多肽修饰的聚酰胺-胺型树枝状聚合物及其制备方法与应用与流程
本发明属于生物矿物界面粘附材料领域,特别涉及多肽修饰的聚酰胺-胺型树枝状聚合物及其制备方法与应用。
背景技术:
聚酰胺-胺型树枝状聚合物(PAMAM)是目前研究最为广泛和深入的树枝状大分子之一,目前有使用羧酸根、阿仑膦酸、磷酸根来修饰聚酰胺-胺型树枝状聚合物并验证了修饰后的聚酰胺-胺型树枝状聚合物对羟基磷灰石具有吸附作用。例如,CN 102166160 B公开了羧基改性聚酰胺-胺型树枝状聚合物作为牙本质再矿化诱导剂的应用,CN 103342819 B公开了端基为磷酸根的聚酰胺-胺型树枝状聚合物、其制备方法以及作为制备牙本质和牙釉质原位再矿化诱导剂的应用。但是,经过上述改性的聚酰胺-胺型树枝状聚合物仅被证实了对羟基磷灰石具有吸附作用,功能较为单一,因此,若能开发出功能更为丰富的改性聚酰胺-胺型树枝状聚合物,例如能够应用于生物矿化领域、蛋白质结晶领域以及组织工程支架材料等多个领域的改性聚酰胺-胺型树枝状聚合物,对于拓展聚酰胺-胺型树枝状聚合物的应用领域将产生积极的意义。
技术实现要素:
本发明的目的在于克服现有技术的不足,提供多肽修饰的聚酰胺-胺型树枝状聚合物及其制备方法,并证明所述多肽修饰的聚酰胺-胺型树枝状聚合物可作为生物矿物界面粘附材料的应用。
本发明提供的多肽修饰的聚酰胺-胺型树枝状聚合物,其结构式如式(Ⅰ)所示:
式(Ⅰ)中,n是所述多肽修饰的聚酰胺-胺型树枝状聚合物的代数,n=1、2、3、4、5、6、7、8或9。
本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
将端基为氨基的聚酰胺-胺型树枝状聚合物溶解于无水二甲基亚砜中形成第一种溶液,所述端基为氨基的聚酰胺-胺型树枝状聚合物为1、2、3、4、5、6、7、8或9代端基为氨基的聚酰胺-胺型树枝状聚合物,所述第一种溶液中端基为氨基的聚酰胺-胺型树枝状聚合物的浓度为1~2g/mL;
②配制第二种溶液
将三乙胺溶解于第一种溶液中形成第二种溶液,所述三乙胺与第一种溶液中的端基为氨基的聚酰胺-胺型树枝状聚合物的氨基的摩尔比为(1~2):1;
③合成
在氩气保护条件下于-5~5℃将丙烯酰氯加入到第二种溶液中,在室温反应至少24h,然后经透析、冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物;所述丙烯酰氯与第二种溶液中的端基为氨基的聚酰胺-胺型树枝状聚合物的氨基的摩尔比为(1~2):1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,将所述溶质溶解于去离子水中形成第三种溶液,所述去离子水的量以溶质能在去离子水中完全溶解为限;
②配制第四种溶液
以结构式如式(Ⅱ)所示的多肽为溶质,将所述溶质溶解于去离子水中形成第四种溶液,所述去离子水的量以溶质能在去离子水中完全溶解为限;
③合成
将二甲基苯基磷加入第三种溶液中并混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温反应至少24h,经透析、冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;
所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为(1~2):1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为(0.000001~0.00001):1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
将氨水、二氧六环、浓度为2~6mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为(15~25):(4~14):(1~6);
②合成
以步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,将所述溶质溶解于第五种溶液中,在密封条件下于室温反应至少24h,然后经透析、冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物;所述第五种溶液的量以溶质能在第五种溶液中完全溶解为限。
上述方法中,所述1、2、3、4、5、6、7、9代端基为氨基的聚酰胺-胺型树枝状聚合物是以乙二胺为起始核心,反复与丙烯酸甲酯通过Michael加成反应后再与乙二胺通过酰胺化反应制备而成的。
本发明还提供了上述多肽修饰的聚酰胺-胺型树枝状聚合物作为生物矿物界面粘附材料的应用。应用时,将所述多肽修饰的聚酰胺-胺型树枝状聚合物配制成水溶液使用,所述水溶液中,多肽修饰的聚酰胺-胺型树枝状聚合物的浓度优选为0.5~5.5mg/mL。实验表明:本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物对羟基磷灰石、磷酸钙、碳酸钙等生物矿物具有牢固的吸附作用,并能在羟基磷灰石、磷酸钙、碳酸钙表面吸附成膜;被本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物包裹的羟基磷灰石、磷酸钙、碳酸钙表面经过进一步接枝全氟酰氯、胶原等功能基团后,可形成新型的表面功能涂层材料,适用于涂覆在生物矿物材料表面使用。本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物能够有效地将生物与矿物粘结在一起,可应用于生物矿化领域、蛋白质结晶领域、表面图章/自清洁/抗污/抗菌材料领域以及组织工程支架材料领域。
与现有技术相比,本发明具有以下有益效果:
1.本发明提供了一类新型结构的改性聚酰胺-胺型树枝状聚合物—多肽修饰的聚酰胺-胺型树枝状聚合物,增加了改性聚酰胺-胺型树枝状聚合物的种类。
2.实验表明:本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物对羟基磷灰石、磷酸钙、碳酸钙等生物矿物具有牢固的吸附作用,并能在羟基磷灰石、磷酸钙、碳酸钙表面吸附成膜,可应用于生物矿化领域(见实施例10~12);被本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物包裹的羟基磷灰石、磷酸钙、碳酸钙表面经过进一步地接枝全氟酰氯、胶原等功能基团后,可形成作为用于生物矿物材料的新型表面功能涂层材料,从而应用于蛋白质结晶领域和组织工程支架材料领域(见实施例13~14)。与现有的改性聚酰胺-胺型树枝状聚合物相比,本发明 所述多肽修饰的聚酰胺-胺型树枝状聚合物的功能更加丰富,可作为生物矿物界面的粘附材料应用。
3.本发明还提供了上述多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,该方法采用常规原料及设备即可实现,易于实现工业化生产。
附图说明
图1为本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物的合成过程示意图;
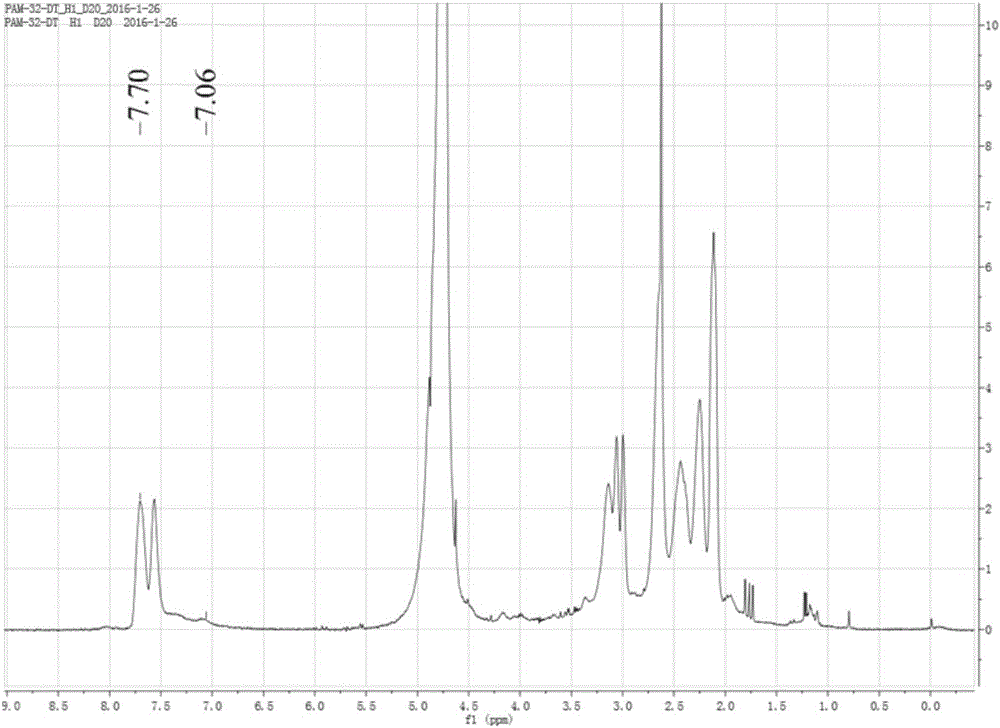
图2是实施例1制备的端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的核磁氢谱;
图3是实施例1制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的核磁氢谱;
图4是实施例1制备的多肽修饰的聚酰胺-胺型树枝状聚合物(SAP-G4)的结构式;
图5是实施例10中不同浓度的SAP-G4在磷酸钙、羟基磷灰石、碳酸钙粉末上的吸附量汇总图;
图6是实施例11中异硫氰酸荧光素标记的SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片上吸附后、异硫氰酸荧光素标记的SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片上吸附后使用二甲基亚砜清洗后以及空白羟基磷灰石片、磷酸钙片、碳酸钙片的激光共聚焦显微镜照片,其中,图6A、6D、6G是异硫氰酸荧光素标记的SAP-G4在羟基磷灰石片、磷酸钙片以及碳酸钙片上的吸附后的激光共聚焦显微镜照片,图6B、6E、6H是异硫氰酸荧光素标记的SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片上吸附后使用二甲基亚砜清洗后的激光共聚焦显微镜照片,图6C、6F、6I是空白羟基磷灰石片、磷酸钙片、碳酸钙片的激光共聚焦显微镜照片;
图7是实施例12中SAP-G4在羟基磷灰石片上吸附成膜后以及空白羟基磷灰石片的扫描电镜照片,其中,图7A~7C是SAP-G4在羟基磷灰石片上聚集成膜后不同放大倍数的扫描电镜照片,图7D~7F是不同放大倍数的羟基磷灰石片的扫面电镜照片;
图8是实施例12中SAP-G4在磷酸钙片上吸附成膜后以及空白磷酸钙片的扫描电镜照片,其中,图8A~8C是SAP-G4在磷酸钙片上聚集成膜后不同放大倍数的扫描电镜照片,图8D~8F是不同放大倍数的磷酸钙片的扫面电镜照片;;
图9是实施例12中SAP-G4在碳酸钙片表面吸附成膜后以及空白碳酸钙片的扫描电镜照片,其中,图9A~9C是SAP-G4在碳酸钙片上聚集成膜后不同放大倍数的扫描电镜照片,图9D~9F是不同放大倍数的碳酸钙片的扫面电镜照片;
图10是实施例13中全氟酰氯改性的SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片,SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片,以及空白空白羟基磷灰石片、 磷酸钙片以及碳酸钙片表面的水接触角测试结果,其中,图10A、10D、10G分别为空白羟基磷灰石片、磷酸钙片以及碳酸钙片表面水接触角测试结果,图10B、10E、10H分别为SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片表面的水接触角测试结果,图10C、10F、10I分别为全氟酰氯改性的SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片表面水接触角测试结果;
图11是实施例13中溶菌酶在全氟酰氯改性的SAP-G4包裹的羟基磷灰石片上结晶后的扫描电镜照片;
图12是实施例14中MG63细胞在胶原修饰的SAP-G4包裹的羟基磷灰石片上的粘附生长后的共聚焦激光显微镜照片。
具体实施方式
以下通过实施例并结合附图对本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物及其制备方法与应用作进一步说明。
下述实施例1~9中,步骤(2)中使用的多肽购买自上海波泰生物科技有限公司,其结构式如式(Ⅱ)所示:
下述实施例1~9中,所采用的1、2、3、4、5、6、7、9代端基为氨基的聚酰胺-胺型树枝状聚合物是以乙二胺为起始核心,反复与丙烯酸甲酯通过Michael加成反应后再与乙二胺通过酰胺化反应制备而成的。
实施例1
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将6.9g 4代端基为氨基的聚酰胺-胺型树枝状聚合物(G4PAMAM)溶解于4mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G4PAMAM的浓度为1.725g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺(TEA)溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G4PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯(Acryloyl chloride)滴加到第二种溶液中,在室温室温、常压条件下反应24h,然后在去离子水中用截留分子量为7000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约14.7g;所述丙烯酰氯与第二种溶液中的G4PAMAM的氨基的摩尔比为1.1:1;该步骤制备的端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的核磁氢谱如图2所示,图2中化学位移5.5~6.2ppm处的峰说明引入了双键。
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的0.86g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以4.57g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷(Dimethylphenylphosphine)加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;该步骤制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的核磁氢谱如图3所示,图3中化学位移7.5~8ppm处的峰说明引入了多肽上面的Fmoc基团。
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G4,其结构式如图4所示。
实施例2
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将5.17g 1代端基为氨基的聚酰胺-胺型树枝状聚合物(G1PAMAM)溶解于5.17mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G1PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G1PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应24h,然后在去离子水中用截留分子量为1000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约5.9g;所述丙烯酰氯与第二种溶液中的G1PAMAM的氨基的摩尔比为1.1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的0.59g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液,所述去离子水的量以溶质能在去离子水中完全溶解为限;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量 为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G1。
实施例3
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将1.4g 2代端基为氨基的聚酰胺-胺型树枝状聚合物(G2PAMAM)溶解于1.4mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G2PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G2PAMAM的氨基的摩尔比为1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应28h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约1.9g;所述丙烯酰氯与第二种溶液中的G2PAMAM的氨基的摩尔比为1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的1.2g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以1.07g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应28h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000001:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为2mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为15:4:3;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应28h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G2。
实施例4
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将3.2g 3代端基为氨基的聚酰胺-胺型树枝状聚合物(G3PAMAM)溶解于3.2mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G3PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一 种溶液中的G3PAMAM的氨基的摩尔比为2:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应30h,然后在去离子水中用截留分子量为4000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约7.1g;所述丙烯酰氯与第二种溶液中的G3PAMAM的氨基的摩尔比为2:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的0.59g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以0.57g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应30h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为2:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.00001:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为6mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为25:14:6;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应30h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G3。
实施例5
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1 所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将14.2g 5代端基为氨基的聚酰胺-胺型树枝状聚合物(G5PAMAM)溶解于7.1mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G5PAMAM的浓度为2g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G5PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约14.7g;所述丙烯酰氯与第二种溶液中的G5PAMAM的氨基的摩尔比为1.1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的1.8g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以8.3g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到 第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G5。
实施例6
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将28.8g 6代端基为氨基的聚酰胺-胺型树枝状聚合物(G6PAMAM)溶解于28.8mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G6PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G6PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应24h,然后在去离子水中用截留分子量为30000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约20.2g;所述丙烯酰氯与第二种溶液中的G6PAMAM的氨基的摩尔比为1.1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的2g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以1.8g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后经然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G6。
实施例7
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将5.8g 7代端基为氨基的聚酰胺-胺型树枝状聚合物(G7PAMAM)溶解于5.8mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G7PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G7PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应24h,然后在去离子水中用截留分子量为60000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约7.2g;所述丙烯酰氯与第二种溶液中的G7PAMAM的氨基的摩尔比为1.1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的4g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以3.6g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G7。
实施例8
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将11.6g 8代端基为氨基的聚酰胺-胺型树枝状聚合物(G8PAMAM)溶解于11.6mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G8PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G8PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应24h,然后在去离子水中用截留分子量为120000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约14.4g;所述丙烯酰氯与第二种溶液中的G8PAMAM的氨基的摩尔比为1.1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的8g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以7.2g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G8。
实施例9
本实施例中,提供多肽修饰的聚酰胺-胺型树枝状聚合物的制备方法,其合成路线如图1所示,步骤如下:
(1)端基为丙烯酰基的聚酰胺-胺型树枝状聚合物的合成
①配制第一种溶液
在室温、常压条件下将2.3g 9代端基为氨基的聚酰胺-胺型树枝状聚合物(G9PAMAM)溶解于2.3mL无水二甲基亚砜中形成第一种溶液;所述第一种溶液中G9PAMAM的浓度为1g/mL;
②配制第二种溶液
在室温、常压条件下将三乙胺溶解于第一种溶液中形成第二种溶液;所述三乙胺与第一种溶液中的G9PAMAM的氨基的摩尔比为1.1:1;
③合成
在0~5℃的冰水浴及氩气保护条件下将丙烯酰氯滴加到第二种溶液中,在室温、常压条件下反应24h,然后在去离子水中用截留分子量为250000道尔顿的透析袋透析1天,再冷冻干燥,即得端基为丙烯酰基的聚酰胺-胺型树枝状聚合物约2.8g;所述丙烯酰氯与第二种溶液中的G9PAMAM的氨基的摩尔比为1.1:1;
(2)带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第三种溶液
以步骤(1)制备的1.6g端基为丙烯酰基的聚酰胺-胺型树枝状聚合物为溶质,在常压、室温条件下将所述溶质溶解于300mL去离子水中形成第三种溶液;
②配制第四种溶液
以1.4g结构式如式(Ⅱ)所示的多肽为溶质,在室温、常压条件下将所述溶质溶解于300mL去离子水中形成第四种溶液;
③合成
将二甲基苯基磷加入第三种溶液中,在室温、常压搅拌0.5h即混合均匀,然后加入第四种溶液并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为2000道尔顿的透析袋透析1天,冷冻干燥,即得带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物;所述第四种溶液的加入量应使第四种溶液中的巯基与第三种溶液中的碳碳双键的摩尔比为1.1:1,所述二甲基苯基磷与第四种溶液中的巯基的摩尔比为0.000004:1;
(3)多肽修饰的聚酰胺-胺型树枝状聚合物的合成
①配制第五种溶液
在室温、常压条件下将氨水、二氧六环、浓度为4mol/L的氢氧化钠水溶液混合均匀得到第五种溶液,第五种溶液中,氨水、二氧六环、氢氧化钠的摩尔比为20:9:1;
②合成
以5g步骤(2)制备的带保护基团的多肽修饰的聚酰胺-胺型树枝状聚合物为溶质,在室温、常压条件下将所述溶质溶解于300mL第五种溶液中并混合均匀,在密封条件下于室温、常压反应24h,然后在去离子水中用截留分子量为3000道尔顿的透析袋透析72h,冷冻干燥,即得多肽修饰的聚酰胺-胺型树枝状聚合物,记作SAP-G9。
实施例10
本实施例中,测定实施例1制备的SAP-G4在羟基磷灰石、磷酸钙、碳酸钙粉末上的吸附情况,所述羟基磷灰石、磷酸钙、碳酸钙粉末的规格为HA/M30,其含水量低于0.1%。
将SAP-G4溶解在去离子水中,配制成SAP-G4浓度分别为0.2mg/mL、0.4mg/mL、0.6mg/mL、0.8mg/mL及1mg/mL的SAP-G4水溶液,用紫外可见分光光度计测量前述5个浓度的SAP-G4水溶液在波长为282nm处的吸光度值,得到SAP-G4浓度与吸光度值之间的线性关系,即标准曲线。
将SAP-G4溶解在去离子水中,配制成SAP-G4浓度分别为0.25mg/mL、0.75mg/mL、1.25mg/mL、1.75mg/mL、2.25mg/mL及2.75mg/mL的SAP-G4水溶液,分别记作1~6#溶液。取量程为4mL的离心PE管12支,分别向其中加入50mg羟基磷灰石、磷酸钙、碳酸钙粉末,然后取1~6#溶液各1mL加入离心PE管中,每个浓度的SAP-G4水溶液设置2支离心PE管,在37℃振荡搅拌12h,然后对各离心PE管中的混合液以10000转/分钟的转速离心4分钟,取离心所得上清液用紫外分光光度计测量上清液中SAP-G4的浓度,对照标准曲线,计算出吸附在羟基磷灰石、磷酸钙、碳酸钙粉末上的SAP-G4的量。结果如图5所示,SAP-G4在羟基磷灰石粉末(50mg)上的饱和吸附量为2.2mg(在浓度为2.25mg/mL时达吸附饱和),在磷酸钙粉末(50mg)上的饱和吸附量为1.8mg(在浓度为2.25mg/mL时达吸附饱和),在碳酸钙粉末(50mg)上的饱和吸附量为0.6mg(在浓度为1.25mg/mL时达吸附饱和)。
实施例11
本实施例中,测定实施例1制备的SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片上的吸附情况,所述羟基磷灰石片、磷酸钙片、碳酸钙片的尺寸为8mm*2mm且含水量低于0.1%。
采用使用激光共聚焦显微镜观察荧光标记了的SAP-G4在羟基磷灰石片、磷酸钙片、碳 酸钙片表面的吸附状况。荧光标记物质选择异硫氰酸荧光素(FITC),标记方法参照VenugantiV V,Sahdev P,Hildreth M,et al.Structure-skin permeability relationship of dendrimers[J].Pharmaceutical research,2011,28(9):2246-2260。向SAP-G4的水溶液中加入等摩尔的FITC乙醇溶液,再加入1mol/L的NaOH溶液和等摩尔的EDC,搅拌3天,过滤后再透析3天,冷冻干燥后得到FITC标记的SAP-G4。用去离子水将FITC标记的SAP-G4配制成浓度为2.25mg/mL的溶液,用移液枪量取1mL FITC标记的SAP-G4溶液浸泡羟基磷灰石片、磷酸钙片、碳酸钙片,浸泡1天后用PBS缓冲液冲洗3次,样品烘干后用激光共聚焦显微镜观察其表面的荧光分布情况,结果如图6所示。
图6A~C、图6D~F、图6G~I分别为羟基磷灰石片、磷酸钙片以及碳酸钙片的激光共聚焦显微镜照片,其中图6A、6D、6G是FITC标记的SAP-G4在上述三种片材上吸附后的激光共聚焦显微镜照片,图6B、6E、6H是FITC标记的SAP-G4在上述三种片材上吸附后使用二甲基亚砜清洗后的激光共聚焦显微镜照片,图6A、6D、6G和图6B、6E、6H中的样品具有明显的绿色荧光,且6A与6B、图6D与6E、图6G和6H的荧光强度相当,图6C、6F、6I是空白的上述三种片材的激光共聚焦显微镜照片,空白的上述片材没有检测到荧光。说明本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物在羟基磷灰石片、磷酸钙片、碳酸钙片上具有良好的吸附性能。
实施例12
本实施例中,测定实施例1制备的SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片上聚集成膜的情况,所述羟基磷灰石片、磷酸钙片、碳酸钙片的尺寸为8mm*2mm且含水量低于0.1%。
采用扫描电镜观察SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片表面的聚集成膜情况。将SAP-G4溶解在去离子水中,配制成SAP-G4浓度为2.25mg/mL的SAP-G4水溶液,用移液枪量取1mLSAP-G4水溶液浸泡羟基磷灰石片、磷酸钙片、碳酸钙片,浸泡1天后用PBS缓冲液冲洗3次,然后烘干后用扫描观察SAP-G4在羟基磷灰石片、磷酸钙片、碳酸钙片表面的聚集成膜情况。
图7为SAP-G4在羟基磷灰石片上吸附成膜后以及空白羟基磷灰石片的扫描电镜照片,图7A~7C是SAP-G4在羟基磷灰石片上聚集成膜后不同放大倍数的扫描电镜照片,图7D~7F是不同放大倍数的羟基磷灰石片的扫面电镜照片;图8为SAP-G4在磷酸钙片上吸附成膜后以及空白磷酸钙片的扫描电镜照片,图8A~8C是SAP-G4在磷酸钙片上聚集成膜后不同放大倍数的扫描电镜照片,图8D~8F是不同放大倍数的磷酸钙片的扫面电镜照片;图9为SAP-G4 在碳酸钙片表面吸附成膜后以及空白碳酸钙片的扫描电镜照片,图9A~9C是SAP-G4在碳酸钙片上聚集成膜后不同放大倍数的扫描电镜照片,图9D~9F是不同放大倍数的碳酸钙片的扫面电镜照片。由图7~9可知,本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物在羟基磷灰石片、磷酸钙片和碳酸钙片上具有良好的聚集成膜性能。
实施例10~12说明本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物可在生物矿化领域应用。
实施例13
本实施例中,测定全氟酰氯改性的SAP-G4包裹羟基磷灰石片、磷酸钙片、碳酸钙片表面的情况,所述羟基磷灰石片、磷酸钙片、碳酸钙片的尺寸为8mm*2mm且含水量低于0.1%。
采用水接触角来直观地反应全氟酰氯改性的SAP-G4包裹羟基磷灰石片、磷酸钙片、碳酸钙片表面的情况。将SAP-G4溶解在去离子水中,配制成SAP-G4浓度为2.25mg/mL的SAP-G4水溶液,用移液枪量取1mLSAP-G4水溶液浸泡羟基磷灰石片、磷酸钙片、碳酸钙片,浸泡1天后用PBS缓冲液冲洗3次,然后烘干,将所得SAP-G4包裹的羟基磷灰石片、磷酸钙片、碳酸钙片在全氟酰氯与三乙胺的乙醚溶液中浸泡4小时(将3mg全氟酰氯溶于3mL乙醚中,然后加入全氟酰氯10倍当量的三乙胺得到),然后在真空干燥箱中干燥1小时,用乙醚冲洗3此后用氮气烘干,进行水接触角测试,结果如图10所示。
图10A、10D、10G分别为空白羟基磷灰石片、磷酸钙片以及碳酸钙片表面水接触角测试结果,图10B、10E、10H分别为SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片表面的水接触角测试结果,图10C、10F、10I分别为全氟酰氯改性的SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片表面水接触角测试结果。图中的ca.表示水接触角(contact angle),由10A、10D、10G、10G、10E、10H可知,空白的以及SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片的水接触角均<90°,说明这些表面是亲水性的,而由图10C、10F、10I可知,全氟酰氯改性的SAP-G4包裹的羟基磷灰石片、磷酸钙片以及碳酸钙片的水接触角均>90°,说明这些表面是疏水性的。
超疏水表面有利于蛋白质结晶(Gao A,Wu Q,Wang D,et al..Advanced Materials,2016,28(3):579-587.),这说明本实施例制备的全氟酰氯改性的SAP-G4包裹的三种疏水表面有利于蛋白质结晶。以下通过实验进行证实:
将100mg溶菌酶溶于1mL醋酸-醋酸盐缓冲液(0.1mol/L,pH=4.5)中得到溶菌酶溶液,取10微升该溶菌酶溶液滴加实施例13制备的全氟酰氯改性的SAP-G4包裹的羟基磷灰石片上,在封闭的充满醋酸的湿润环境中于4℃反应4小时,然后在室温自然干燥并使用扫描电 镜观察溶菌酶在全氟酰氯改性的SAP-G4包裹的羟基磷灰石片上的结晶情况,结果如图11所示,由图10可知,全氟酰氯改性的SAP-G4包裹的羟基磷灰石片能诱导蛋白质晶体生成,说明,本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物经全氟酰氯改性后,可用于蛋白质结晶领域。
实施例14
将SAP-G4溶解在去离子水中,配制成SAP-G4浓度为2.25mg/mL的SAP-G4水溶液,用移液枪量取1mLSAP-G4水溶液浸泡羟基磷灰石片,浸泡1天后用PBS缓冲液冲洗3次,然后烘干,得到SAP-G4包裹的羟基磷灰石片。将所得SAP-G4包裹的羟基磷灰石片在胶原、羟基丁二酰亚胺、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的磷酸缓冲液中浸泡4小时,然后分别用磷酸缓冲液和去离子水冲洗3次,再用氮气吹干,得到胶原-SAP-G4包裹的羟基磷灰石片。将MG63细胞接种在胶原-SAP-G4包裹的羟基磷灰石片的表面,接种浓度大概为5000细胞/片,然后孵育1小时,用PBS缓冲液冲洗去除未贴壁的细胞,接着用4%的多聚甲醛固定30分钟,用PBS缓冲液冲洗,细胞用鬼笔环肽染色30分钟,然后用PBS和去离子水冲洗,最后采用共聚焦激光显微镜观察,结果如图12所示。
由图12可知,在胶原-SAP-G4包裹的羟基磷灰石片出现了明显的细胞粘附生长,说明本发明所述多肽修饰的聚酰胺-胺型树枝状聚合物在组织工程支架材料领域也具有应用前景。
- 还没有人留言评论。精彩留言会获得点赞!