酰胺系弹性体发泡颗粒、其制造方法、发泡成形体和发泡成形体的制造方法与流程
本发明涉及酰胺系弹性体发泡颗粒、其制造方法、发泡成形体和发泡成形体的制造方法(酰胺系弹性体发泡颗粒、其制造方法、发泡成形体和其制造方法)。更特别地,本发明涉及酰胺系弹性体发泡颗粒,可由其得到恢复性(recoverability)和回弹性(resilience)优异、发泡性高且具有微细均一的泡孔结构(cell structure)的发泡成形体,该酰胺系弹性体发泡颗粒的制造方法,恢复性和回弹性优异、发泡性高且具有微细均一的泡孔结构的发泡成形体,和其制造方法。由于本发明的发泡成形体的恢复性和回弹性优异,因此可以用于诸如工业领域、床的芯材、缓冲垫(cushion)的填料、缓冲材料、座垫(新干线和飞行器的座板的缓冲垫)、和汽车构件(汽车内部材料等)等广泛的用途。
背景技术:
过去,聚苯乙烯发泡成形体已经广泛用作缓冲材料和包装材料。因此,发泡成形体可通过如下来得到:将发泡性颗粒如发泡性聚苯乙烯颗粒加热至发泡(预发泡)而得到发泡颗粒(预发泡颗粒)、将所得发泡颗粒填充进模具的模腔中、其后将发泡颗粒二次发泡从而通过热熔合而将它们一体化。
已知的是由于作为原材料的单体是苯乙烯,所以聚苯乙烯发泡成形体具有高的刚性、但是低的恢复性和回弹性。出于该原因,存在的问题在于在反复压缩的用途中、和在要求挠性的用途中难以使用聚苯乙烯发泡成形体。
为了解决上述问题,在专利文献1中,提出了使用包括由具有结晶性聚酰胺链段和聚醚链段的嵌段聚合物组成的热塑性弹性体树脂的交联处理产物的发泡颗粒的发泡成形体。其中据称该发泡成形体显示高的橡胶弹性。
现有技术文献
专利文献
专利文献1:日本审查的专利申请平4-17977号公报
技术实现要素:
发明要解决的问题
然而,专利文献1的发泡颗粒在其表面也是交联的。出于该原因,由于成形性因成形时表面的伸长和颗粒之间的熔合的缺陷而劣化,因此存在的问题是恢复性和回弹性差。此外,由于该缺陷而存在的问题是形状自由度劣化。另外,由于发泡制品的泡孔(平均泡孔直径为320至650μm)粗大且不均一,因此存在的问题是外观劣化。
用于解决问题的方案
本发明的发明人发现了,即使在其中非交联的酰胺系弹性体是基材树脂的情况下,只要其具有规定的肖氏D硬度和平均泡孔直径,则可以提供能够在良好的成形性下赋予发泡成形体以优异的恢复性和回弹性的发泡颗粒,得到了本发明。
因而,本发明提供包括具有65以下的肖氏D硬度的非交联的酰胺系弹性体作为基材树脂且具有20至250μm的平均泡孔直径的酰胺系弹性体发泡颗粒。
另外,本发明提供酰胺系弹性体发泡颗粒的制造方法,所述方法包括以下步骤:
将发泡剂含浸于包括非交联的酰胺系弹性体的树脂颗粒中从而得到发泡性颗粒;和
将所述发泡性颗粒发泡。
此外,本发明提供了通过上述酰胺系弹性体发泡颗粒的模内发泡(in-mold expanding)得到的发泡成形体。
另外,提供了包括将上述酰胺系弹性体发泡颗粒模内发泡的发泡成形体的制造方法。
发明的效果
根据本发明的酰胺系弹性体发泡颗粒,可以提供能够良好成形恢复性和回弹性优异的发泡成形体的发泡颗粒。此外,高发泡是可行的,并且可以实现微细均一的泡孔结构。
另外,当酰胺系弹性体发泡颗粒具有0.015至0.5g/cm3的体积密度时,可以提供能够良好成形恢复性和回弹性更优异的发泡成形体的发泡颗粒。
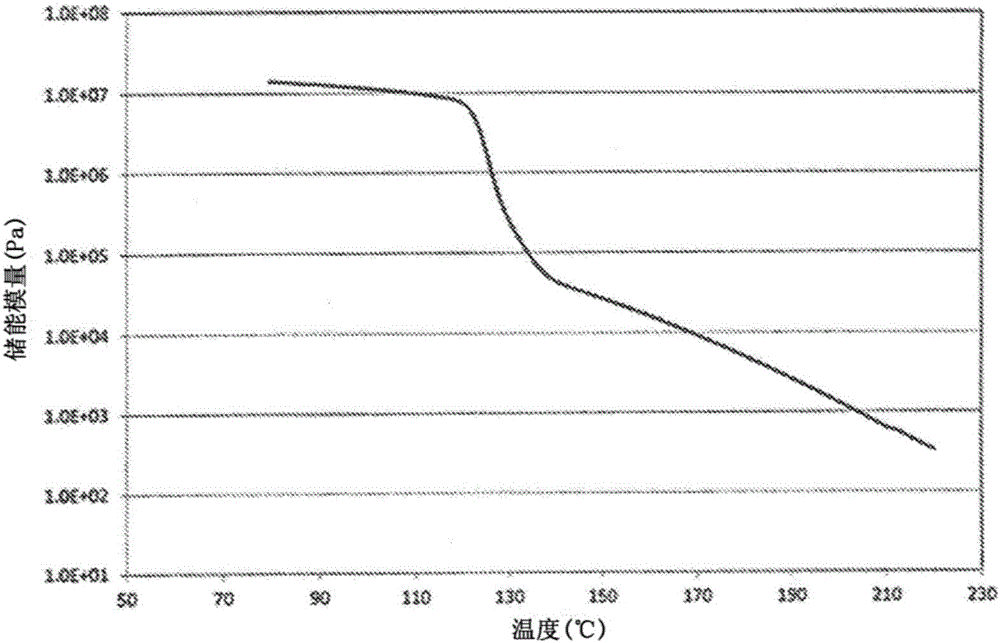
此外,当非交联的酰胺系弹性体为在结晶化温度-10℃的温度下的储能模量和在结晶化温度-15℃的温度下的储能模量均在4×106至4×107Pa的范围内的弹性体时,可以提供能够良好成形恢复性和回弹性更优异的发泡成形体的发泡颗粒。
另外,当非交联的酰胺系弹性体在通过将储能模量的对数表示为y轴和将温度表示为x轴得到的图中、通过由结晶化温度下测量的储能模量和对应于比结晶化温度低5℃的温度下的储能模量得到的等式y=αx+β表示时的α的绝对值为0.08以上的弹性体时,可以提供能够良好成形恢复性和回弹性更优异的发泡成形体的发泡颗粒。
此外,当酰胺系弹性体发泡颗粒具有大于5mm且15mm以下的平均粒径时,可以提供能够良好成形恢复性和回弹性更优异的发泡成形体的发泡颗粒。
另外,当酰胺系弹性体发泡颗粒具有1.5至5mm的平均粒径时,更高的发泡是可行的,并且可以实现微细均一的泡孔结构。
此外,当酰胺系弹性体发泡颗粒具有截面中显示平均泡孔直径为20至150μm的最外表层时,更高的发泡是可行的,并且可以实现微细均一的泡孔结构。
另外,酰胺系弹性体发泡颗粒可以通过将发泡剂含浸于包含非交联的酰胺系弹性体的树脂颗粒中从而得到发泡性颗粒的步骤和将发泡性颗粒发泡的步骤而容易地制造。
当树脂颗粒包含100质量份非交联的酰胺系弹性体和0.02至1质量份泡孔调节剂(cell adjusting agent)时,酰胺系弹性体发泡颗粒可以更容易地制造。
此外,当泡孔调节剂是脂肪酸酰胺系有机物质时,酰胺系弹性体发泡颗粒可以更容易地制造。
另外,当发泡性颗粒通过将发泡剂在水的存在下含浸于树脂颗粒中得到并且以基于100重量份树脂颗粒为0.5至4重量份使用水时,酰胺系弹性体发泡颗粒可以更容易地制造。
此外,本发明可以提供恢复性和回弹性优异的发泡成形体。此外,可以提供具有高发泡性且具有微细均一的泡孔结构的发泡成形体。
当发泡成形体具有10%以下的压缩永久变形和50以上的回弹系数(restitution coefficient)时,可以提供恢复性和回弹性更优异的发泡成形体。
另外,根据本发明的发泡成形体的制造方法,当使用利用在0.27MPa的表压下的水蒸气加热20秒时显示1.5至4.0倍的二次发泡倍率的酰胺系弹性体发泡颗粒进行模内发泡时,可以在更好的成形性下提供上述的发泡成形体。
附图说明
图1为示出实施例1a的酰胺系弹性体的温度与储能模量之间的关系的图。
图2为示出实施例2a的酰胺系弹性体的温度与储能模量之间的关系的图。
图3为示出实施例3a的酰胺系弹性体的温度与储能模量之间的关系的图。
图4为示出实施例4a的酰胺系弹性体的温度与储能模量之间的关系的图。
图5为示出比较例1a的酰胺系弹性体的温度与储能模量之间的关系的图。
图6为示出比较例4a的酰胺系弹性体的温度与储能模量之间的关系的图。
图7为实施例1b的发泡颗粒的截面照片。
图8为比较例1b的发泡颗粒的截面照片。
图9为实施例1c的发泡颗粒的截面照片。
图10为实施例2c的发泡颗粒的截面照片。
图11为用于测量最外表层的平均泡孔直径的方法的示意性说明图。
具体实施方式
(酰胺系弹性体发泡颗粒)
酰胺系弹性体发泡颗粒(下文中,也称为发泡颗粒)包括非交联的酰胺系弹性体作为基材树脂。本说明书中,非交联意指在可溶解的有机溶剂中不溶的凝胶的分数为3.0质量%以下。凝胶分数(gel fraction)可以采取3.0质量%、2.5质量%、2.0质量%、1.5质量%、1.0质量%、0.5质量%、和0质量%。
这里,酰胺系弹性体发泡颗粒的凝胶分数依据下述概要测量。
测量酰胺系弹性体发泡颗粒的质量W1。然后,将酰胺系弹性体发泡颗粒浸渍在130℃下的溶剂(100毫升的3-甲氧基-3-甲基-1-丁醇)中24小时。
然后,将溶剂中的残留物使用80目金属网过滤,并且将该金属网上留下的残余物在130℃下在真空干燥器中干燥1小时,测量该金属网上留下的残余物的质量W2,并且可以基于下式计算酰胺系弹性体发泡颗粒的凝胶分数。
凝胶分数(质量%)=100×W2/W1
(1)非交联的酰胺系弹性体
非交联的酰胺系弹性体具有65以下的肖氏D硬度。当硬度大于65时,在发泡时的软化是困难的,并且不会得到期望的发泡倍率。肖氏D硬度可以采取65、60、55、50、45、40、35、30、25、20、15、10、5、和0。优选的肖氏D硬度为20至60,并且更优选的肖氏D硬度为30至60。
优选的是非交联的酰胺系弹性体具有均在4×106至4×107Pa的范围内的在结晶化温度-10℃的温度下的储能模量和在结晶化温度-15℃的温度下的储能模量。当储能模量大于4×107Pa时,在发泡时的软化是困难的,并且不会得到期望的发泡倍率。当储能模量小于4×106Pa时,发泡形状不会保持并且发泡颗粒在发泡后的冷却过程中会收缩。储能模量可以采取4×106Pa、6×106Pa、8×106Pa、1×107Pa、和4×107Pa。更优选的是储能模量的范围为6×106至2×107Pa。此外,优选的是结晶化温度-10℃的温度与结晶化温度-15℃的温度之间的储能模量都在4×106至4×107Pa的范围内。
优选的是,在通过将储能模量的对数表示为y轴和将温度表示为x轴得到的图中,通过由结晶化温度下测量的储能模量和对应于比结晶化温度低5℃的温度下的储能模量得到的等式y=αx+β表示时的α的绝对值为0.05以上。当该绝对值低于0.05时,发泡颗粒在发泡后的冷却过程中会收缩。α的绝对值可以采取0.05、0.08、0.10、0.15、0.20、0.25、0.30、0.35、0.40、0.45、0.50、0.55、0.60、0.65、和0.70等。更优选的是α的绝对值为0.08以上。优选的是该绝对值的上限为1.0。此外,更优选的是该绝对值在0.08至0.50的范围内。
在通过将储能模量的对数表示为y轴和将温度表示为x轴得到的图中,优选的是通过由对应于比结晶化温度低15℃的温度下的储能模量和对应于比结晶化温度低20℃的温度下的储能模量得到的等式y=α’x+β’表示时的α’与α的比α’/α为0.5以下。当该比超过0.5时,发泡颗粒在发泡后的冷却过程中会收缩。α’/α比的下限优选为0.01。α’/α比可以采取0.01、0.05、0.10、0.15、0.20、0.25、0.30、0.35、0.40、0.45、和0.50。
在非交联的酰胺系弹性体中,可以使用具有聚酰胺嵌段(硬链段)和聚醚嵌段(软链段)的共聚物。
聚酰胺嵌段的实例包括源自聚ε癸酰胺(尼龙6)、聚己二酰丁二胺(尼龙46)、聚己二酰己二胺(尼龙66)、聚癸二酰己二胺(尼龙610)、聚十二烷酰己二胺(尼龙612)、聚己二酰十一烷二胺(尼龙116)、聚十一酰胺(尼龙11)、聚十二酰胺(尼龙12)、聚间苯二甲酰己二胺(尼龙6I)、聚对苯二甲酰己二胺(尼龙6T)、聚对苯二甲酰壬二胺(尼龙9T)、和聚己二酰间苯二甲胺(尼龙MXD6)等的聚酰胺结构。聚酰胺嵌段可以是构成这些聚酰胺结构的单元的组合。
聚醚嵌段的实例包括源自聚乙二醇(PEG)、聚丙二醇(PPG)、聚四亚甲基二醇(PTMG)、和聚四氢呋喃(PTHF)等的聚醚结构。聚醚嵌段可以是构成这些聚醚结构的单元的组合。
聚酰胺嵌段和聚醚嵌段可以随机分散。
优选的是聚酰胺嵌段的数均分子量Mn为300至15,000。聚酰胺嵌段的数均分子量Mn可以采取300、3,000、6,000、9,000、12,000、和15,000。优选的是聚醚嵌段的数均分子量Mn为100至6,000。聚醚嵌段的数均分子量Mn可以采取100、1,000、2,000、3,000、4,000、5,000、和6,000。更优选的是聚酰胺嵌段的数均分子量Mn为600至5,000。更优选的是聚醚嵌段的数均分子量Mn为200至3,000。
优选的是非交联的酰胺系弹性体具有120至180℃的熔点。当熔点低于120℃时,弹性体在当暴露于发泡后的室温下时会收缩,而当熔点超过180℃时,到期望的发泡倍率的发泡会变得困难。熔点可以采取120℃、125℃、130℃、135℃、140℃、145℃、150℃、155℃、160℃、165℃、170℃、175℃、和180℃。更优选的熔点为125至175℃。
非交联的酰胺系弹性体的熔点依照JIS K7121:1987用差示扫描量热计(DSC)测量。此外,熔点是再次升温过程中的吸热峰的温度。
优选的是非交联的酰胺系弹性体具有90至140℃的结晶化温度。当结晶化温度低于90℃时,弹性体在当暴露于发泡后的室温下时会收缩,而当结晶化温度超过140℃时,到期望的发泡倍率的发泡会变得困难。结晶化温度可以采取90℃、95℃、100℃、105℃、120℃、125℃、130℃、135℃、和140℃。更优选的结晶化温度为100至130℃。
非交联的酰胺系弹性体的结晶化温度依照JIS K7121:1987用差示扫描量热计(DSC)测量。此外,结晶化温度是2℃/min的降温速率的降温过程中的放热峰温度。
非交联的酰胺系弹性体中,也可以使用美国专利No.4,331,786、美国专利No.4,115,475、美国专利No.4,195,015、美国专利No.4,839,441、美国专利No.4,864,014、美国专利No.4,230,838、和美国专利No.4,332,920中记载的酰胺系弹性体。
作为非交联的酰胺系弹性体,优选通过将具有反应性端基的聚酰胺嵌段和具有反应性端基的聚醚嵌段的共缩聚得到的非交联的酰胺系弹性体。该共缩聚的实例特别地包括以下:
1)具有二胺链端的聚酰胺嵌段和具有二羧酸链端的聚氧化烯嵌段的共缩聚,
2)通过将称为聚醚二醇的脂族二羟基化α,ω-聚氧化烯单元氰乙基化和氢化得到的具有二羧酸链端的聚酰胺单元和具有二胺链端的聚氧化烯单元的共缩聚,和
3)具有二羧酸链端的聚酰胺单元和聚醚二醇的共缩聚(在该情况下得到的特别地称为聚(醚酯酰胺))。
产生具有二羧酸链端的聚酰胺嵌段的化合物的实例包括通过二羧酸和二胺在α,ω-氨基羧酸、内酰胺或二羧酸的链调整剂(chain regulating agent)的存在下的缩合得到的化合物。
在1)的共缩聚的情况下,例如,通过将聚醚二醇、内酰胺(或α,ω-氨基酸)和作为链限制剂(chain-limiting agent)的二酸在少量水的存在下反应,可以得到非交联的酰胺系弹性体。非交联的聚酰胺系弹性体可以具有各种长度的聚醚嵌段和聚酰胺嵌段,另外,它可以通过各组分的无规反应而分散在聚合物链中。
在上述的共缩聚时,可直接使用聚醚二醇的嵌段,或者可将其羟基与具有羧基末端基团的聚酰胺嵌段共聚而使用,或者在其羟基胺化而使聚醚二醇转变为聚醚二胺之后,将胺化的产物与具有羧基末端基团的聚酰胺嵌段缩合,并且可以使用缩合产物。或者,通过将具有聚酰胺前体的聚醚二醇的嵌段和链限制剂混合并且将混合物进行共缩聚,也可以得到包含随机分散的聚酰胺嵌段和聚醚嵌段的聚合物。
基材树脂除了非交联的酰胺系弹性体之外,还可以在不阻碍本发明的效果这样的范围内包含其他树脂,如酰胺系树脂、聚醚树脂、交联酰胺系弹性体、苯乙烯弹性体、烯烃系弹性体、和酯系弹性体。其他树脂也可以是已知的热塑性树脂或热固性树脂。
(2)发泡颗粒的形状等
优选的是发泡颗粒具有在0.015至0.5g/cm3的范围内的体积密度。当体积密度小于0.015g/cm3时,由于在所得发泡成形体中产生收缩,所以外观不会变得良好,并且发泡成形体的机械强度会降低。当体积密度大于0.5g/cm3时,发泡成形体的轻量性会劣化。体积密度可以采取0.015g/cm3、0.05g/cm3、0.1g/cm3、0.15g/cm3、0.2g/cm3、0.25g/cm3、0.3g/cm3、0.35g/cm3、0.4g/cm3、0.45g/cm3、和0.5g/cm3。更优选的体积密度为0.02至0.2g/cm3,进一步优选的体积密度为0.05至0.1g/cm3。
发泡颗粒的形状没有特别限定,但是其实例包括真球状、椭球状(卵状)、柱状、棱柱状、类丸粒状(pellet-like shape)、或粒状(granular shape)等。
发泡颗粒具有20至250μm的平均泡孔直径。当平均泡孔直径小于20μm时,发泡成形体会收缩。当平均泡孔直径大于250μm时,会造成成形制品的外观的劣化和熔合(fusion)的不良。平均泡孔直径可以采取20μm、40μm、50μm、100μm、150μm、200μm、和250μm。平均泡孔直径更优选为20至200μm,并且进一步优选为40至150μm。
发泡颗粒中,为了阻止发泡成形体的泡孔结构变得不均一,优选的是具有超过250μm的泡孔直径的泡孔的数量少。具体地,具有超过250μm的泡孔直径的泡孔的比例相对于全部泡孔优选为30%以下,更优选为20%以下,并且进一步优选为10%以下。
发泡颗粒可以具有大于5mm且15mm以下的平均粒径。当平均粒径为5mm以下时,成形时的二次发泡性(secondary expandability)会劣化。当平均粒径大于15mm时,在通过模内成形制备发泡成形体时的模具填充性会劣化。平均粒径可以采取5.5mm、8mm、10mm、12mm、14mm、和15mm。更优选的平均粒径为7至12mm。
发泡颗粒可以具有1.5至5mm的平均粒径。当平均粒径小于1.5mm时,发泡颗粒本身的制造会是困难的,并且制造成本会增加。当平均粒径大于5mm时,在通过模内成形制备发泡成形体时,会难以得到具有复杂形状的发泡成形体。平均粒径可以采取1.5mm、2mm、3mm、4mm、和5mm。更优选的平均粒径为2.0至4.0mm。
发泡颗粒可以直接用作缓冲垫的填料,或者可以用作模内发泡用发泡成形体的原材料。当用作发泡成形体用原材料时,通常地,发泡颗粒称为“预发泡颗粒”,并且用于得到它们的发泡称为“预发泡”。
(酰胺系弹性体发泡颗粒的制造方法)
酰胺系弹性体发泡颗粒可通过将发泡剂含浸于包括非交联的酰胺系弹性体的树脂颗粒中从而得到发泡性颗粒的步骤(含浸步骤)和将发泡性颗粒发泡的步骤(发泡步骤)来得到。
(1)含浸步骤
(a)树脂颗粒
树脂颗粒可以使用已知的制造方法和制造设备来得到。
例如,树脂颗粒可以通过将非交联的酰胺系弹性体树脂使用挤出机熔融混炼,然后,通过挤出、水中切割、或线料切割(strand cutting)等进行造粒来制造。在熔融混炼时的温度、时间和压力等可以与待使用的原材料和制造设备相适应而酌情设定。
熔融混炼时挤出机中的熔融混炼温度优选为170至250℃,其为充分地软化非交联的酰胺系弹性体的温度,并且更优选为200至230℃。熔融混炼温度意指通过用热电偶型温度计测量接近挤出机头的熔融混炼产物流路的中心部的温度而得到的挤出机中的熔融混炼产物的温度。
树脂颗粒的形状例如是真球状、椭球状(卵状)、柱状、棱柱状、类丸粒状、或粒状。
树脂颗粒是当其长度由L表示和平均直径由D表示时L/D优选为0.8至3的树脂颗粒。当树脂颗粒的L/D小于0.8或超过3时,到成形模具中的填充性会劣化。此外,树脂颗粒的长度L是指在挤出方向上的长度,平均直径D是指与长度L的方向大致上正交的树脂颗粒的截面的直径。
树脂颗粒的平均直径D优选为0.5至1.5mm。当平均直径小于0.5mm时,发泡剂的保持性(holdability)会劣化,从而劣化发泡性颗粒的发泡性。当平均直径大于1.5mm时,发泡颗粒的到成形模具中的填充性会劣化,与此同时,当制造板状发泡成形体时,发泡成形体的厚度可能无法减少。
树脂颗粒可以包含泡孔调节剂。
泡孔调节剂的实例包括高级脂肪酸酰胺、高级脂肪酸双酰胺、高级脂肪酸盐、和无机泡孔成核剂等。这些泡孔调节剂的多种可以组合。
高级脂肪酸酰胺的实例包括硬脂酸酰胺、和12-羟基硬脂酸酰胺等。
高级脂肪酸双酰胺的实例包括亚乙基双(硬脂酸酰胺)、亚乙基双-12-羟基硬脂酸酰胺、和亚甲基双(硬脂酸酰胺)等。
高级脂肪酸盐的实例包括硬脂酸钙等。
无机泡孔成核剂的实例包括滑石、硅酸钙、和合成或天然存在的二氧化硅等。
泡孔调节剂中,优选高级脂肪酸酰胺和高级脂肪酸双酰胺的脂肪酸酰胺系有机物质。
泡孔调节剂的量优选为0.02至1质量份,基于100质量份非交联的酰胺系弹性体。当泡孔调节剂的量小于0.02质量份,由于泡孔调节剂引起的泡孔的微细化效果会劣化。该劣化使通过将发泡性颗粒发泡得到的发泡颗粒的泡孔粗大,结果,在模内发泡成形时发泡颗粒会破裂且收缩,并且不能得到发泡成形体,或者即使得到发泡成形体,该劣化也会导致外观劣化的产生,如在其表面上凹凸的产生。当该量大于1质量份时,效果达到平台,反之,会引起诸如发泡成形体之间的熔合不足和成本增加等的问题。泡孔调节剂的量更优选为0.02至0.3质量份,并且进一步优选为0.05至0.1质量份。
泡孔调节剂可以与树脂在挤出机中混合,或者可以附着至树脂颗粒。
树脂颗粒可以额外地包含阻燃剂,如六溴环十二烷和三烯丙基异氰脲酸酯6溴化物;着色剂,如炭黑、铁氧化物和石墨;等等。
(b)发泡性颗粒
树脂颗粒含浸有发泡剂从而制造发泡性颗粒。此外,作为将发泡剂含浸于树脂颗粒中的要领,可以使用已知的要领。其实例包括:
·树脂颗粒分散在水中的方法,通过将树脂颗粒、分散剂和水供给至高压釜来搅拌从而制造分散液,将发泡剂在加压下进给至该分散液中,并且将发泡剂含浸于树脂颗粒中(湿式含浸法);和
·将发泡剂在加压下进给至高压釜内的树脂颗粒中,并且将发泡剂含浸于树脂颗粒中的方法(干式含浸法)。
湿式含浸法中的分散剂没有特别限定,但是其实例包括难水溶性的无机物质如磷酸钙、焦磷酸镁、焦磷酸钠和氧化镁,以及表面活性剂如十二烷基苯磺酸钠。
干式含浸法中,由于不使用大量的水,所以水处理是不必要的,因而具有可以减少制造成本的优势。干式含浸法没有特别限定,但是可以在已知的条件下进行。例如,含浸温度可以为40至140℃。
干式含浸法中,优选的是在少量水的存在下含浸发泡剂。通过使少量水存在,可以防止发泡剂从发泡颗粒的表面逸散。本发明人认为,通过将少量的水定位在发泡性颗粒的表面上,阻止发泡性颗粒中的发泡剂的逸散。水的量优选为0.5至4质量份,基于100质量份树脂颗粒。当水的量小于0.5质量份时,水的展开不充分,不能防止发泡剂的逸散,并且发泡颗粒的表层中的泡孔会粗大化。当水的量超过4质量份时,在从干式含浸装置中提取出时产生桥连(bridging),并且可能无法取出发泡性颗粒。另外,即使当可以取出发泡性颗粒时,由于时间对于取出是必要的,结果发泡剂逸散,并且表层泡孔会粗大化。更优选的水的量为1至3质量份。
作为发泡剂,使用通用的发泡剂,其实例包括无机气体如空气、氮气和二氧化碳(碳酸气体)和氩气;脂族烃如丙烷、丁烷和戊烷;和卤代烃,并且优选无机气体和脂族烃。此外,发泡剂可以单独使用,或者其两种以上可以同时使用。
优选的是含浸于树脂颗粒中的发泡剂的量为4质量份以上,基于100质量份树脂颗粒。此外,该量优选为12质量份以下。当该量小于4质量份时,发泡能力降低,并且在高发泡倍率下难以进行良好发泡。当发泡剂的含量超过12质量份时,容易产生泡孔膜的破坏,增塑效果变得太大,发泡时的粘度容易降低,并且收缩变得容易出现。发泡剂的更优选的量为5质量份以上。发泡剂的进一步优选的量为6至8质量份。在该范围内,发泡能力可充分地提高,甚至在高发泡倍率下,颗粒可以进一步良好地发泡。当发泡剂的含量为8质量份以下时,抑制泡孔膜的破坏,由于增塑效果不变得太大,所以抑制了发泡时粘度的过分降低,并且抑制收缩。此外,发泡剂的量可以为5至30质量份,或者可以为10至20质量份,基于100质量份树脂颗粒。
相对于100质量份树脂颗粒已含浸的发泡剂的含量(含浸量)如下测量。
(在无机气体的情况下)
测量在树脂颗粒放置于压力容器中之前的质量Xg。在压力容器中,在树脂颗粒含浸有发泡剂之后,测量从压力容器中取出含浸产物之后的质量Yg。通过下式,得到相对于100质量份树脂颗粒已含浸的发泡剂的含量(含浸量)。
发泡剂的含量(质量%)=((Y-X)/X)×100
(在无机气体以外的气体的情况下)
精确称量约15mg量的树脂颗粒,放在由Shimadzu Corporation制造的热解器PYR-1A的分解炉入口处,并且用氮气吹扫约15秒从而使在样品安放时混合于其中的气体排出。关闭之后,将样品插入在200℃下的炉芯中,加热120秒以排出气体,并且使用由Shimadzu Corporation制造的气相色谱GC-14B(检测器:FID)定量确定该排出的气体,由此得到残留气体的量。测量条件为柱使用Shimalite 60/80 NAW(Squalane 25%)3m×3φ,使用柱温(70℃),载气(氮气),载气流量(50ml/min),注入口温度(110℃),和检测器温度(110℃)。
当发泡剂含浸于树脂颗粒中的温度低时,将发泡剂含浸于树脂颗粒中所必要的时间将变长,并且生产效率会降低。另一方面,当该温度高时,树脂颗粒相互熔合从而生成结合的颗粒。含浸的温度优选为-15至120℃,更优选在0至110℃或60至120℃的范围内,并且进一步优选为70至110℃。发泡助剂(增塑剂)可以与发泡剂一起使用。发泡助剂(增塑剂)的实例包括己二酸二异丁酯、甲苯、环己烷、和乙苯等。
(2)发泡步骤
发泡步骤中,发泡温度和加热介质没有特别限定,只要发泡性颗粒可以发泡而得到发泡颗粒即可。
在发泡步骤中,优选的是无机组分添加至发泡性颗粒。无机组分的实例包括如碳酸钙和氢氧化铝等无机化合物的颗粒。无机组分的添加量优选为0.03质量份以上,更优选为0.05质量份以上,优选为0.2质量份以下,更优选为0.1质量份以下,基于100质量份发泡性颗粒。
当在高压蒸气下进行发泡时,如果使用有机聚结防止剂,它在发泡时熔融。另一方面,无机聚结防止剂如碳酸钙甚至在高压蒸气加热下也具有充分的聚结防止效果。
无机组分的粒径优选为5μm以下。无机组分的粒径的最小值为0.01μm左右。当无机组分的粒径不大于上限时,无机组分的添加量可降低,并且无机组分变得难以负面影响(阻害)(inhibit)后面的成形步骤。
此外,发泡之前,粉末状金属皂如硬脂酸锌;碳酸钙;和氢氧化铝可以涂布在树脂颗粒的表面上。该涂布可以减轻发泡步骤中树脂颗粒之间的结合。或者,可以涂布表面处理剂如抗静电剂和展开剂。抗静电剂的实例包括聚氧乙烯烷基酚醚、和硬脂酸甘油单酯等。展开剂的实例包括聚丁烯、聚乙二醇、和硅油等。
(发泡成形体)
发泡成形体通过将发泡颗粒模内成形而得到,并且包括多个发泡颗粒的熔合体(fused body)。例如,发泡成形体可以通过将发泡颗粒填充至具有大量小孔的密闭模具中,将发泡颗粒用加压水蒸气加热发泡从而填充发泡颗粒之间的间隙,并且与此同时,将发泡颗粒相互地熔合从而使其一体化来得到。于是,例如,发泡成形体的密度可以通过调节要填充到模具中的发泡颗粒的量等来调节。
优选的是发泡成形体具有0.015至0.5g/cm3的密度。在该范围内,可以使压缩永久变形和机械物理特性在良好的平衡下兼容。更优选的密度为0.02至0.2g/cm3。
优选的是使用当用在0.27MPa的表压下的水蒸气加热20秒时显示1.5至4.0倍的二次发泡倍率的上述酰胺系弹性体发泡颗粒进行模内发泡。在该二次发泡倍率的范围内,可得到其中发泡颗粒更加熔合的发泡成形体。
另外,发泡颗粒可以含浸有非活性气体以改善发泡颗粒的发泡能力。通过改善发泡能力,发泡颗粒之间的相互熔合性(fusibility)在模内成形时得以改善,并且发泡成形体具有进一步优异的机械强度。此外,非活性气体的实例包括二氧化碳、氮气、氦气和氩气等。
将非活性气体含浸于发泡颗粒中的方法的实例包括:通过将发泡颗粒放置在具有大气压以上的压力下的非活性气体的气氛下而将非活性气体含浸有发泡颗粒的方法。发泡颗粒可以在填充至模具之前含浸有非活性气体,或者在将发泡颗粒填充至模具之后,发泡颗粒可以通过将它们在非活性气体气氛下与模具放置在一起而含浸有非活性气体。此外,当非活性气体为氮气时,优选的是使发泡颗粒放置在0.1至2.0MPa的氮气气氛下20分钟至24小时。
当发泡颗粒已含浸有非活性气体时,发泡颗粒可以当它们在模具中时加热和发泡,或者在将发泡颗粒填充至模具之前,将发泡颗粒加热和发泡以得到高发泡倍率的发泡颗粒,其后,发泡颗粒可以填充至模具中,并且加热和发泡。通过使用这样的高发泡倍率的发泡颗粒,可得到高发泡倍率的发泡成形体。
此外,当在制造发泡颗粒时使用聚结防止剂时,在制造发泡成形体时,可以在聚结防止剂附着至发泡颗粒的同时进行成形。此外,为了促进发泡颗粒的相互熔合,聚结防止剂可以在成形步骤之前洗涤从而除去,或者在除去或没有除去聚结防止剂的情况下,作为熔合促进剂的硬脂酸可以在成形时添加。
本发明的发泡成形体可以用于例如,工业领域、床的芯材、缓冲垫的填料、缓冲材料、座垫(新干线和飞行器的座板的缓冲垫)、和汽车构件(汽车内部材料等)等。特别地,发泡成形体可以用于其中要求压缩永久变形的改善的预期用途。
实施例
然后,将通过实施例的方式进一步详细地解释本发明,但是本发明不限于这些。
<肖氏D硬度>
肖氏D硬度是指依照JIS K6253-3:2012的试验方法测量的硬度。具体地,将D-型硬度计垂直地推向已经调节为100mm×100mm×10mm厚的样品,并且测量1秒之后的数值。然后,在距离样品外端12mm以上内侧的测量位置处进行测量,确保测量点之间10mm间隔,对于一个样品在5点处进行测量,并且平均值定义为肖氏D硬度。
<储能模量>
动态粘弹性用粘弹性测量装置Physica MCR301(由Anton Paar制造)和温度控制系统CTD450测量。首先,将树脂样品用热压机在180℃温度的条件下成形为具有25mm直径和3mm厚度的圆盘状试验片。将试验片用真空干燥器在120℃、减压条件下干燥4小时。然后,将试验片安放在于测量温度下加热的粘弹性测量装置的板上,并且在氮气气氛下加热和熔融5分钟。其后,将试验片用具有25mm直径的平行板挤压直到2mm的间隔,并且除去从板中突出的树脂。另外,在温度达到测量温度±1℃并且试验片加热5分钟之后,在动态应变为1%、频率为1Hz、测量模式为振动测量(温度依赖性)和从220℃冷却的冷却速率为2℃/min的情况下,在氮气气氛下测量动态粘弹性,并且测量储能模量的温度依赖性。
<结晶化温度>
结晶化温度通过JIS K7121:1987“塑料的转变温度的测试方法(Testing Methods for Transition Temperatures of Plastics)”中记载的方法来测量。条件是采样方法和温度条件如下进行。使用差示扫描量热计DSC6220型(由SII Nano Technology Inc.制造),将约6mg的样品填充至铝制测量容器的底部而没有间隙;当在20mL/min的氮气流量下温度从30℃升高至220℃(加热),保持10分钟,温度从220℃降低至-40℃(冷却)时,获得DSC曲线。另外,升温速率为10℃/min,和降温速率为2℃/min。作为基准物质,使用氧化铝。此外,结晶化温度是通过使用装置附带的分析软件,读取在冷却过程中观察到的最高温度侧的结晶峰顶的温度得到的值。
<熔点>
熔点通过JIS K7121:1987“塑料的转变温度的测试方法”中记载的方法来测量。条件是采样方法和温度条件如下进行。使用差示扫描量热计DSC6220型(由SII Nano Technology Inc.制造),将约6mg的样品填充至铝制测量容器的底部而没有间隙;当在20mL/min的氮气流量下温度从30℃降低至-40℃,保持10分钟,温度从-40℃升高至220℃(第一次加热),保持10分钟,温度从220℃降低至-40℃(冷却),保持10分钟,温度从-40℃升高至220℃(第二次加热)时,获得DSC曲线。另外,升温和降温全部以10℃/min的速度进行,并且作为基准物质,使用氧化铝。此外,熔点是通过使用装置附带的分析软件,读取在第二次加热过程中观察到的最高温度侧的熔融峰顶的温度得到的值。
<发泡颗粒的体积密度>
首先,Wg发泡颗粒收集作为测量样品,将该测量样品自然落下至量筒中,轻轻振荡以使样品的表观体积(V)cm3恒定,测量其质量和体积,并且基于下式测定发泡颗粒的体积密度。
体积密度(g/cm3)=测量样品的质量(W)/测量样品的体积(V)
<发泡颗粒的平均粒径>
使用Ro-Tap型振筛机(由SIEVE FACTORY IIDA CO.,LTD制造),将约50g发泡颗粒利用筛目开孔为16.00mm、13.20mm、11.20mm、9.50mm、8.00mm、6.70mm、5.60mm、4.75mm、4.00mm、3.35mm、2.80mm、2.36mm、2.00mm、1.70mm、1.40mm、1.18mm、和1.00mm的JIS标准筛分级5分钟。测量筛网上的样品的质量,并且基于从结果中得到的累积质量分布曲线,累积质量变为50%时的粒径(中值直径)定义为平均粒径。
<平均泡孔直径和泡孔的比例>
发泡颗粒的平均泡孔直径是指依照ASTM D2842-69的试验方法测量的平均泡孔直径。具体地,将发泡颗粒切割为大致两个相等的部分,将切出的截面使用扫描电子显微镜(产品名“S-3000N”,由Hitachi,Ltd.制造)放大100倍且拍摄。拍摄的图像的各4个图像打印在A4纸张上,在任意的地方画出一条具有60mm长度的直线,并且从该直线上存在的泡孔数,通过下式计算泡孔的平均弦长(t)。
平均弦长t=60/(泡孔数×照片的放大倍率)
此外,当在画出直线时,直线与泡孔点接触,则该泡孔也包括在泡孔数内,另外,当直线的两端部处在泡孔内的位置而没有贯穿泡孔的状态时,直线的两端部所处于的泡孔也包括在泡孔数内。另外,在拍摄的图像的任意5个地方,以与如上所述相同的要领计算平均弦长,并且这些平均弦长的算术平均值定义为发泡颗粒的泡孔的平均泡孔直径。
另外,通过用于上述平均泡孔直径的测量方法中的图像,测量全体泡孔数、具有超过250μm的泡孔直径的泡孔数、具有150μm以上且小于250μm的泡孔直径的泡孔数、和具有小于150μm的泡孔直径的泡孔数。计算各泡孔直径的数相对于全体泡孔的比。
<用于确定熔合性的标准>
○:发泡成形体中,当形成深度5mm的缺口,并且发泡制品沿着缺口弯曲时,将不会断裂。
×:发泡成形体中,当形成深度5mm的缺口,并且发泡制品沿着缺口弯曲时,断裂。
<压缩永久变形>
压缩永久变形依照压缩永久变形试验(JIS K6767:1999)通过以下计算方法测量。
·试验片:50W×50L×25T(mm)(单侧片(one side skin))
·压缩比:25(%)
·试验数:3
压缩永久变形的计算方法
压缩永久变形(%)={(原始厚度(mm)–试验后的厚度(mm))/原始厚度(mm)×100}
<回弹性>
回弹性是指依照JIS K6400-3:2011的试验方法测量的回弹性。具体地,使用由KOBUNSHI KEIKI CO.,LTD.制造的泡沫回弹性试验机FR-2型,具有5/8英寸直径和16.3g质量的刚玉(corundum)从500mm的高度落到已经成形为50mm厚度的成形制品上,并且读取刚玉达到最高回弹时的高度。重复总计5次的相同操作,并且从平均值计算回弹性。
回弹性的计算方法
回弹性(%)=刚玉达到最高回弹时的高度(mm)/500(mm)×100
实施例1a
(1)含浸步骤
将作为其中尼龙12为硬链段和聚四亚甲基二醇为软链段的酰胺系弹性体的Pebax 5533(由Arkema制造)的颗粒(平均直径3mm)100质量份密封在压力容器中,压力容器的内部气氛用碳酸气体置换,并且将碳酸气体在加压下进给直到4.0MPa的含浸压力。使颗粒放置在20℃的环境下,经过24小时的含浸时间,其后,压力容器的内部气氛经5分钟缓慢减压。通过这样做,酰胺系弹性体含浸有碳酸气体从而得到发泡性颗粒。当通过上述方法测量已经含浸于发泡性颗粒中的发泡剂的含浸量时,该量求得为6.2质量%。
(2)发泡步骤
在上述(1)含浸步骤中减压之后即刻,将发泡性颗粒从压力容器中取出,其后,添加碳酸钙0.08质量份,并且混合材料。其后,使用水蒸气,将上述含浸产物用水蒸气在高压发泡浴中发泡,同时在136℃的发泡温度下搅拌。发泡之后,将颗粒从高压发泡浴中取出,用氯化氢水溶液除去碳酸钙,其后,使用气流输送式干燥机进行干燥从而得到发泡颗粒。当所得发泡颗粒的体积密度通过上述方法测量时,求得其为0.05g/cm3。此外,当测量所得发泡颗粒中的平均泡孔直径时,求得其为180μm。
(3)成形步骤
使所得发泡颗粒放置在室温(23℃)下1天,并且密封在压力容器中,压力容器的内部气氛用氮气置换,将氮气在加压下进给直到1.0MPa的含浸压力(表压)。使颗粒放置在20℃的环境下,并且进行加压调制(pressure curing)8小时。取出颗粒,并且用0.22MPa下的水蒸气加热20秒以便测量二次发泡倍率,二次发泡倍率求得为3.5倍。将与已经测量了二次发泡倍率的发泡颗粒不同的发泡颗粒填充至23mm×300mm×400mm的成形模具中,用0.27MPa下的水蒸气加热40秒,然后冷却直到发泡成形体的最大表面压力降至0.01MPa,由此得到了发泡成形体。发泡颗粒和发泡成形体的评价在表1中记载。
实施例2a
除了弹性体变为其中尼龙12为硬链段和聚醚为软链段的酰胺系弹性体的Pebax 4033(由Arkema制造)之外,以与实施例1a中相同的方式进行含浸、发泡和成形。发泡颗粒和发泡成形体的评价在表1中记载。
实施例3a
除了弹性体变为其中尼龙12为硬链段和聚醚为软链段的酰胺系弹性体的UBESTA9040X1(由Ube Industries,Ltd.制造)之外,以与实施例1a中相同的方式进行含浸、发泡和成形。发泡颗粒和发泡成形体的评价在表1中记载。
实施例4a
除了弹性体变为其中尼龙12为硬链段和聚醚为软链段的酰胺系弹性体的UBESTA9048X1(由Ube Industries,Ltd.制造)之外,以与实施例1a中相同的方式进行含浸、发泡和成形。发泡颗粒和发泡成形体的评价在表1中记载。
实施例5a
将Pebax 5533(由Arkema制造)的颗粒(平均直径3mm)100质量份、水100质量份、作为分散剂的磷酸三钙0.5质量份、具有25%纯度的十二烷基苯磺酸钠0.1质量份、作为发泡剂的丁烷12质量份和作为泡孔调节剂的亚乙基双(硬脂酸酰胺)0.2质量份密封在压力容器中,在搅拌的同时在2℃/min的升温速率下加热至110℃。将材料保持在110℃下5小时,并且进行发泡剂的含浸处理。冷却至20℃之后,从容器中取出含浸产物,并且以与实施例1a中相同的方式进行发泡和成形。
比较例1a
除了弹性体变为其中尼龙12为硬链段和聚醚为软链段的酰胺系弹性体的Pebax 7033(由Arkema制造)之外,以与实施例1a中相同的方式进行含浸。在146℃的发泡温度下进行发泡,但是发泡没有成功。
比较例2a
除了从压力容器中取出含浸产物,使其放置在室温下直到经过20分钟,并且将颗粒中包含的碳酸气体的量减少至4.5%之外,以与实施例1a中相同的方式进行比较例2a。当测量发泡颗粒的泡孔直径时,求得为270μm。发泡颗粒和发泡成形体的评价在表2中记载。
比较例3a
将Pebax 5533(由Arkema制造)的颗粒(平均直径3mm)100质量份、水100质量份、作为分散剂的磷酸三钙0.5质量份、具有25%纯度的十二烷基苯磺酸钠0.1质量份、和作为交联剂的过氧化二枯基0.6质量份密封在压力容器中,在搅拌的同时在2℃/min的升温速率下加热至130℃。将材料保持在130℃下5小时,并且进行交联处理。冷却至40℃之后,以与实施例1a中相同的方式进行含浸。在146℃的发泡温度下进行发泡,但是发泡没有成功。
比较例4a
除了弹性体变为其中尼龙12为硬链段和聚醚为软链段的酰胺系弹性体的Pebax 2533(由Arkema制造)之外,以与实施例1a中相同的方式进行含浸。在120℃的发泡温度下进行发泡,但是发泡之后出现收缩,并且不能得到发泡颗粒。
实施例1a至5a的结果示于表1中,比较例1a至4a的结果示于表2中。表1和表2中,成形制品的外观基于以下标准确定。
○:颗粒之间没有间隙,并且成形制品的表面是平坦的。
×:颗粒之间产生间隙,并且成形制品的表面的平滑性差。
此外,显示实施例1a(实施例5a)、实施例2a至4a、比较例1a(比较例2a和3a)、和比较例4a的酰胺系弹性体的温度与储能模量之间的关系的图示于图1至6中。
[表1]
[表2]
从实施例1a至5a和比较例1a至4a中,可见的是通过包含作为基材树脂的具有65以下的肖氏D硬度的非交联的酰胺系弹性体、并且具有0.015至0.5g/cm3的体积密度和20至250μm的平均泡孔直径,可以提供熔合性、外观、压缩永久变形和回弹性优异的发泡成形体。
然后,将通过其他实施例的方式进一步详细地解释本发明,但是本发明不限于这些。
(酰胺系弹性体的树脂颗粒的制造例)
首先,将其中尼龙12为硬链段和聚四亚甲基二醇为软链段的非交联的(结晶性)酰胺系弹性体(产品名“Pebax 5533”,由Arkema制造,凝胶分数3质量%以下,肖氏D硬度55)供给至具有65mm口径的单螺杆挤出机,并且熔融混炼。此外,在单螺杆挤出机中,将酰胺系弹性体初始在180℃下熔融混炼,其后在温度升高至220℃的同时熔融混炼。
随后,在将熔融状态下的酰胺系弹性体冷却之后,酰胺系弹性体通过安装在单螺杆挤出机的前端的多喷嘴模头的各个喷嘴挤出。此外,多喷嘴模头具有40个喷嘴,其中出口部具有0.7mm的直径,并且所有的喷嘴的出口部以相等的间隔分布在假定位于多喷嘴模头的前端面上的具有139.5mm直径的虚拟圆上。多喷嘴模头保持在220℃下。
4个旋转叶片沿旋转轴的周向以相等的间隔一体地分布在旋转轴的后端部的外周面上,并且将各旋转叶片配置为使其在始终接触多喷嘴模头的前端面的状态下在虚拟圆上移动。
另外,提供具有由正面圆形状的前部和从该前部的外周缘向后延伸且具有315mm内径的圆柱状周壁部构成的冷却鼓的冷却构件。冷却水通过供给管和鼓的供给口而供给至冷却鼓,并且在周壁部的整个内表面上,20℃的冷却水沿着该内表面向前螺旋地流动。
分布在多喷嘴模头的前端面上的旋转叶片以3,440rpm的转数旋转,并且将已经通过多喷嘴模头的各喷嘴的出口部挤出的酰胺系弹性体挤出物用旋转叶片切割从而制造大致球形状的酰胺系弹性体的树脂颗粒。
此外,在制造树脂颗粒时,首先,旋转轴没有安装在多喷嘴模头中,并且将冷却构件从多喷嘴模头中缩回。在该状态下,酰胺系弹性体从挤出机中挤出。然后,将旋转轴安装在多喷嘴模头中,并且将冷却构件在规定位置配置,其后,旋转轴旋转,并且酰胺系弹性体用旋转叶片在喷嘴的出口部的开口端切割,从而制造树脂颗粒。
树脂颗粒通过由于旋转叶片引起的切割应力而向外或向前飞扬,并且与沿着冷却构件的冷却鼓的内表面流动的冷却水碰撞从而使其立即冷却。
冷却的树脂颗粒与冷却水一起通过冷却鼓的排出口排出,并且用脱水机与冷却水分离。所得树脂颗粒具有1.2至1.7mm的颗粒长度L和0.8至0.9mm的颗粒直径D。
(实施例1b)
<发泡性颗粒的制备>
将具有内容积5升的搅拌器的高压釜装填有2.3kg上述树脂颗粒、2.0kg蒸馏水、6.0g焦磷酸镁和0.81g十二烷基苯磺酸钠,并且将材料在320rpm的搅拌下悬浮。
然后,向0.3kg蒸馏水中添加0.9g焦磷酸镁、0.11g十二烷基苯磺酸钠和1.2g作为泡孔调节剂的亚乙基双(硬脂酸酰胺)(基于100质量份树脂颗粒为0.05质量份),将材料用均质混合器搅拌从而制备悬浮液,并且将该悬浮液添加至反应容器中。其后,将温度升高至110℃,460g作为发泡剂的丁烷(异丁烷:正丁烷=35:65(质量比))在加压下进给至其中,并且将混合物保持在110℃下6小时,冷却至20℃,取出,洗涤,脱水,和干燥而得到发泡性颗粒。发泡性颗粒的整个表面均匀地覆盖有基于100质量份所得发泡性颗粒为0.1质量份的碳酸钙。
<发泡颗粒的制备>
将发泡性颗粒放在具有50升体积量的圆柱状间歇式加压预发泡机中,并且用蒸气加热,由此得到了发泡颗粒。发泡颗粒具有0.1g/cm3的体积密度、116μm的平均泡孔直径、1.9mm的平均粒径。发泡颗粒的截面照片示于图7中。从图7中,可见的是发泡颗粒具有微细均一的泡孔结构。
<发泡成形体的制备>
将发泡颗粒放在密闭容器中,将氮气在0.5MPa的压力(表压)下加压进给至该密闭容器,并且使该容器放置在室温下6小时,从而将氮气含浸于发泡颗粒中。
发泡颗粒从密闭容器中取出,填充至具有400mm×300mm×30mm的模腔尺寸的成形模具的模腔,并且用0.25MPa下的水蒸气加热和成形35秒从而得到具有0.1g/cm3的密度的发泡成形体。所得发泡成形体具有微细均一的泡孔结构。
(实施例2b)
除了添加2.3g(基于100质量份树脂颗粒为0.1质量份)亚乙基双(硬脂酸酰胺)之外,以与实施例1b中相同的方式得到。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.08g/cm3的体积密度、162μm的平均泡孔直径、和1.9mm的平均粒径。
(实施例3b)
除了添加23g(基于100质量份树脂颗粒为1质量份)亚乙基双(硬脂酸酰胺)之外,以与实施例1b中相同的方式得到。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.2g/cm3的体积密度、82μm的平均泡孔直径、和1.7mm的平均粒径。
(实施例4b)
除了添加2.3g(基于100质量份树脂颗粒为0.1质量份)聚乙烯蜡之外,以与实施例1b中相同的方式得到。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.07g/cm3的体积密度、181μm的平均泡孔直径、和1.9mm的平均粒径。
(实施例5b)
除了添加2.3g(基于100质量份树脂颗粒为0.1质量份)亚乙基双-12-羟基硬脂酸酰胺之外,以与实施例1b中相同的方式得到。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.17g/cm3的体积密度、181μm的平均泡孔直径、和1.7mm的平均粒径。
(实施例6b)
除了使用其中尼龙12为硬链段和聚四亚甲基二醇为软链段的非交联的(结晶性)酰胺系弹性体(产品名“UBESTA9040”,由Ube Industries,Ltd.制造,凝胶分数3质量%以下)之外,以与制造例和实施例1b中相同的方式得到发泡成形体。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.1g/cm3的体积密度、120μm的平均泡孔直径、和1.9mm的平均粒径。
(实施例7b)
除了在酰胺系弹性体的树脂颗粒的制造例中,以基于100质量份酰胺系弹性体为0.5质量份供给作为泡孔调节剂的碳酸钠柠檬酸系化学发泡剂(产品名“Fine Cell Master PO410K”,由Dainichiseika Color&Chemicals Mfg.Co.,Ltd.制造)之外,以与实施例1b中相同的方式得到。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.1g/cm3的体积密度、131μm的平均泡孔直径、和1.9mm的平均粒径。
(实施例8b)
以与实施例7b中相同的方式得到具有0.16g/cm3的体积密度的发泡颗粒。然后,将发泡颗粒放在密闭容器中,将氮气在0.8MPa的压力(表压)下加压进给至该密闭容器,并且使该容器放置在室温下6小时,从而将氮气含浸于发泡颗粒中
发泡颗粒从密闭容器中取出,放在具有50升体积量的圆柱状间歇式加压预发泡机中,并且用蒸气加热从而得到具有0.04g/cm3的体积密度的发泡颗粒。发泡颗粒具有189μm的平均泡孔直径和3.2mm的平均粒径。
以与实施例1b中相同的方式制造发泡成形体。所得发泡成形体具有微细均一的泡孔结构。
(比较例1b)
除了未添加亚乙基双-12-羟基硬脂酸酰胺之外,以与实施例1b中相同的方式得到发泡性颗粒。然后,将发泡性颗粒放在预发泡机中,并且用蒸气加热,产生泡孔的聚结,并且不能得到发泡成形体。发泡颗粒具有282μm的平均泡孔直径。其中产生泡孔的聚结的发泡颗粒的截面照片示于图8中。
(比较例2b)
除了添加0.12g(基于100质量份树脂颗粒为0.005质量份)亚乙基双-12-羟基硬脂酸酰胺之外,以与实施例1b中相同的方式得到发泡性颗粒。然后,将发泡性颗粒放在预发泡机中,并且用蒸气加热,产生泡孔的聚结,并且不能得到发泡成形体。发泡颗粒具有296μm的平均泡孔直径。
(参考例1b)
除了添加250g(基于100质量份树脂颗粒为10质量份)亚乙基双-12-羟基硬脂酸酰胺之外,以与实施例1b中相同的方式得到。所得发泡成形体具有微细均一的泡孔结构。发泡颗粒具有0.1g/cm3的体积密度、112μm的平均泡孔直径、1.9mm的平均粒径。
实施例和比较例的物性一起记载于表3中。表中,A意指Pebax 5533,B意指UBASTA9040,EBSA意指亚乙基双(硬脂酸酰胺),PEW意指聚乙烯蜡,EBHSA意指亚乙基双-12-羟基硬脂酸酰胺。表3中,目视评价发泡成形体的外观。发泡成形体表面上的发泡颗粒相互连接的边界部分是平滑的情况表示为○,产生泡孔的聚结,并且不能得到发泡颗粒和发泡成形体的情况表示为×。
[表3]
从表3中,可见的是通过具有在特定范围内的平均泡孔直径和平均粒径的发泡颗粒,得到了具有良好外观的发泡成形体。
将通过又一些其他实施例的方式更详细地解释本发明,但是本发明不限于这些。
(聚酰胺系弹性体小颗粒的制造)
首先,将100质量份结晶性酰胺系弹性体(产品名“Pebax 5533”,由Arkema制造)和0.2质量份作为泡孔调节剂的亚乙基双(硬脂酸酰胺)(产品名“KAO WAX EB-FF”,由Kao Corporation制造)供给至具有65mm口径的单螺杆挤出机,并且熔融混炼。此外,在单螺杆挤出机中,将酰胺系弹性体初始在180℃下熔融混炼,其后在温度升高至220℃的同时熔融混炼。
随后,在将熔融状态下的酰胺系弹性体冷却之后,酰胺系弹性体通过安装在单螺杆挤出机的前端的多喷嘴模头的各个喷嘴挤出。此外,多喷嘴模头具有40个喷嘴,其中出口部具有0.7mm的直径,并且所有的喷嘴的出口部以相等的间隔分布在假定位于多喷嘴模头的前端面上的具有139.5mm直径的虚拟圆上。多喷嘴模头保持在220℃下。
4个旋转叶片沿旋转轴的周向以相等的间隔一体地分布在旋转轴的后端部的外周面上,并且将各旋转叶片配置为使其在始终接触多喷嘴模头的前端面的状态下在虚拟圆上移动。
另外,提供具有由正面圆形状的前部和从该前部的外周缘向后延伸且具有315mm内径的圆柱状周壁部构成的冷却鼓的冷却构件。冷却水通过供给管和鼓的供给口而供给至冷却鼓,并且在周壁部的整个内表面上,20℃的冷却水沿着该内表面向前螺旋地流动。
分布在多喷嘴模头的前端面上的旋转叶片以3,440rpm的转数旋转,并且将已经通过多喷嘴模头的各喷嘴的出口部挤出的酰胺系弹性体挤出物用旋转叶片切割从而制造大致球形状的酰胺系弹性体的树脂颗粒。
此外,在制造树脂颗粒时,首先,旋转轴没有安装在多喷嘴模头中,并且将冷却构件从多喷嘴模头中缩回。在该状态下,酰胺系弹性体从挤出机中挤出。然后,将旋转轴安装在多喷嘴模头中,并且将冷却构件在规定位置配置,其后,旋转轴旋转,并且酰胺系弹性体用旋转叶片在喷嘴的出口部的开口端切割,从而制造树脂颗粒。
树脂颗粒通过由于旋转叶片引起的切割应力而向外或向前飞扬,并且与沿着冷却构件的冷却鼓的内表面流动的冷却水碰撞从而使其立即冷却。
冷却的树脂颗粒与冷却水一起通过冷却鼓的排出口排出,并且用脱水机与冷却水分离。所得树脂颗粒具有1.2至1.7mm的颗粒长度和0.8至0.9mm的颗粒直径。
(实施例1c)
<发泡性颗粒的制备>
向具有50升内容积的耐压可密闭V-型混合机中放入15kg(使其为100质量份)所得树脂颗粒、3质量份水、和0.25质量份作为聚结防止剂的碳酸钙,关闭混合机,并且搅拌材料。在搅拌的同时,将12质量份丁烷在加压下进给至其中。丁烷在加压下进给至其中之后,将混合机的内部保持在60℃下2小时,并且冷却至30℃,由此取出发泡性颗粒。
<发泡颗粒的制备>
将发泡性颗粒放在具有50升体积量的圆柱状间歇式加压预发泡机中,并且用蒸气加热,由此得到了发泡颗粒。发泡颗粒具有0.1g/cm3的密度、68μm的平均泡孔直径、118μm的最外表层泡孔直径、1.7mm的平均粒径。发泡颗粒的截面照片示于图9中。从图9中,可见的是发泡颗粒具有微细均一的泡孔结构。
<发泡成形体的制备>
将发泡颗粒放在密闭容器中,将氮气在1.0MPaG的压力(表压)下加压进给至该密闭容器,并且使该容器放置在室温下12小时,从而将氮气含浸于发泡颗粒中。
发泡颗粒从密闭容器中取出,填充至具有400mm×300mm×30mm的模腔尺寸的成形模具的模腔,并且用0.25MPa下的水蒸气加热和成形35秒从而得到具有0.1g/cm3的密度的发泡成形体。所得发泡成形体具有60%的熔合率(fusion ratio),并且具有微细均一的泡孔结构。
(实施例2c)
除了在干式含浸时添加1质量份水之外,以与实施例1c中相同的方式得到具有0.1g/cm3的密度的发泡成形体。发泡颗粒具有0.1g/cm3的体积密度、84μm的平均泡孔直径、131μm的表层平均泡孔直径、1.9mm的平均粒径。所得发泡成形体具有60%的熔合率,并且具有微细均一的泡孔结构。发泡颗粒的截面照片示于图10中。从图10中,可见的是发泡颗粒具有微细均一的泡孔结构。
<表层平均泡孔直径(最外表层的平均泡孔直径)>
发泡颗粒的平均泡孔直径是指依照ASTM D2842-69的试验方法测量的平均泡孔直径。具体地,将发泡颗粒切割为大致两个相等的部分,将切出的截面使用扫描电子显微镜(产品名“S-3000N”,由Hitachi,Ltd.制造)放大且拍摄。拍摄的图像的每4个图像打印在A4纸上,例如如图11中用白线所示的,用直线连接这些颗粒的截面的最外表层中存在的泡孔。从连接表层中所有泡孔的直线的总长度和这些直线上存在的泡孔数,通过下式计算泡孔的平均弦长(t)。此外,这里的最外表层中的泡孔意指在发泡颗粒的截面中,距离颗粒表层的10%半径中包含的泡孔。
平均弦长t=直线的总长度/(泡孔数×照片的放大倍率)
另外,对于5个颗粒,以与如上所述相同的要领计算平均弦长,并且这些平均弦长的算术平均值定义为发泡颗粒的泡孔的平均泡孔直径。
- 还没有人留言评论。精彩留言会获得点赞!