的第一反应混合物;
c)将溶剂和/或未反应的环亚氨醚单体与所述经聚合的第一反应混合物中含有的聚合材料分离;
d)将分离的未反应的环亚氨醚单体和/或分离的溶剂与其他组分结合,以提供含有环亚氨醚单体和溶剂的第二液体反应混合物;以及
e)通过阳离子开环聚合将在第二反应混合物中的环亚氨醚单体进行聚合以制得聚合度(dp)至少为100的均匀的聚合物。
本文使用的术语“环亚氨醚”指的是取代的或非取代的杂环有机化合物,这些有机化合物的环成员包括一个氧原子和一个氮原子,且由一个碳原子隔开。环亚氨醚的实例为噁唑啉(含5元环)和噁嗪(oxazines)(含6元环)。
除非特别指明,本文使用的术语“聚合度”指的是聚合的数均程度(number-averageddegree)。
除非特别指明,本文使用的术语“分散度”指的是分散指数θ。其可使用方程式:θdp=mw/mn计算得出,其中mw是聚合物的重均分子量,mn是数均分子量。
除非特别指明,本文使用的术语“链断裂杂质(chainbreakingimpurities)”指的是能够引起在第二反应混合物中使用的环亚氨醚单体的阳离子开环聚合初步终止或链转移反应的物质。
除非特别指明,本文使用的术语“低温蒸馏”指的是采用真空帮助从通过低温冷却浓缩的挥发物中蒸发出挥发物的蒸馏过程。
根据本发明所述的方法可适用于由噁唑啉、噁嗪和其他环亚氨醚制备高摩尔质量聚合物。根据优选的实施方式,本发明使用的环亚氨醚单体是任选取代的噁唑啉,优选任选2-取代2-噁唑啉。
适用于本发明的方法作为单体的2-取代2-噁唑啉的具体实例包括2-甲基-2-噁唑啉、2-乙基-2噁唑啉、2-正丙基-2噁唑啉、2-异丙基-2-噁唑啉、2-丁基-2-噁唑啉、2-(3-丁烯基)-2-噁唑啉、2-(甲氧基羧乙基)-2-噁唑啉(2-(methoxycarboxyethyl)-2-oxazoline)及其组合。
所述第一反应混合物的聚合典型地是在0-100℃、优选10-60℃以及最优选30-50℃下的温度进行。该反应可在真空或惰性气体中进行。
所述第一反应混合物应含有一定量的活性聚合物(livingpolymer)以形成足量的低聚物的或聚合的活性聚合物链,以便与存在的所有链断裂杂质反应。根据本发明的方法,所述第一反应混合物通常地含有10-100wt.%的环醚单体。更优选地,所述第一反应混合物含有15-80wt.%、最优选20-50wt.%的环亚氨醚单体。
本发明方法可适合地使用无溶剂的第一反应混合物。优选地,所述第一反应混合物含有5-90wt.%的溶剂。更优选地,所述第一反应混合物的溶剂的含量为20-80wt.%,最优选为50-80wt.%。
本发明的发明人发现互不干扰的、不反应的溶剂(极性或非极性)是最适合进行第一反应混合物的聚合的媒介。根据本发明的方法,所述第一反应混合物的溶剂优选选自任选取代的芳香烃、环丁砜(sulfolane)、二甲基乙酰胺及其混合物。更优选地,所述溶剂为任选取代的芳香烃,最优选为氯苯。
为了确保所述第二反应混合物中进行的实际聚合步骤不受分散性-提高副反应(dispersity-increasingsidereactions)的阻碍,重要的是在第一反应混合物中的牺牲聚合(sacrificialpolymerization)期间,所有链断裂杂质已经与活性聚(环亚氨醚)链充分反应。为了实现这一点,所述第一反应混合物中含有聚合引发剂是有用的。典型地,所述第一反应混合物含有的引发剂的浓度为0.1-10wt.%,优选为0.5-5wt.%,以及最优选为1-2wt.%。
所述第一反应混合物中的牺牲聚合产生了聚合材料。剩下的溶剂和/或未反应的环亚氨醚单体基本上完全不含有链断裂杂质,在将它们用于第二聚合步骤之前,需要将它们与聚合材料进行分离。
在本发明优选的实施方式中采用蒸馏、优选采用低温蒸馏将溶剂和/或未反应的环亚氨醚单体与经聚合的第一反应混合物中的聚合材料分离。
本发明的方法步骤c)中获得的分离出的溶剂和分离出的未反应的环亚氨醚单体基本不含有链断裂杂质。本发明的优点在于可实现将分离出的溶剂或分离出的单体包含于第二液体反应混合物中。自然地,所述第二液体反应混合物中同时使用分离的溶剂和分离的单体,也可实现本发明这些优点。
根据本发明的方法的一种实施方式,从聚合第一反应混合物中,与所述经聚合的第一反应混合物中的聚合材料分离的溶剂随后与环亚氨醚单体结合以制得第二液体反应混合物。
为了使分离的溶剂中链断裂杂质的浓度最小化,重要的是应当将溶剂与聚合材料以这样的方式分离,使得分离后溶剂中基本上不含聚合材料。优选地,分离的溶剂含有小于10ppm的聚合材料,更优选小于5ppm,以及甚至更优选小于1ppm。最优选地,分离的溶剂中不含有聚合材料。
在本发明方法的另一种优选的实施方式中,在步骤c)溶剂与聚合材料分离之前,在至少95wt.%的环亚氨醚单体已经聚合之后,将额外的溶剂添加到第一液体反应混合物中,然后继续阳离子开环聚合,额外的溶剂大于第一液体反应混合物中使用的溶剂的80wt.%。该实施方式的优点在于,牺牲聚合步骤的效率将显著增加,因为初始聚合速率大幅地高于在聚合开始之前将所有溶剂引入第一反应混合物中的情况。
根据本发明方法的另外一种优选的实施方式,从第一反应混合物中,将未反应的环亚氨醚单体与所述经聚合的第一反应混合物中的聚合材料分离并随后将分离出的未反应的环亚氨醚单体与溶剂结合以制得第二液体反应混合物。
为了使分离的单体中链断裂杂质的浓度最小化,应当将未反应的环亚氨醚单体与聚合材料以这样的方式分离,使得分离后单体中基本上不含聚合材料。优选地,分离的未反应的环亚氨醚含有小于10ppm、更优选小于5ppm以及甚至更优选小于1ppm的聚合材料。最优选地,分离的未反应的环亚氨醚单体不含有聚合材料。
根据本发明方法的特别优选的实施方式,将分离的溶剂和分离的未反应的环亚氨醚单体都与其他组分结合以制得第二液体反应混合物。
在本发明方法的另一种实施方式中,将溶剂和未反应的环亚氨醚单体与所述经聚合的第一反应混合物中的聚合材料分离并与其他组分结合以制得第二液体反应混合物。为了使分离的单体中链断裂杂质的浓度最小化,应当将溶剂和未反应的环亚氨醚单体与聚合材料以这样的方式分离,使得分离后溶剂和单体中基本上不含聚合材料。优选地,分离的溶剂和未反应的环亚氨醚的组合含有小于10ppm、更优选小于5ppm以及甚至更优选小于1ppm的聚合材料。最优选地,分离的溶剂和未反应的环亚氨醚单体不含有聚合材料。
本发明的方法中,第一反应混合物和第二反应混合物中使用的环亚氨醚单体可以是相同的,也可以是不同的。尤其是,如果在第二反应混合物使用分离的未反应的环亚氨醚单体的情况下,优选第一反应混合物和第二反应混合物中使用同样的环亚氨醚单体。
根据本发明的方法,第二反应聚合物的聚合优选在相对低的温度下进行。原因在于,在相对低的温度下,环亚氨醚更少易于互变异构至其烯胺形式。重要的是保持第二反应混合物中烯胺的浓度尽可能低,因为聚合时烯胺将引起链转移反应,而每个链转移会得到一条终止的聚合物链以及一条新引的聚合物链,这将增加所得聚合物链的摩尔质量的不均匀性。
因此,根据本发明的方法,典型地,所述第二反应混合物的聚合在不高于60℃的温度、优选不高于50℃的温度下进行。典型地,所述第二反应混合物的聚合在至少0℃、优选至少20℃以及最优选30℃的温度下进行。
典型地,所述第二反应混合物含有10-95wt.%、优选20-80wt.%以及最优选20-50wt.%的环亚氨醚单体。
所述第二反应混合物的溶剂含量优选在5-90wt.%的范围内,更优选为20-80wt.%,最优选为50-80wt.%。
发现极性的不反应的溶剂是进行所述第二反应混合物的聚合的最适宜的媒介。优选地,第二反应混合物中的溶剂选自任选取代的芳香烃、环丁砜、二甲基乙酰胺及其混合物。更优选地,所述溶剂是任选取代的芳香烃。最优选地,所述溶剂是氯苯。
所述第二反应混合物优选含有引发剂。更优选地,以聚合前存在的环亚氨醚单体总量为基准,所述第二反应混合物含有的引发剂的浓度为0.00001-0.05mol.%,甚至更优选为0.0005-0.01mol.%。
根据一种特别优选的实施方式,第一反应混合物和第二反应混合物中使用的引发剂在环境压力下具有的沸点大于150℃,优选大于200℃,最优选大于250℃。使用高沸点的引发剂的优势在于,当单体和/或溶剂从第一反应器低温蒸馏到第二反应器时,引发剂仍保留在第二烧瓶里。
本发明的方法还可以使用沸点小于250℃的挥发性引发剂。使用挥发性引发剂的情况下,优选将这些挥发性引发剂与液体组分(如:溶剂)一起注射入第一反应混合物和/或第二反应混合物中。
根据本发明的方法优选的实施方式,所述引发剂为盐。甚至更优选地,聚合前的第一反应混合物和第二反应混合物中含有的引发剂为噁唑啉鎓盐(oxazoliniumsalt)。最优选地,所述噁唑啉鎓盐是晶体且通常为2-取代2-噁唑啉(比如2-苯基-2-噁唑啉)和强酸(比如三氟甲基磺酸(triflic)、四氟硼酸(tetrafluroboric)或六氟磷酸(hexafluorophoric))或烷基化试剂的加合物。
本发明方法中第一反应混合物和/或第二反应混合物中使用的引发剂还可以包括一种或多种功能引发剂。功能引发剂含有功能基团且其能够在聚合物的链末端引入该功能基团。可使用的功能引发剂的实例包括由功能烷基化试剂(functionalalkylatingagent,比如炔丙基三氟甲基磺酸(propargyltriflate))制得的噁唑啉鎓类、在噁唑啉鎓的2-取代基带有功能性的噁唑啉鎓类(例如基于2-甲氧基羧乙基-2-噁唑啉或2-(3-丁烯基)-2-噁唑啉)、或者功能烷基化试剂(比如炔丙基三氟甲基磺酸)。
在本发明的方法优选的实施方式中,环亚氨醚单体总量的一部分通过牺牲聚合与在第一反应混合物中的链断裂杂质反应了,剩余的环亚氨醚单体则用于第二反应混合物中以制备均匀的高摩尔质量聚合物。
因此,本发明的方法中,优选在第二反应混合物中使用大多数的环亚氨醚单体。典型地,与分离的溶剂组合的环亚氨醚单体的量占在本方法中使用的环亚氨醚单体的总量的至少80mol.%,更优选地为至少90mol.%。
根据本发明的方法,第一反应混合物和/或第二反应混合物中的聚合反应可以通过将活性聚合物与适合的亲核终止剂进行封端反应(end-cappingreaction)而终止。在一种优选的实施方式中,当达到期望的分子量时,使用适合的亲核终止剂对第二反应混合物中的聚合进行封端。因此,封端反应可制备出所需分子量和分散性的良好界定的聚合物。
适于高摩尔质量的均匀聚合物封端的终止剂的实例包括:氨,氢氧化钠或氢氧化钾的甲醇溶液,伯胺、仲胺或叔胺(包括哌啶和哌啶的衍生物),硫醇和膦类化合物。
本发明方法的优点在于可以制造高摩尔质量的均匀的聚合物。优选地,本方法制备的均匀的聚合物具有至少200、优选至少250、甚至更优选至少300以及最优选至少400的聚合度。代表性地,该均匀的聚合物的聚合度不超过10,000,尤其是不超过4,000,甚至优选不超过2,000。
在本发明方法中,均匀聚合物的分散度仍然随着分子量的增加而增加。典型地,本发明方法得到的均匀的聚合物具有250的聚合度,并且小于θmax的分散度,其中θmax通过下列方程(方程1)计算得出:
当250≤dp<900时,θmax=1.2+(dp-250)/8000;
当900≤dp<1000时,θmax=1.28125+(dp-900)/500;
当1000≤dp时,θmax=1.48125+(dp-1000)/9770。
优选地,本发明方法得到的均匀的聚合物的分散度小于θmax,其中θmax通过下列方程(方程2)计算得出:
当250≤dp<900时,θmax=1.15+(dp-250)/8000;
当900≤dp<1000时,θmax=1.23125+(dp-900)/500;
当1000≤dp时,θmax=1.43125+(dp-1000)/9770。
最优选地,本发明方法得到的均匀的聚合物的分散度小于θmax,其中θmax通过下列方程(方程)计算得出:
当250≤dp<900时,θmax=1.1+(dp-250)/8000;
当900≤dp<1000时,θmax=1.18125+(dp-900)/500;
当1000≤dp时,θmax=1.38125+(dp-1000)/9770。
下表中,θmax值是根据方程1、2和3计算不同的dp聚合物值得到的。
代表性地,本发明方法得到的均匀的聚合物具有小于1.25、优选小于1.2、更优选小于1.15、更优选小于1.1及最优选地小于1.08的分散度。
本发明聚合物的分散度可以通过尺寸排阻色谱法(sizeexclusionchromatography)并使用含有三氟乙酸钾(3g/l)的六氟异丙醇(hexafluoroisopropanol)作为洗脱液来测得。
本发明的方法可以应用于制备均聚物、嵌段共聚物和统计聚合物(statisticalpolymers)。
优选地,本发明方法制备的均匀的聚合物是均聚物或者为至少50%、优选至少70%以及最优选至少90%的重复单元衍生至环亚氨醚单体的共聚物。最优选地,所述均匀的聚合物是单独由2-噁唑啉单体组成的均聚物或共聚物。
根据本发明的方法可以任选包括额外的步骤以功能化通过第二反应混合物聚合得到的均匀的聚合物。比如,可以在聚合物结构中引入功能基团以使得聚合物可与活性基团(比如治疗基团、靶向基团和/或诊疗基团)形成缀合物。这些缀合物可适用于治疗性治疗(therapeutictreatment)、预防性治疗或者疾病或紊乱的诊断。
所述均匀的聚合物还可以通过将亲电的和/或亲核的基团引入至聚合物的侧基或末端来功能化。亲电的和/或亲核的基团的引入可以使聚合物与其他功能化的聚合物和/或交联剂发生交联反应。由此获得的交联材料(比如聚合物缀合物和固体分散体/固溶体)可适用于多种医疗用途和设备。
根据本发明的方法优选制得了均匀的聚噁唑啉聚合物,如下文所定义的。
本发明另一方面涉及本文所述的方法制备的聚合物。
然而本发明另一方面涉及聚噁唑啉聚合物,其含有至少50wt.%、优选至少60wt.%、最优选至少80wt.%衍生自2-取代2-噁唑啉单体的重复单元,所述2-取代2-噁唑啉单体选自2-甲基-2-噁唑啉、2-乙基-2-噁唑啉、2-正丙基-2-噁唑啉、2-异丙基-2-噁唑啉及其组合,所述聚合物具有至少250的dp和小于θmax的分散度,其中θmax是通过下列方程计算得出:
当250≤dp<900时,θmax=1.2+(dp-250)/8000;
当900≤dp<1000时,θmax=1.28125+(dp-900)/500;
当1000≤dp时,θmax=1.48125+(dp-1000)/9770。
优选地,所述聚噁唑啉聚合物的分散度小于θmax,其中θmax通过下列方程计算得出:
当250≤dp<900时,θmax=1.15+(dp-250)/8000;
当900≤dp<1000时,θmax=1.23125+(dp-900)/500;
当1000≤dp时,θmax=1.43125+(dp-1000)/9770。
最优选地,所述聚噁唑啉聚合物的分散度小于θmax,其中θmax通过下列方程计算得出:
当250≤dp<900时,θmax=1.1+(dp-250)/8000;
当900≤dp<1000时,θmax=1.18125+(dp-900)/500;
当1000≤dp时,θmax=1.38125+(dp-1000)/9770。
本发明的聚噁唑啉聚合物优选地通过本文描述的聚合方法获得。
所述聚噁唑啉聚合物代表性地具有小于1.25、优选小于1.2、更优选小于1.15、更优选小于1.1及最优选小于1.08的聚合度。
根据一种特别优选的实施方式,根据本发明的聚噁唑啉聚合物含有至少50wt.%、优选至少60wt.%以及最优选至少80wt.%的衍生自2-乙基-2-噁唑啉的重复单元。
根据本发明的一种实施方式,所述聚噁唑啉聚合物是均聚物,最优选是2-乙基-2-噁唑啉的均聚物。
根据本发明的另一种实施方式,本发明的聚合物是至少50wt.%、优选至少60wt.%以及最优选至少80wt.%的重复单元衍生自2-c1-3-烷基-2-噁唑啉的共聚物。
根据本发明的聚噁唑啉聚合物的特点在于其同时具有高聚合度和低(聚合)分散度。
根据优一种选的实施方式,根据本发明的聚合物具有至少350、更优选至少400的dp。
本发明的聚噁唑啉聚合物的结构上可以包含功能基团以使得聚合物可与活性基团(比如治疗基团、靶向基团和/或诊疗基团)形成缀合物。根据本发明的聚合物的这些缀合物由此便可治疗性治疗、预防性治疗或者疾病或紊乱的诊断。
本发明的聚噁唑啉聚合物还可以含有亲电的和/或亲核的基团以使聚噁唑啉聚合物与其他聚合物和/或其他交联剂发生交联。由此获得的交联结构可以适合作为多种医疗用途和设备的材料。
本发明的聚噁唑啉聚合物优选通过前文所述的均匀的聚合物的制备方法获得。
本发明另一方面涉及含有前文所述的聚噁唑啉聚合物的药物组合物或医疗设备。根据本发明的药物组合物的实例包括与药物、肽或蛋白质形成的聚合物缀合物、固体分散体和固溶体,以及与药物、肽或蛋白质形成的其他配方。
本发明包括的医疗设备的实例包括组织粘合剂材料(tissueadhesivematerials)、水凝胶材料。
下面通过实施例进一步说明本发明提供的方法,但本发明并不因此而受到任何限制。
实施例:
材料和方法
将2-乙基-2-噁唑啉(etox)进行分馏(fractionallydistilled)并收集128℃沸腾中心馏分(boilingcentrecut)。然后将其与钠搅拌数天,脱气并蒸馏至干净的储罐中。以类似的方式纯化2-甲基-2-噁唑啉(meox)。
通过witte-seeliger环化缩合(witte-seeligercyclocondensation)合成2-异丙基-2-噁唑啉和2-正丙基-2-噁唑啉,并如对etox描述的那样进行纯化。
通过酰基氯(acidchloride)和氯乙醇胺(ethanolaminechloride)的一锅两步反应合成2-甲氧基羧乙基-2-噁唑啉(mestox),然后与氧化钡蒸馏纯化。
将氯苯使用浓硫酸(被洗涤体积的10%)洗涤三次,饱和碳酸氢钠溶液(体积的10%)洗涤三次,再用水洗涤三次。用硫酸镁进行干燥,过滤然后分馏,再然后与五氧化二磷或氢化钙搅拌至少过夜以进行干燥,最后进行蒸馏。
环丁砜通过添加的高锰酸钾且随后添加甲醇,过滤二氧化锰且随后分馏以进行纯化。分馏的环丁砜与氢化钙蒸馏且而后与苯基噁唑啉鎓四氟硼酸盐(phenyloxazoliniumtetrafluoroborate)进行蒸馏,并在干燥箱中处理。
通过使新蒸馏的2-苯基-2-噁唑啉与过量的合适的酸(三氟甲基磺酸、四氟硼酸或六氟磷酸)反应并将产物重结晶至少一次,以制备引发剂盐。
聚合步骤
将干净且干燥的蒸馏桥(distillationbridge)通过锥形接口(conesocket)连接到真空管线上,将schlenk烧瓶连接到一侧,并将安装有三通头(three-waytap)的圆底烧瓶连接到另一侧。将玻璃的或特氟龙(teflon)涂覆的搅拌棒放置在每一侧上。将该体系排空并重新填充上氩气。相对氩气逆流,由schlenk烧瓶上的三通头加入2-5ml三甲基甲硅烷基氯(trimethylsilylchloride,tmscl),并隔离该体系。通过在液氮中浸泡使tmscl冷却,并排空体系。解冻后,tmscl变为基本的气体成分(primaryatmosphericcomponent)并与玻璃上的羟基反应以使体系疏水。在至少30分钟后,用氩气吹扫该体系,除去tmscl,用水、丙酮和石油醚冲洗烧瓶,然后在空气流中干燥。
将引发剂(约100mg)加入schlenk烧瓶中,与所有链断裂杂质反应(通过最终反应混合物的着色消失判断)。将另一部分的引发剂加入到接收烧瓶(该烧瓶中进行主要的聚合)中,其用量取决于所需的聚合物摩尔质量。然后将体系排空16小时。
为了引发聚合,用干燥的氩气吹扫该体系,将溶剂和单体注入schlenk烧瓶中,并搅拌1小时。然后使用用于冷冻的液氮经过冷冻-泵-解冻(freeze-pump-thaw)使溶液脱气三次,并蒸馏到聚合烧瓶中。在聚合烧瓶解冻并用干燥氩气吹扫后,通过关闭三通头隔离该烧瓶,并与真空管线分离,置于42-60℃的预热油浴中。
为了终止聚合,将烧瓶(通过丁基橡胶软管)连接到干燥氩气源,并且(在少量取样后)添加大量过量溶解于甲醇中的氢氧化钾(如果需要羟基端基)或氨气(如果需要胺基端基)。通过在二乙醚中沉淀,然后抽滤(对于较高mw的聚合物,该过程需要重复2-3次以除去动力学上限制(kineticallytrapped)的单体和溶剂),并在真空烘箱或干燥器中干燥过夜以回收聚合物。然后将水溶性聚合物溶于水中并冻干,得到最终的聚合物产物(白色粉末)。
聚合物分析
使用多个不同的色谱柱体系确定分散度。分辨率越高聚合物分布越窄。在所有情况下,聚合物与pmma标准品一样窄。
1.2xplhfipgel和防护装置–低分辨率,中等范围(默认,除非另有说明)
2.2xpsspfg
3.2xpsspfg线性xl和防护装置–中等分辨率和较大范围(规格100-3,000,000da)
一系列合成的聚合物的绝对mw通过粘度测定法测定,然后相对测量的mp进行绘图以导出hfip柱组1中petox和校准标准品(pmma)之间的校正因子0.777。
分散度通过使用含有三氟乙酸钾(3g/l)的六氟异丙醇作为洗脱液的尺寸排阻色谱(sec)测定。由于陡峭的校准曲线(梯度=-1.03167min/logm)引起的仪器致宽,需要使用比较方法修正分散度值,使用最接近的校准物作为对比参照,使用下述方程式:
其中,σ=未知物(1)和校准物(2)的方差,及
实施例1
通过聚合至接近量变(nearquantitativeconversion)以制备具有端胺基的40kda标称重量的聚(2-乙基-2-噁唑啉)。
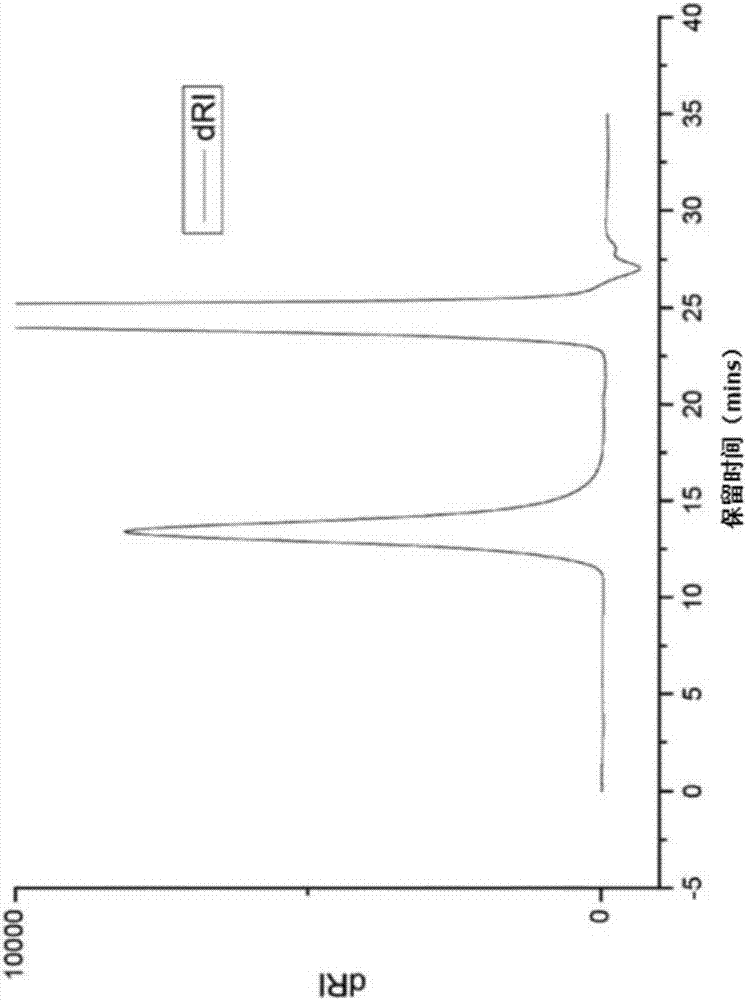
向反应体系中,加入121.9mgh.phox.bf4到清洗容器(cleaningvessel)中,并在聚合容器中加入57.9mg(246μmols)h.phox.bf4。相对干燥的氩气逆流,将氯苯(20ml)和2-乙基-2-噁唑啉(10ml,99mmols,m:i的比率=403)注射到清洁容器中。反应混合物在引发剂结晶溶解后立即变黄。搅拌1小时后,将液体脱气并静态地蒸馏到聚合容器中并密封,在聚合容器中没有颜色变化。将聚合容器在42℃加热21天,然后(取样后)用含有7n氨的甲醇(1ml)进行终止,然后在乙醚中沉淀,得到8.75g(89%产率)白色聚合物,即为具有端胺基的聚(2-乙基-2-噁唑啉)。
如图1所示的,hfipsec显示出与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(含有ktfa的洗脱剂hfip):mp=44,400g/mol,mn=46,100g/mol,
实施例2
带有端胺基的非常高分子量(大约100kda)的聚(2-乙基-2-噁唑啉)的制备如下。
如前文的实施例1,95mgh.phox.bf4加入到清洗容器中,并在聚合容器中加入10.41mg(44.3μmols)h.phox.bf4。将氯苯(20ml)和2-乙基-2-噁唑啉(10ml,m:i=2,236)注射到清洁容器中,然后如实施例1那样处理液体。定期取聚合样品,并且在42℃下保持28天后,在74%转化时终止。沉淀(3个循环)生成5.10g(70%产率)的白色聚合物,即为其具有端胺基的petox。
如图2所示的,hfipsec显示出与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(含有ktfa的洗脱剂hfip):mp=111,200g/mol,mn=94,100g/mol,
实施例3
按照实施例1的步骤,制备具有不同分子质量的一系列petox,结果如表1所示。这些数据是通过在线粘度法分析得到的,并且hfip的校正因子测定为0.777。类似于pmma标准品的宽度,hfip的洗脱峰确实为泊松分布(poisson)。
实施例4
带有端胺基的(2-乙基-2-噁唑啉)(2-异丙基-2-噁唑啉)的嵌段共聚物的制备如下。
如前文的实施例,将64.5mgh.phox.bf4加入到清洁容器中,并向聚合容器中加入10.19mg(43.4μmols)的h.phox.bf4。加入氯苯(5ml)和2-乙基-2-噁唑啉(5ml,m:i=1,141),并且将液体蒸馏到聚合容器中且加热到42℃。45小时后,对反应进行取样,将聚合容器连接到真空系统,脱气并蒸馏出液体,随后进行强烈抽吸(hardpumping)。将氯苯(15ml)和2-异丙基-2-噁唑啉(5ml)从105.1mg的h.phox.bf4蒸馏入其中,并将混合物在42℃下聚合110.75小时,然后用含有7n氨的甲醇(0.5ml)终止并沉淀,得到0.98g白色嵌段共聚物。
如图3所示的,均聚物和二嵌段共聚物的hfipsec显示出与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(具有ktfa的洗脱剂hfip):单嵌段:mp=10,100g/mol,mn=10,100g/mol,
实施例5
带有端胺基的2-乙基-2-噁唑啉和2-甲基羧乙基-2-噁唑啉(mestox)的统计共聚物的制备如下。
如前文的实施例,将96.4mgh.phox.bf4加入到清洁容器中,并向聚合容器中加入11.14mg(47.4μmols)的h.phox.bf4。加入氯苯(10ml)、2-乙基-2-噁唑啉(9ml)和2-甲基羧乙基-2-噁唑啉(1ml,1.16g,7.4mmols)(进料比=1,881:152:1),并且如前面的实施例所述的进行处理。12天后,如前述实施例所述的终止反应混合物,得到2.783g白色粉末,其1h-nmr显示具有13.1%mestox和82.9%etox。
如图4所示,hfipsec显示出与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(含有ktfa的洗脱剂hfip):mp=84,200g/mol,mn=74,200g/mol,
实施例6
如下在60℃制备带有端胺基的100kda聚(2-乙基-2-噁唑啉)。
如前文的实施例,将114.0mgh.phox.bf4加入到清洁容器中,并向聚合容器中加入23.2mg(98.7μmols)的h.phox.bf4。加入氯苯(30ml)和2-乙基-2-噁唑啉(10.2ml;m/i=1024),并且同前述实施例那样进行处理。60℃下反应549小时后,使用过量的甲醇氨(methanolicammonia)终止。在用乙醚沉淀后,溶解在水中并冻干,分离出8.8g(91%)的蓬松白色粉末。
如图5所示,hfipsec显示出与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(具有ktfa的洗脱剂hfip,柱组3):mp=111,100g/mol(未校正),
实施例7
如下制备聚(2-正丙基-2-噁唑啉)(2-乙基-2-噁唑啉)(2-正丙基-2-噁唑啉)三嵌段共聚物。
如前文的实施例,将92.4mgh.phox.bf4加入到清洁容器中,并向聚合容器中加入39.4mg的h.phox.bf4。加入1,3亚苯基二噁唑啉鎓双四氟硼酸盐(1,3phenylenebisoxazoliniumbistetrafluoroborate)(100μmol;双功能引发剂)、氯苯(20ml)和2-乙基-2-噁唑啉(10ml;m/i=1010),并如前述实施例那样进行处理。在60℃下反应114.5小时后,对反应取样并将聚合容器连接到真空管线上,通过蒸馏和抽吸除去挥发物。将氯苯(20ml)和2-正丙基-2-噁唑啉(10ml;88.5mmol)蒸馏到聚合容器中,继续在42℃下聚合139.67小时,然后用甲醇氨终止。在用乙醚沉淀后,溶于水并冻干,分离出13.1g蓬松白色粉末。1h-nmr显示组成为聚(nprox)80(etox)500(nprox)80。
如图6所示,hfipsec显示出均聚物和三嵌段共聚物都具有与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(具有ktfa的hfip,柱组3)显示第一嵌段为mp=106,200g/mol(未校正),
实施例8
如下在60℃下制备带有端胺基的218kda聚(2-乙基-2-噁唑啉)。
按照前文的实施例,将75.6mgh.phox.bf4加入清洗容器中,并向聚合容器加入5.10mg(21.7μmols)的h.phox.bf4。如前述实施例那样注入氯苯(30ml)和2-乙基-2-噁唑啉(10.2ml;m/i=4750)。在60℃下反应672小时(54%转化率)后,用过量的甲醇氨终止反应。在用乙醚沉淀后,溶于水并冻干,分离出4.63g蓬松的白色粉末。
如图7所示,hfipsec显示出与用于校准色谱柱的pmma标准品一样窄的单峰。
sec(具有ktfa的hfip):mp=218,000g/mol(校正的),
实施例9
如下制备10kda聚(2-正丙基-2-噁唑啉)。
如前文实施例所述,加入185.0mgh.phox.bf4至清洁容器,并向聚合容器加入197.2mg(0.84mmols)h.phox.bf4。如前述实施例那样注入氯苯(30ml)和2-乙基-2-噁唑啉(10.2ml;m/i=107)。在42℃下进行反应192小时后,用过量的甲醇氨终止反应。在用乙醚沉淀后,溶于水并冻干,分离9.5g蓬松白色粉末。
hfipsec显示出与用于校准色谱柱的pmma标准物一样窄的单峰,如图8所示。
sec(具有ktfa的hfip):mp=13,200g/mol(校正的)vsp(etox),
实施例10
如下制备带有端胺基的50kda聚(2-甲基-2-噁唑啉)。
如前文实施例所述,加入12.0mgh.phox.bf4至清洁容器,并且向聚合容器加入106.7mg(0.45mmols)h.phox.bf4。同前述实施例注入2-甲基-2-噁唑啉(25ml;m/i=653),并蒸馏到聚合容器中。然后将聚合容器装入干燥箱中,加入环丁砜(83.7ml)。在60℃下进行反应216小时后,用过量的甲醇氨终止反应。在用乙醚沉淀后,溶于水,透析并冻干,分离出15g蓬松的白色粉末。
hfipsec显示出与用于校准色谱柱的pmma标准物一样窄的单峰,如图9所示。
sec(具有ktfa的hfip):mp=104,800g/mol(未校正),
- 还没有人留言评论。精彩留言会获得点赞!