一种7‑氯‑2‑氧代庚酸乙酯的纯化方法与流程
本发明属于有机化学领域,具体涉及一种西司他丁钠(CilastatinSodium)关键中间体7-氯-2-氧代庚酸乙酯的纯化方法。
背景技术:
:西司他丁钠,结构式如(I)所示,是由美国默克公司在研发β-碳青霉烯类抗生素亚胺培南(imipenem)时研发的一种肾脱氢二肽酶抑制剂,与亚胺培南配伍使用,能有效抑制肾脱氢二肽酶对亚胺培南的降解,并能降低亚胺培南的肾毒性。亚胺培南/西司他丁钠,已作为一种复合广谱抗菌制剂被广泛使用,其在美国的商品名为“Primaxin”。现有技术已经出现多种西司他丁钠(I)的制备方法,其中以专利US5147868报道的方法最为常用且已实现大规模工业化生产,其工艺路线如下所示:上述方法中,结构式(II)的Z-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸是制备西司他丁钠(I)的关键中间体。在同一篇专利文件中报道了以结构式(III)的7-氯-2氧代庚酸乙酯为起始原料来合成该关键中间体的方法,工艺路线如下所示:此外,现有技术中关于7-氯-2-氧代庚酸乙酯(III)也已经出现了多种制备方法的报道,其中最主要的是以下三种方法:I.专利EP48301报道的格氏试剂法,合成路线如下所示:II.文献J.Med.Chem,1987,30,1074报道的二噻烷法。合成路线如下所示:III.专利US5268501报道的亚硝酰硫酸-甲醛法,合成路线如下所示:由于西司他丁钠(I)为静脉注射用药,临床制剂对其纯度要求很高。但西司他丁钠(I)为无定形态,本身不易纯化。因此,高纯度的各中间体,如7-氯-2-氧代庚酸乙酯(III)等能有效减少带入终产品西司他丁钠(I)中的杂质。但是,无论是7-氯-2-氧代庚酸乙酯(III),还是上面结构式(IV)的中间体,亦或是制备7-氯-2-氧代庚酸乙酯(III)的起始原料和各中间体,都是液体油状物,通常只能通过精馏或柱层析的方法进行纯化。工业化精馏或柱层析设备投资大、占地大、能耗高且不易得到高纯度产物。因此,精馏或柱层析都不是理想的工业化方法。专利US6348617报道了一种7-氯-2-氧代庚酸乙酯的纯化方法:先将7-氯-2-氧代庚酸乙酯溶于甲苯等有机溶剂,然后与亚硫酸氢钠的水溶液反应,得到7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐,然后在酸的作用下分解,得到含量提高的7-氯-2-氧代庚酸乙酯。但是试验发现该方法得到的7-氯-2-氧代庚酸乙酯仍然含有较多杂质。为了提高西司他丁钠的纯度、保证临床用药安全,有必要对7-氯-2-氧代庚酸乙酯(III)的纯化方法进行深入研究。技术实现要素:针对上述问题,本发明的一个目的在于提供一种7-氯-2-氧代庚酸乙酯的纯化方法。该方法操作简便、适合工业化生产,得到的7-氯-2-氧代庚酸乙酯纯度高,GC检测纯度和含量可达98%以上,总杂不高于2%。为了实现上述发明目的,本发明采用了如下技术方案:一种7-氯-2-氧代庚酸乙酯的纯化方法,包含下述步骤:1)将结构式(III)的7-氯-2-氧代庚酸乙酯粗品(油状物)直接在一定温度下与亚硫酸氢盐溶液反应,得到结构式(VI)的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体;或者将结构式(III)的7-氯-2-氧代庚酸乙酯粗品(油状物)溶于与水相混溶的有机溶剂A,在一定温度下与亚硫酸氢盐溶液反应,得到结构式(VI)的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体;2)将步骤1)得到的结构式(VI)的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体过滤,用有机溶剂B洗涤所得固体,干燥;3)将步骤2)得到的所述结构式(VI)的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体溶于水,在一定温度下加入酸或碱,使7-氯-2-氧代庚酸乙酯的亚硫酸氢盐分解为结构式(III)的7-氯-2-氧代庚酸乙酯;4)向步骤3)的反应体系加入与水不相溶的有机溶剂C萃取,萃取得到的有机相经脱水后蒸干,得到纯度提高的7-氯-2-氧代庚酸乙酯。上述纯化方法的反应式,如下所示:其中,M=Na,K,NH4。优选的,所述步骤1)中,所述亚硫酸氢盐选自亚硫酸氢钠、亚硫酸氢钾或亚硫酸氢铵,更优选亚硫酸氢钠。还优选的,所述步骤1)中,所述亚硫酸氢盐与7-氯-2-氧代庚酸乙酯的摩尔比可以为1.0~3.0∶1.0,更优选1.2~2.0∶1.0。优选的,所述步骤1)中,所述亚硫酸氢盐溶液为饱和溶液,通过如下方法制备:将所述亚硫酸氢盐溶于计算量的水;或将所述亚硫酸氢盐溶于计算量的水和与水相混溶的有机溶剂A按照一定比例构成的混合溶剂中;所述与水相溶的有机溶剂A选自甲醇、乙醇或四氢呋喃等,更优选乙醇;当优选乙醇时,水和乙醇的体积比为1.0~5.0∶1.0,更优选2.0~3.0∶1.0。优选的,所述步骤1)中,7-氯-2-氧代庚酸乙酯粗品或7-氯-2-氧代庚酸乙酯粗品溶于所述与水相混溶的有机溶剂A得到的溶液采用滴加的方式加入所述亚硫酸氢盐溶液中;所述与水相溶的有机溶剂A选自甲醇、乙醇或四氢呋喃等,更优选为乙醇,乙醇与7-氯-2-氧代庚酸乙酯粗品的体积重量比(ml:g)为1.0~5.0∶1.0,优选1.0~2.0∶1.0。优选的,所述步骤1)中,反应温度为0~50℃,更优选为0~30℃。优选的,所述步骤2)中,所述有机溶剂B选自甲醇、乙醇、乙酸乙酯、甲基叔丁醚、二氯甲烷、甲苯、1,2-二氯乙烷和氯仿中的一种或多种;更优选为乙醇或乙酸乙酯。优选的,所述步骤3)中,所述酸与7-氯-2-氧代庚酸乙酯的亚硫酸氢盐的摩尔比可以为1.0~2.0∶1.0,更优选为1.0~1.5∶1.0。进一步优选的,所述酸选自无机酸,更优选为盐酸、硫酸和磷酸中的一种或多种,最优选为盐酸。所述酸可以为浓酸也可以为稀酸。最优选的,所述酸为浓度1.0~6.0mol/L的盐酸。优选的,所述步骤3)中,所述碱与7-氯-2-氧代庚酸乙酯的亚硫酸氢盐的摩尔比为1.0~5.0∶1.0,更优选1.0~3.0∶1.0。进一步优选的,所述碱选自无机碱,更优选为碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、氢氧化钠和氢氧化钾中的一种或多种,最优选为碳酸钠。所述碱可以为浓碱也可以为稀碱。最优选的,所述碱为碳酸钠;碳酸钠水溶液的重量百分比浓度为5%~10%。优选的,所述步骤3)中,所述温度为0~60℃,更优选为45~55℃。优选的,上述步骤4)中,所述水不相溶的有机溶剂C选自甲苯、乙酸乙酯、甲基叔丁醚、甲基异丁基甲酮、二氯甲烷、1,2-二氯乙烷和氯仿等中的一种或多种,更优选为甲苯。作为一个可以替代的方案,所述水不相溶的有机溶剂C可以在所述步骤3)中,分解反应开始之前加入7-氯-2-氧代庚酸乙酯的亚硫酸氢盐的水溶液。为了进一步提高7-氯-2-氧代庚酸乙酯的纯度,本发明所述的纯化方法还包括7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体的重结晶:在步骤3)之前,将步骤2)得到的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体重结晶一次或多次,得到的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐的精制品再进入步骤3)。优选的,所述重结晶方法为:将步骤2)得到的7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体溶于水中,向所得溶液中滴加与所述水相溶的有机溶剂A,使7-氯-2-氧代庚酸乙酯的亚硫酸氢盐析出,过滤,干燥,即得7-氯-2-氧代庚酸乙酯的亚硫酸氢盐的精制品。优选的,所述重结晶步骤中,7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体重量(g)与水的体积(ml)比为1.0∶0.5~5.0,更优选为1.0∶0.5~1.5。所述与水相溶的有机溶剂A选自甲醇、乙醇和四氢呋喃中的一种或多种;更优选为乙醇。所述有机溶剂A与水的体积比为1.0~5.0∶1.0,更优选为2.0~3.0∶1.0。上述重结晶步骤可以重复多次,直至7-氯-2-氧代庚酸乙酯的亚硫酸氢盐固体的纯度达到要求。作为一个优选的实施方式,本发明提供一种7-氯-2-氧代庚酸乙酯的纯化方法,包含下述步骤:1)将7-氯-2-氧代庚酸乙酯粗品滴加到亚硫酸氢钠饱和溶液中,或者将7-氯-2-氧代庚酸乙酯粗品先溶于乙醇,再滴加到亚硫酸氢钠饱和溶液中,乙醇体积(ml)与7-氯-2-氧代庚酸乙酯粗品的重量(g)比为1.0~5.0∶1.0;0~50℃下反应得到7-氯-2-氧代庚酸乙酯的亚硫酸氢钠固体;其中,亚硫酸氢钠与7-氯-2-氧代庚酸乙酯的摩尔比为1.0~3.0∶1.0;亚硫酸氢钠饱和溶液通过如下方法制备:将所述亚硫酸氢钠溶于计算量的水;或将所述亚硫酸氢钠溶于计算量的水和乙醇按照体积比为1.0~5.0∶1.0构成的混合溶剂中;2)将步骤1)得到的7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐过滤,用乙醇或乙酸乙酯洗涤所得固体,干燥;3)将步骤2)所得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠固体溶于水中,向所得溶液中滴加乙醇,使7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐析出,过滤,干燥;得到精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐;其中,7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐固体重量(g)与水的体积(ml)比为1.0:0.5~5.0;乙醇与水的体积比为1.0~5.0:1.0;如有必要,重复上述操作直至所述7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐纯度达到要求;4)将步骤3)得到的精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐溶于水,在0~60℃下,加入酸浓度为1.0~6.0mol/L的盐酸或重量百分比浓度为5%~10%的碳酸钠水溶液,使7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐分解为7-氯-2-氧代庚酸乙酯;用甲苯萃取一次以上,合并萃取得到的有机相,脱水后蒸干,得到GC检测纯度和含量≥95%的7-氯-2-氧代庚酸乙酯;其中,盐酸与7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐的摩尔比为1.0~2.0:1.0;碳酸钠与7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐的摩尔比为1.0~5.0:1.0;每次萃取甲苯的体积(ml)与7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐的重量(g)的比例为0.5~5.0:1.0。作为一个更优选的实施方式,本发明提供一种7-氯-2-氧代庚酸乙酯的纯化方法,包含下述步骤:1)将7-氯-2-氧代庚酸乙酯粗品滴加到亚硫酸氢钠饱和溶液中,或者将7-氯-2-氧代庚酸乙酯粗品先溶于乙醇,再滴加到亚硫酸氢钠饱和溶液中,乙醇体积(ml)与7-氯-2-氧代庚酸乙酯粗品的重量(g)比为1.0~2.0∶1.0;0~30℃下反应得到7-氯-2-氧代庚酸乙酯的亚硫酸氢钠固体;其中,亚硫酸氢钠与7-氯-2-氧代庚酸乙酯的摩尔比为1.2~2.0∶1.0;亚硫酸氢钠饱和溶液通过如下方法制备:将所述亚硫酸氢钠溶于计算量的水;或将所述亚硫酸氢钠溶于计算量的水和乙醇按照体积比为2.0~3.0∶1.0构成的混合溶剂中;2)将步骤1)得到的7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐过滤,用乙醇或乙酸乙酯洗涤所得固体,干燥;3)将步骤2)所得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠固体溶于水中,向所得溶液中滴加乙醇,使7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐析出,过滤,干燥;得到精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐;其中,7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐固体的重量(g)与水的体积(ml)比为1.0:0.5~1.5;乙醇与水的体积比为2.0~3.0:1.0;如有必要,重复上述操作直至所述7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐纯度达到要求;4)将步骤3)得到的精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐溶于水,在45~55℃下,加入酸浓度为1.0~6.0mol/L的盐酸或重量百分比浓度为5%~10%的碳酸钠,使7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐分解为7-氯-2-氧代庚酸乙酯;用甲苯萃取一次以上,合并萃取得到的有机相,脱水后蒸干,得到GC检测纯度和含量≥95%的7-氯-2-氧代庚酸乙酯;其中,盐酸与7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐的摩尔比为1.0~1.5:1.0;碳酸钠与7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐的摩尔比为1.0~3.0:1.0;每次萃取甲苯的体积(ml)与7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐的重量(g)的比例为1.0~2.0:1.0。上述方法中,7-氯-2-氧代庚酸乙酯粗品可以按照现有技术的报道制备,例如
背景技术:
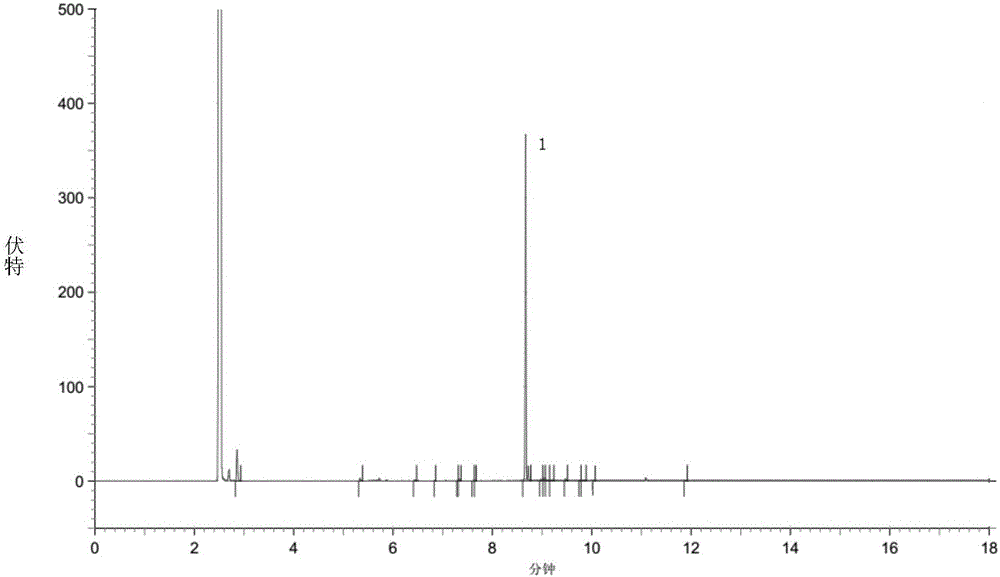
中提到的格氏试剂法(专利EP48301)、二噻烷法(文献J.Med.Chem,1987,30,1074)或亚硝酰硫酸-甲醛法(专利US5268501)。通过本发明的上述方法制备的7-氯-2-氧代庚酸乙酯的纯度和含量可达97%以上,甚至可以高达98%以上、总杂不高于2%。而现有技术中。专利US6348617制备的7-氯-2-氧代庚酸乙酯的纯度为85.9%,总杂为13.9%(实施例14)。在下面的具体实施方式部分的对比例1中,发明人重复该现有技术的纯化方法,得到的7-氯-2-氧代庚酸乙酯的纯度也仅为84.7%(归一化法),与文献报道一致。可见,本发明的方法可以显著提高7-氯-2-氧代庚酸乙酯的纯度,减少杂质的含量,并提高下步产物的纯度。总之,本发明提供了一种操作简便、适合工业化生产且能获得高纯度7-氯-2-氧代庚酸乙酯的方法,为制备高纯度的西司他丁钠静脉注射剂提供了必要的物质基础,从而可以更好地保证临床用药安全。附图说明以下,结合附图来详细说明本发明的实施方案,其中:图1为实施例1得到的高纯度7-氯-2-氧代庚酸乙酯的GC色谱图,其中1号峰为7-氯-2-氧代庚酸乙酯的吸收峰。图2为对比例1得到的7-氯-2-氧代庚酸乙酯的GC色谱图,其中1号峰为7-氯-2-氧代庚酸乙酯的吸收峰。具体实施方式以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。各实施例所用的7-氯-2-氧代庚酸乙酯的粗品(油状物)可以按照专利US5268501报道的亚硝酰硫酸-甲醛法、专利EP48301报道的格氏试剂法或文献J.Med.Chem,1987,30,1074报道的二噻烷法制备。高效液相色谱(HPLC)条件:色谱柱以十八烷基硅烷键合硅胶为填充剂,规格4.6μm×25cm,以磷酸调节0.05mol/L磷酸氢二钾溶液pH值至10,加入乙腈配成50/50(V/V)的溶液,以该溶液为流动相,流速1.0ml/min,检测波长为237nm;气相色谱(GC)条件:取样品适量,加乙腈溶解,HP-1型气相色谱柱;气化室温度250℃,检测器温度280℃,梯度升温,初温60℃,保留3min,再30℃/min,升至230℃,保留10min,分流比30∶1,流速1.2ml/min,进样量0.2μl。此条件下7-氯-2-氧代庚酸乙酯的保留时间为8.67min。实施例17-氯-2-氧代庚酸乙酯的纯化1)室温下,将50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,GC归一含量60.6%,外标含量58.3%,相当于含有7-氯-2-氧代庚酸乙酯29.2g,0.14mol)缓慢滴入由21.9g亚硫酸氢钠(0.21mol)溶于37ml水配成的饱和溶液中,控制滴加速度,使反应液温度维持室温。滴加完毕后,室温下搅拌,约0.5h后析出白色固体。2)将步骤1)得到的反应混合物于0~5℃继续搅拌2h后过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)40.1g。3)室温下,将步骤2)所得40.1g白色固体溶于25ml水中,向所得溶液中滴加60ml无水乙醇,逐渐析出白色固体。滴毕,于0~5℃继续搅拌2h,过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)39.0g,熔点170~172℃,HPLC显示其归一含量为98.1%。4)步骤3)所得39.0g白色固体(0.13mol)溶于50ml水中,加入50ml甲苯,加热至50~55℃,然后缓慢滴入6mol/L盐酸25ml(0.15mol),滴毕,继续于50~55℃反应,每隔1h取样HPLC监测反应进程,约2h反应完毕。将反应液冷至室温,分液,分出上层甲苯层,向下层水层加入甲苯萃取2次,每次50ml,合并甲苯层;甲苯萃取液加入100ml饱和盐水洗涤一次,加入无水硫酸钠干燥,过滤,以少量甲苯洗涤滤渣,收集洗涤液,与干燥后的甲苯萃取液合并,减压浓缩回收甲苯,得7-氯-2-氧代庚酸乙酯油状物25.0g,收率86.5%,气相色谱(GC)归一含量98.6%(气相色谱面积归一化结果,见表1),外标含量98.3%。表1实施例1的气相色谱面积归一化结果表保留时间峰高面积分离度(USP)面积百分比8.35520341182980.000.2658.6675552464681514811.0798.6128.982302512793710.950.4049.07942966496613.490.719总计56460226911044—100.00实施例27-氯-2-氧代庚酸乙酯的纯化1)室温下,将18.7g亚硫酸氢钠(0.18mol)溶于32ml水,然后加入11ml无水乙醇,搅拌均匀;将50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,GC归一含量64.3%,外标含量62.4%,相当于含有7-氯-2-氧代庚酸乙酯31.2g,0.15mol)缓慢滴入,使反应液温度维持室温。滴加完毕后,室温下搅拌,约0.5h后析出白色固体。2)将步骤1)得到的反应混合物于0~5℃继续搅拌3h后过滤,以适量乙酸乙酯洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)43.9g。3)室温下,将步骤2)所得43.9g白色固体溶于30ml水中,向所得溶液中滴加90ml无水乙醇,逐渐析出白色固体。滴毕,于0~5℃继续搅拌2h,过滤,以适量乙酸乙酯洗涤滤饼,抽干,干燥,得精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)42.1g,HPLC显示其归一含量98.3%。4)将上步得到的42.1g白色固体(0.14mol)溶于50ml水中,加热至45~50℃,然后缓慢滴入6mol/L盐酸30ml(0.18mol),滴毕,于45~50℃继续反应,每隔1h取样HPLC监测反应进程,约2h反应完毕。将反应液冷至室温,加入50ml甲苯,分液,分出甲苯层(上层),向水层(下层)加入甲苯萃取2次,每次50ml,合并甲苯层,加入100ml饱和盐水洗涤一次,加入5无水硫酸钠干燥2h,过滤,以少量甲苯洗涤滤渣,收集甲苯洗涤液,与甲苯萃取液合并,减压浓缩回收甲苯,得7-氯-2-氧代庚酸乙酯油状物26.9g,收率86.2%,气相色谱(GC)归一含量98.7%,外标含量98.5%。实施例37-氯-2-氧代庚酸乙酯的纯化1)室温下,将50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,GC归一含量50.8%,外标含量49.6%,相当于含有7-氯-2-氧代庚酸乙酯24.8g,0.12mol)溶于50ml无水乙醇,然后缓慢滴入由16.2g亚硫酸氢钠(0.16mol)溶于28ml水配成的饱和溶液中,控制滴加速度,使反应液温度维持室温。滴加完毕后,室温下搅拌,约0.5h后析出白色固体。2)将步骤1)得到的反应混合物于0~5℃继续搅拌2h后过滤,以适量甲苯洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠(白色固体)34.7g。3)室温下,将步骤2)所得34.7g白色固体溶于30ml水中,向所得溶液中滴加90ml无水乙醇,逐渐析出白色固体。滴毕,于0~5℃继续搅拌2h,过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠(白色固体)33.3g,HPLC显示其归一含量93.6%。4)室温下,将步骤3)所得33.3g白色固体溶于20ml水中,向所得溶液中滴加60ml无水乙醇,逐渐析出白色固体。滴毕,于0~5℃下继续搅拌2h,过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠精制品(白色固体)32.1g,HPLC显示其归一含量98.8%。5)将步骤4)所得32.1g白色固体(0.11mol)溶于30ml水中,加入30ml甲苯,加热至50~55℃,然后缓慢滴入6mol/L盐酸28ml(0.17mol),滴毕,继续于50~55℃反应,每隔1h取样HPLC监测反应进程,约2h反应完毕。将反应液冷至室温,分液,分出上层甲苯层,向下层水层加入甲苯萃取2次,每次50ml,合并甲苯层,加入100ml饱和盐水洗涤一次,加入无水硫酸钠干燥2h,过滤,以少量甲苯洗涤滤渣,收集甲苯洗涤液,与甲苯萃取液合并,减压浓缩回收甲苯,得7-氯-2-氧代庚酸乙酯油状物20.6g,收率83.2%,气相色谱(GC)归一含量98.5%,外标含量98.3%。实施例47-氯-2-氧代庚酸乙酯的纯化1)室温下,将50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,GC归一含量60.6%,外标含量58.3%,相当于含有7-氯-2-氧代庚酸乙酯29.2g,0.14mol)缓慢滴入由21.9g亚硫酸氢钠(0.21mol)溶于37ml水配成的饱和溶液中,控制滴加速度,使反应液温度维持室温。滴加完毕后,室温下搅拌,约0.5h后析出白色固体。2)将步骤1)得到的反应混合物于0~5℃继续搅拌2h后过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)40.1g,HPLC显示其归一含量为89.3%。3)将上步反应所得40.1g白色固体溶于50ml水中,加入50ml甲苯,加热至50~55℃,然后缓慢滴入6mol/L盐酸25ml(0.15mol),滴毕,继续于50~55℃反应,每隔1h取样HPLC监测反应进程,约2h反应完毕。将反应液冷至室温,分液,分出上层甲苯层,向下层水层加入甲苯萃取2次,每次50ml,合并甲苯层,加入100ml饱和盐水洗涤一次,加入无水硫酸钠干燥,过滤,以少量甲苯洗涤滤渣,收集甲苯洗涤液,与甲苯萃取液合并,减压浓缩回收甲苯,得7-氯-2-氧代庚酸乙酯油状物25.0g,收率86.5%,气相色谱(GC)归一含量88.6%,外标含量87.5%。实施例57-氯-2-氧代庚酸乙酯的纯化1)室温下,将50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,气相色谱(GC)归一含量60.6%,外标含量58.3%,相当于含有7-氯-2-氧代庚酸乙酯29.2g,0.14mol)缓慢滴入由21.9g亚硫酸氢钠(0.21mol)溶于37ml水配成的饱和溶液中,控制滴加速度,使反应液温度维持室温。滴加完毕后,室温下搅拌,约0.5h后析出白色固体。2)将步骤1)得到的反应混合物于0~5℃继续搅拌2h后过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)40.1g。3)室温下,将步骤2)所得40.1g白色固体溶于25ml水中,向所得溶液中滴加60ml无水乙醇,逐渐析出白色固体。滴毕,于0~5℃继续搅拌2h,过滤,以适量无水乙醇洗涤滤饼,抽干,干燥,得精制7-氯-2-氧代庚酸乙酯的亚硫酸氢钠盐(白色固体)39.0g,HPLC显示其归一含量为98.6%。4)将步骤3)所得39.0g白色固体(0.13mol)溶于50ml水中,加入50ml甲苯,加热至50~55℃,然后缓慢滴入10%(wt%)碳酸钠溶液413.4g(0.39mol),滴毕,继续于50~55℃反应,每隔1h取样HPLC监测反应进程,约2h反应完毕。将反应液冷至室温,分液,分出上层甲苯层,向下层水层加入甲苯萃取2次,每次50ml,合并甲苯层,加入100ml饱和盐水洗涤一次,加入无水硫酸钠干燥,过滤,以少量甲苯洗涤滤渣,收集甲苯洗涤液,与甲苯萃取液合并,减压浓缩回收甲苯,得7-氯-2-氧代庚酸乙酯油状物24.4g,收率83.7%,气相色谱(GC)归一含量98.2%,外标含量97.6%。实施例67-氯-2-氧代庚酸乙酯的纯化1)室温下,50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,GC归一含量60.6%,外标含量58.3%,相当于含有7-氯-2-氧代庚酸乙酯29.2g,0.14mol)溶于30ml四氢呋喃,将该溶液缓慢滴入由20.4g亚硫酸氢钠(0.20mol)溶于35ml水配成的饱和溶液中,控制滴加速度,使反应液温度维持室温。滴加完毕后,室温下搅拌,约0.5h后析出白色固体。2)将步骤1)得到的反应混合物于0~5℃继续搅拌2h后过滤,以适量四氢呋喃洗涤滤饼,抽干,干燥,得7-氯-2-氧代庚酸乙酯的亚硫酸氢钠(白色固体)41.8g。3)室温下,将步骤2)所得41.8g白色固体溶于30ml水中,向所得溶液中滴加90ml四氢呋喃,逐渐析出白色固体。滴毕,于0~5℃继续搅拌2h,过滤,以适量四氢呋喃洗涤滤饼,抽干,干燥,得精制的7-氯-2-氧代庚酸乙酯的亚硫酸氢钠(白色固体)40.1g,HPLC显示其归一含量98.6%。4)将步骤3)所得40.1g白色固体(0.13mol)溶于50ml水中,加入50ml甲基叔丁醚,加热至45~50℃,然后缓慢滴入10%(wt%)碳酸氢钠溶液252.0g(0.30mol),滴毕,于50~55℃继续反应,每隔1h取样HPLC监测反应进程,约2h反应完毕。将反应液冷至室温,分液,分出上层甲基叔丁醚层,向下层水层加入甲基叔丁醚萃取2次,每次50ml,合并甲基叔丁醚层,加入100ml饱和盐水洗涤一次,加入无水硫酸钠干燥2h,过滤,以少量甲基叔丁醚洗涤滤渣,收集甲基叔丁醚洗涤液,与甲基叔丁醚萃取液合并,减压浓缩回收甲基叔丁醚,得7-氯-2-氧代庚酸乙酯油状物24.1g,收率82.6%,气相色谱(GC)归一含量98.4%,外标含量97.5%。对比例17-氯-2-氧代庚酸乙酯的纯化取7-氯-2-氧代庚酸乙酯油状物粗品(气相色谱(GC)归一含量60.6%,外标含量58.3%),按照专利US6348617报道的方法纯化。具体操作如下:室温下,50.0g7-氯-2-氧代庚酸乙酯粗品(油状物,GC归一含量60.6%,外标含量58.3%,相当于含有7-氯-2-氧代庚酸乙酯29.2g,0.14mol)溶于50ml甲苯,将该溶液缓慢滴入由17.5g亚硫酸氢钠(0.17mol)溶于70ml水的溶液中,控制滴加速度,使反应液温度维持室温。滴加完毕后,室温下搅拌2h,然后向反应混合物中加入50ml甲苯,分液,收集水层I,待用。向甲苯层中加入8.7g亚硫酸氢钠(0.084mol)溶于35ml水的溶液,室温下搅拌1h,分液,收集水层II,水层II与水层I合并,得到水溶液。向上步所得的水溶液中加入50ml甲苯,将反应混合物加热至50~55℃,然后缓慢滴入浓盐酸42ml(0.51mol),约1h滴加完毕。滴毕,保温50~55℃搅拌40min,分液,加入50ml水洗涤甲苯层,收集甲苯层,加入无水硫酸钠干燥,过滤,收集滤液,减压浓缩回收甲苯,得黄色油状物,气相色谱(GC)外标法显示其中含7-氯-2-氧代庚酸乙酯27.1g(收率93.5%),归一含量84.7%(气相色谱面积归一化结果,见表2),总杂15.3%。表2对比例1的气相色谱结果表保留时间峰高面积分离度(USP)面积百分比2.8612518543721930.0010.1195.335218352655269.220.7226.437113911418334.000.3866.8382556247313.390.0677.3032679250517.570.0687.33515207157101.190.4277.619323836749.710.1007.648149915341.000.0428.6692816926311653135.8984.7318.972108251270810.670.3459.04322210229102.540.6239.1057508172001.530.4689.1999146119892.230.3269.47218391219098.700.5969.758343538249.500.1049.8527920222861.700.60610.042342439293.410.10711.8783484605747.070.165总计32135283678167—100.00试验例1Z-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸的制备以本发明实施例1所得的纯化的7-氯-2-氧代庚酸乙酯,参考专利WO2008138228报道的方法制备Z-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸(II)。具体操作为:将实施例1所得的25.0g7-氯-2-氧代庚酸乙酯(GC归一含量98.3%,外标含量98.5%),13.5g(+)-S-2,2-二甲基环丙烷甲酰胺和0.15g对甲苯磺酸加入125ml甲苯中,加热回流,以甲苯与水共沸带出反应产生的水,带反应完毕后,降温至室温,先后分别以10%的稀盐酸(100ml×3)和10%的亚硫酸氢钠溶液(100ml×3)洗涤反应液三次。洗涤完毕后,分出甲苯层,加入无水硫酸钠干燥。过滤,减压浓缩回收甲苯,得36.4g棕色粘稠液体,产品不作分离直接用于下步反应。将上一步反应所得36.4g棕色粘稠液体加入60ml乙醇和75g10%的氢氧化钠溶液中,加热至45~50℃,保温搅拌使其反应,HPLC监控反应过程,约10h反应完毕;然后加入叔丁基醚洗涤反应液三次,每次加入叔丁基醚100ml,弃去有机层,向水层中加浓盐酸酸化,调节pH至3~3.5,加入乙酸乙酯萃取酸化液三次,每次加入乙酸乙酯的量为100ml,弃去水层,向乙酸乙酯层加入无水硫酸钠干燥,然后过滤,减压浓缩滤液并回收乙酸乙酯,获得31.6g棕色粘稠液体。室温下将31.6g棕色粘稠液体溶于80ml二氯甲烷中,然后加入240ml甲苯,搅拌均匀后将所得溶液置于0℃静置12h,过滤,抽干,收集溶液中析出的大量固体,真空干燥,得(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸(白色固体)15.6g。同时将结晶后得母液减压浓缩至恒重,得13.6g棕色粘稠溶液,依照上述方法,在室温下将13.6g棕色粘稠溶液加入30ml二氯甲烷和75ml甲苯,0℃静置12h,收集溶液中析出的固体,真空干燥,得(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸(白色固体)3.2g。前后两次共得(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸(白色固体)18.8g,HPLC测定其纯度为98.8%,以7-氯-2-氧代庚酸乙酯计算的(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸的收率为57.8%。试验例2Z-7-氯-2-(2,2-二甲基环丙基甲酰胺基)-2-庚烯酸的制备以对比例1纯化得到的7-氯-2-氧代2庚酸乙酯,参考专利WO2008138228报道的方法制备Z-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸。具体操作如下:将对比例1所得的7-氯-2-氧代庚酸乙酯(GC归一含量84.8%,总杂15.2%,含7-氯-2-氧代庚酸乙酯27.1g)油状物,14.8g(+)-S-2,2-二甲基环丙烷甲酰胺和0.16g对甲苯磺酸加入145ml甲苯中,加热回流,以甲苯与水共沸带出反应产生的水,带反应完毕后,降温至室温,先后分别以10%的稀盐酸(100ml×3)和10%的亚硫酸氢钠溶液(100ml×3)洗涤反应液三次。洗涤完毕后,分出甲苯层,加入无水硫酸钠干燥。过滤,减压浓缩回收甲苯,得38.1g棕色粘稠液体,产品不作分离直接用于下步反应。将上一步反应所得38.1g棕色粘稠液体加入60ml乙醇和70g10%的氢氧化钠溶液中,加热至45~50℃,保温搅拌使其反应,HPLC监控反应过程,约10h反应完毕;然后加入叔丁基醚洗涤反应液三次,每次加入叔丁基醚90ml,弃去有机层,向水层中加浓盐酸酸化,调节pH至3~3.5,加入乙酸乙酯萃取酸化液三次,每次加入乙酸乙酯的量为90ml,弃去水层,向乙酸乙酯层加入无水硫酸钠干燥,然后过滤,减压浓缩滤液并回收乙酸乙酯,获得33.2g棕色粘稠液体。室温下将33.2g棕色粘稠液体溶于75ml二氯甲烷中,然后加入225ml甲苯,搅拌均匀后将所得溶液置于0℃静置12h,过滤,抽干,收集溶液中析出的大量固体,真空干燥,得(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸(白色固体)13.2g,HPLC测定其纯度为93.5%。将结晶后得母液减压浓缩至恒重,得14.9g棕色粘稠溶液,依照上述方法,在室温下将14.9g棕色粘稠溶液加入30ml二氯甲烷和75ml甲苯,0℃静置12h,溶液中出现粘稠油状物,未能析出白色固体。后尝试多种重结晶方法,都未能析出固体。与试验例1相同的方法和条件下,本试验例以对比例1纯化得到的7-氯-2-氧代2庚酸乙酯制备Z-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸,通过简单重结晶手段只能得到13.2g白色固体,HPLC测定其纯度为93.5%,以7-氯-2-氧代庚酸乙酯计算的(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸(II)的收率为36.8%。本试验例所得(Z)-7-氯-2-((S)-2,2-二甲基环丙基甲酰胺基)-2-庚烯酸的纯度和收率均低于试验例1。正是由于本发明提供的纯化方法提高了油状7-氯-2-氧代2庚酸乙酯的纯度,才显著提高后续反应产物的纯度和产率。以上对本发明具体实施方式的描述并不限制本发明,本领域技术人员可以根据本发明作出各种改变或变形,只要不脱离本发明的精神,均应属于本发明所附权利要求的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1