硫醚化合物及其合成方法与流程
本发明涉及有机合成和自由基聚合领域,特别是涉及一类硫醚化合物的结构及其合成方法。
背景技术:
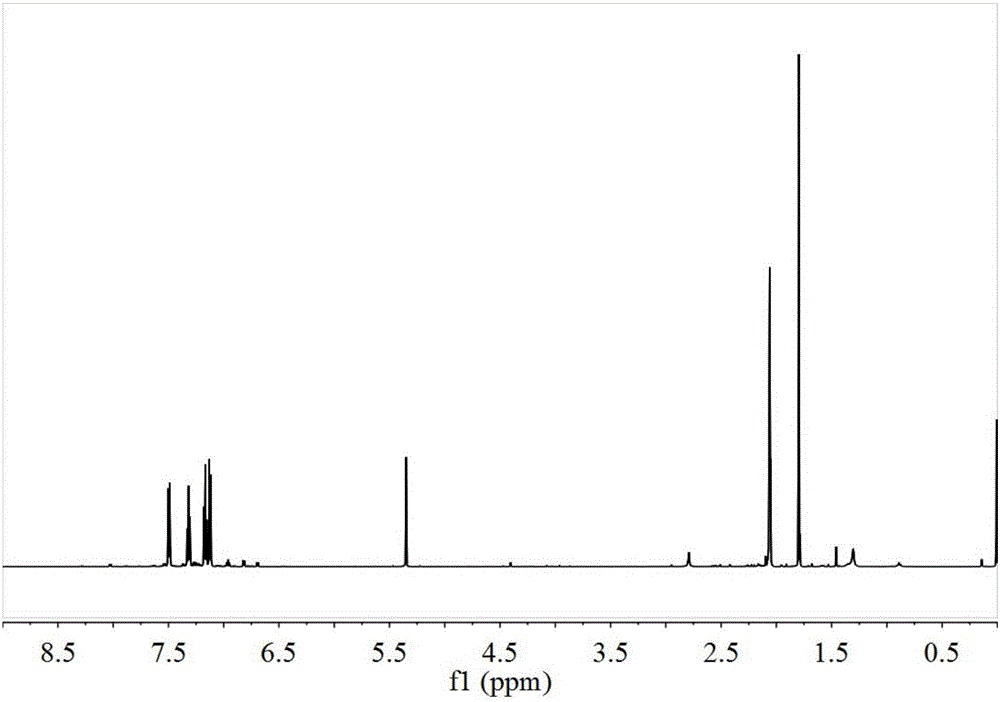
:自“活性”/可控自由基聚合概念提出以来,已经出现NMP、ATRP、Iniferter、RAFT、GTP、TKMP等自由基聚合体系,每一种聚合体系都有自己代表性的化合物在聚合过程中发挥作用,使聚合物分子量随单体转化率不断提高,并保持较窄的分子量多分散系数。基于“活性”/可控自由基聚合的最基本原理和我们的实验结果,我们提出了一类全新的对自由基聚合同时具有引发和调控能力的化合物,即以9H-氧杂蒽-9-硫酮加成初级自由基后的小分子硫醚结构为模板设计合成的一系列硫醚化合物。与其他可控自由基聚合方法相比,使用本发明中硫醚化合物没有毒性、没有气味,得到的聚合产物几乎没有颜色,且没有引入金属离子,不需要加入其他配体以提高溶解性。该发明在可控自由基聚合领域具有较好前景技术实现要素:本发明提供一类硫醚化合物以及其合成方法,该类硫醚化合物的组成为结构如式I所示化合物。式I中,X选自O、S、Se、Te、CR’R”、羰基、N-R’或P-R’,其中R’和R”是C1-10的烷基,且当式I中X为O,同时R1为苯基时,R2不是苄基。式I中R1是C1-15烷基,苯基、苄基、1-苯乙基、2-苯乙基、2-甲基苯甲基、3-甲基苯甲基、4-甲基苯甲基、2-萘基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基中的一种。式I中R2是氢、C1-10烷基、苯基、苄基、1-(2-甲基苯)基、1-(3-甲基苯)基、1-(4-甲基苯)基、1-苯乙基、2-苯乙基、2-甲基苯甲基、3-甲基苯甲基、4-甲基苯甲基、2-萘基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-乙酸甲酯基、2-乙酸乙酯基、2-异丁酸甲酯基、2-异丁酸乙酯基中的一种。式I中R3和R7中有一个为C1-10烷基、C1-10烷氧基中的一种时,则另一个为氢。式I中R4、R5、R6、R8、R9、R10各自独立的选自氢、C1-10烷基、C1-10烷氧基、C7-16苯烷基、苯氧基、苯硫基。式I化合物是通过如下式的反应路径合成的,包括步骤1)~4)四个步骤分别为加氢、硫醚化、拔氢和烷基化;其中加氢反应过程为:在甲醇、乙醇、乙二醇、异丙醇、四氢呋喃、DMF或其他对加氢还原剂和底物溶解和加氢有利的溶剂中,并在冰水浴或室温条件下,通过硼氢化钠、硼氢化钾、氢化铝锂、异丙醇铝、锌等加氢还原剂中的一种或组合,对a化合物的酮羰基进行加氢还原,形成b化合物。加氢还原剂对底物的当量比应在1至20倍之间,可在反应过程中通过TLC检测反应终点。反应后的产物需要柱层析或重结晶方法予以分离;第二步硫醚化反应可遵循以下步骤实施,即使用卤化试剂将b化合物的羟基置换为卤原子,再将形成的卤化物与R1基团对应的硫醇钠盐R1SNa反应,脱除生成的盐,得到c化合物。卤化反应可根据b化合物和卤化试剂活性而定,卤化试剂选自盐酸、氢溴酸、氢碘酸、Lucas试剂、三氯化磷、三溴化磷、DMF-五氯化磷、三苯基膦卤化物、亚磷酸三苯酯卤化物、二氯亚砜、DMF-二氯亚砜、二溴亚砜、DMF-二溴亚砜、NCS、NBS。反应产物需用柱层析或重结晶方式分离提纯。另外当硫醚化合物的R1是含有5个以上碳原子的烷基或苯基以及苯基烷基时,则硫醚化反应过程还可以按如下方式实施,即在与水能够形成共沸物,并能够溶解b化合物、R1对应硫醇R1SH以及高氯酸的溶剂中,在共沸温度以及高氯酸的催化条件下,将b化合物与R1SH反应形成硫醚,并通过共沸作用从体系中脱除生成的水,从而制备c化合物。高氯酸的加入量为,每1mol底物,加入0.1ml。反应产物需用柱层析或重结晶方式分离提纯。拔氢反应过程为:在适当的溶剂和低温条件下,向c化合物的溶液中加入过量的甲基锂、乙基锂、丙基锂、异丙基锂、正丁基锂、叔丁基锂、苯基锂或二异丙基胺基锂中的一种,拔除与硫醚相连的碳原子上的氢,生成含有碳负离子的d化合物。该反应应选择四氢呋喃等不与锂试剂发生反应的溶剂,反应前应当进行冷冻抽排或惰性气体吹扫,以出去体系中的氧和二氧化碳,并保证反应在惰性气体气氛中进行。反应在干冰-丙酮浴,或乙酸乙酯凝固浴中进行,以保持低温减少副反应。锂试剂的加入量应当为底物c当量的1-1.5倍;烷基化反应过程为:向拔氢反应的产物d化合物中直接加入R2对应的卤化物R2Y,即获得最终的产物。其中Y选自氯、溴、碘。该步反应应当紧接上一步拔氢反应进行,在加入锂试剂后30分钟后,即可向体系中注入卤化物。卤化物在注入前也应进行冷冻抽排或氮气吹扫以排氧、排二氧化碳。最终产物通过柱层析或重结晶获得;经过以上过程合成的硫醚化合物在部分乙烯基单体的溶液和本体聚合当中可以起到引发和调控自由基聚合的作用。这些乙烯基单体包括甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸丁酯、甲基丙烯酸异丁酯、甲基丙烯酸异辛酯、甲基丙烯酸缩水甘油酯、甲基丙烯酸羟乙酯、甲基丙烯酸月桂酯、甲基丙烯酸羟丙酯、甲基丙烯酸环己酯、甲基丙烯酸乙二醇酯、甲基丙烯酸异冰片酯、甲基丙烯酸二甲基氨基乙酯、α-甲基苯乙烯、丙烯酸甲酯、丙烯酸乙酯、丙烯酸丁酯、丁二烯、异戊二烯、丙烯酸、苯乙烯和马来酸酐等;自由基聚合按如下技术方案实施:一种或多种乙烯基单体与溶剂组成的混合物中加入本发明中的硫醚化合物和引发剂,在惰性气体保护下,加热至40-100℃,保持此条件持续聚合1-100小时。硫醚化合物与单体的摩尔配比为1:200-1:1000,引发剂与硫醚化合物的摩尔配比为1:0.5-1:6,溶剂与单体的体积配比为0-5:1。附图说明图1是实施例1和实施例4中的9-苯硫基-9H-氧杂蒽的1H-NMR谱图。图2是实施例2和实施例3中9-甲硫基-9H-氧杂蒽的1H-NMR谱图。图3是实施例5、实施例6、实施例7和实施例8中2-异丙基-9-苯硫基-9H-硫杂蒽的1H-NMR谱图。图4是实施例1中化合物II的1H-NMR谱图。图5是实施例1中化合物II的13C-NMR谱图。图6是实施例2中化合物III的1H-NMR谱图。图7是实施例2中化合物III的13C-NMR谱图。图8是实施例3中化合物IV的1H-NMR谱图。图9是实施例3中化合物IV的13C-NMR谱图。图10是实施例4中化合物V的1H-NMR谱图。图11是实施例4中化合物V的13C-NMR谱图。图12是实施例5中化合物VI的1H-NMR谱图。具体实施方式实施例1如式II结构的硫醚化合物的合成方法如下:第一步,将10g氧杂蒽酮加入到100ml甲醇中,在室温下加入将20g硼氢化钠加入到溶液中。反应过程有氢气放出,通过TLC监测反应进程。至氧杂蒽酮完全反应后,蒸馏掉溶剂甲醇,然后加入二氯甲烷溶解,并用去离子水洗涤有机相数次,至水相pH值降低到7.0。分液收集有机相并浓缩,待占吨醇结晶后分离。反应和后处理过程应当注意要在惰性气氛中进行,以防止占吨醇氧化。此步反应产率为82%。第二步,将上一步获得的占吨醇5g溶解于100mL甲苯中,加入3.06g苯硫酚,并滴加0.25ml70%质量浓度的高氯酸水溶液。将体系加热至沸腾,并用分水器将生成的水从体系中分离。待出水量达到理论值,即反应完全后,水洗有机相至水相pH升高到7.0,蒸馏掉甲苯后获得粗产品。用二氯甲烷/石油醚体积比为1/3的展开剂通过柱层析的方法获得的9-苯硫基-9H-氧杂蒽。此步反应产率为61%,生成的产物的1H-NMR谱图如图1所示。第三步,将第二步获得的9-苯硫基-9H-氧杂蒽5g溶解于无水的四氢呋喃中,将反应器密闭,冷冻抽排三次除去氧气和二氧化碳,并以氮气保护。将反应器加入到经过液氮处理的乙酸乙酯凝固浴中(-84℃)降温。通过注射器向不断搅拌的体系中逐滴注入2.5M的正丁基锂的己烷溶液8.27ml,正丁基锂注入量为底物9-苯硫基-9H-氧杂蒽的1.2倍当量。反应半小时。第四步,将经过冷冻抽排过的2.82mL的(1-溴乙基)苯注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式III的最终产物。此步反应产率为73%,生成的产物II的1H-NMR谱图如图4所示,13C-NMR谱图如图5所示。实施例2第一步与实施例1中相同。第二步,将第一步的占吨醇5g溶解于20mL二氯亚砜当中,反应在冰水浴中进行,并给予氮气保护。待占吨醇完全溶解后,溶液变为橙红色,蒸馏掉二氯亚砜。将产物用二氯甲烷重新溶解,并在氮气保护下加入1.77g甲硫醇钠,立即有盐析出。过滤掉形成的盐,并将溶液过一个短的硅胶柱,蒸馏掉溶剂即获得的9-甲硫基-9H-氧杂蒽。此步反应产率为43%,生成的产物的1H-NMR谱图如图2所示。第三步,将第二步获得的9-甲硫基-9H-氧杂蒽5g溶解于无水的四氢呋喃中,将反应器密闭,冷冻抽排三次除去氧气和二氧化碳,并以氮气保护。将反应器加入到经过液氮处理的乙酸乙酯凝固浴中(-84℃)降温。通过注射器向不断搅拌的体系中逐滴注入2.5M的正丁基锂的己烷溶液10.51ml,正丁基锂注入量为底物9-甲硫基-9H-氧杂蒽的1.2倍当量。反应半小时。第四步,将经过冷冻抽排过的3.13mL苄溴注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式III的最终产物。此步反应产率为58%,生成的产物IV的1H-NMR谱图如图6所示,13C-NMR谱图如图7所示。实施例3第一步、第二步与第三步的操作方法与实施例3中相同。第四步,将经过冷冻抽排过的3.59mL苄溴注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式IV的最终产物。此步反应产率为56%,生成的产物V的1H-NMR谱图如图8所示,13C-NMR谱图如图9所示。实施例4第一步与第二步的操作方法与实施例1中相同。第三步,将第二步获得的9-苯硫基-9H-氧杂蒽5g溶解于无水的四氢呋喃中,将反应器密闭,冷冻抽排三次除去氧气和二氧化碳,并以氮气保护。将反应器加入到经过液氮处理的乙酸乙酯凝固浴中(-84℃)降温。通过注射器向不断搅拌的体系中逐滴注入2.0M的二异丙基胺基锂的四氢呋喃溶液10.33ml,二异丙基胺基锂注入量为底物9-苯硫基-9H-氧杂蒽的1.2倍当量。反应半小时。第四步,将经过冷冻抽排过的3.34mL2-溴异丁酸甲酯注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙红色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/3的展开剂,通过柱层析法分离提纯出如式V的最终产物。此步反应产率为17%,生成的产物VI的1H-NMR谱图如图10所示,13C-NMR谱图如图11所示。实施例5第一步,将10g的2-异丙基硫杂蒽酮加入到100ml甲醇中,在室温下加入将15g硼氢化钠加入到溶液中。反应过程有氢气放出,通过TLC监测反应进程。至2-异丙基硫杂蒽酮完全反应后,蒸馏掉溶剂甲醇,然后加入二氯甲烷溶解,并用去离子水洗涤有机相数次,至水相pH值降低到7.0。分液收集有机相并浓缩,待羟基化产物结晶后分离。反应和后处理过程应当注意要在惰性气氛中进行,以防止羟基化产物氧化。第二步,将上一步获得的羟基化产物5g溶解于100mL甲苯中,加入2.36g苯硫酚,并滴加0.20ml70%质量浓度的高氯酸水溶液。将体系加热至沸腾,并用分水器将生成的水从体系中分离。待出水量达到理论值,即反应完全后,水洗有机相至水相pH升高到7.0,蒸馏掉甲苯后获得粗产品。用二氯甲烷/石油醚体积比为1/4的展开剂通过柱层析的方法获得的2-异丙基-9-苯硫基-9H-硫杂蒽。此步反应产率为55%,生成的产物的1H-NMR谱图如图3所示。第三步,将第二步获得的2-异丙基-9-苯硫基-9H-硫杂蒽5g溶解于无水的四氢呋喃中,将反应器密闭,冷冻抽排三次除去氧气和二氧化碳,并以氮气保护。将反应器加入到经过液氮处理的乙酸乙酯凝固浴中(-84℃)降温。通过注射器向不断搅拌的体系中逐滴注入2.5M的正丁基锂的己烷溶液6.31ml,正丁基锂注入量为底物2-异丙基-9-苯硫基-9H-硫杂蒽的1.1倍当量。反应半小时。第四步,将经过冷冻抽排过的1.87mL苄溴注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式VI的最终产物。此步反应产率为21%,生成的产物VII的1H-NMR谱图如图12所示。实施例6第一步、第二步与第三步的操作方法与实施例6中相同。第四步,将经过冷冻抽排过的2.15mL(1-溴乙基)苯注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式VII的最终产物。此步反应产率为19%。实施例7第一步的操作方法与实施例6相同。第二步,将第一步的羟基化产物5g溶解于20mL二氯亚砜当中,反应在冰水浴中进行,并给予氮气保护。待占吨醇完全溶解后,溶液变为橙红色,蒸馏掉二氯亚砜。将产物用二氯甲烷重新溶解,并在氮气保护下加入1.37g甲硫醇钠,立即有盐析出。过滤掉形成的盐,并将溶液过一个短的硅胶柱,蒸馏掉溶剂即获得的9-甲硫基-9H-氧杂蒽。第三步,将第二步获得的2-异丙基-9-甲硫基-9H-硫杂蒽5g溶解于无水的四氢呋喃中,将反应器密闭,冷冻抽排三次除去氧气和二氧化碳,并以氮气保护。将反应器加入到经过液氮处理的乙酸乙酯凝固浴中(-84℃)降温。通过注射器向不断搅拌的体系中逐滴注入2.5M的正丁基锂的己烷溶液7.68ml,正丁基锂注入量为底物2-异丙基-9-甲硫基-9H-硫杂蒽的1.1倍当量。反应半小时。第四步,将经过冷冻抽排过的2.28mL苄溴注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式VIII的最终产物。此步反应产率为17%。实施例8第一步、第二步与第三步的操作方法与实施例7中相同。第四步,将经过冷冻抽排过的2.62mL(1-溴乙基)苯注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/6的展开剂,通过柱层析法分离提纯出如式IX的最终产物。此步反应产率为18%。实施例9第一步,将10g10-甲基-9(10H)-吖啶酮加入到100ml甲醇中,在室温下加入将20g硼氢化钠加入到溶液中。反应过程有氢气放出,通过TLC监测反应进程。至10-甲基-9(10H)-吖啶酮完全反应后,蒸馏掉溶剂甲醇,然后加入二氯甲烷溶解,并用去离子水洗涤有机相数次,至水相pH值降低到7.0。分液收集有机相并浓缩,待羟基化产物结晶后分离。反应和后处理过程应当注意要在惰性气氛中进行,以防止羟基化产物氧化。此步反应产率为68%。第二步,将上一步获得的羟基化产物5g溶解于100mL甲苯中,加入2.87g苯硫酚,并滴加0.23ml70%质量浓度的高氯酸水溶液。将体系加热至沸腾,并用分水器将生成的水从体系中分离。待出水量达到理论值,即反应完全后,水洗有机相至水相pH升高到7.0,蒸馏掉甲苯后获得粗产品。用二氯甲烷/石油醚体积比为1/3的展开剂通过柱层析的方法获得的苯硫醚化N-甲基吖啶。此步反应产率为56%。第三步,将第二步获得的苯硫醚化N-甲基吖啶5g溶解于无水的四氢呋喃中,将反应器密闭,冷冻抽排三次除去氧气和二氧化碳,并以氮气保护。将反应器加入到经过液氮处理的乙酸乙酯凝固浴中(-84℃)降温。通过注射器向不断搅拌的体系中逐滴注入2.5M的正丁基锂的己烷溶液7.91ml,正丁基锂注入量为底物苯硫醚化N-甲基吖啶的1.2倍当量。反应半小时。第四步,将经过冷冻抽排过的2.35mL苄溴注入到第三步的反应半小时后的体系当中,此时溶液颜色由不透明的深红色变为透明的橙黄色,将体系逐步升温至室温,经过半小时反应完成。负压脱除四氢呋喃,用二氯甲烷重新溶解,过滤掉析出的盐,并使用二氯甲烷/石油醚体积比为1/5的展开剂,通过柱层析法分离提纯出如式X的最终产物。此步反应产率为45%。实施例10化合物II调控St单体在甲苯中的溶液聚合反应。在25ml单口瓶中,加入化合物II0.0318g,St4g,及甲苯6g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应34小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大、分散系数在1.9至2.2之间。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数13.1221001.948276001.71717.2293001.721123.1316001.842431.5386002.143433.9416002.24实施例11化合物II调控MMA单体在甲苯中的溶液聚合反应。在25ml单口瓶中,加入化合物II0.0231g,MMA3g,及甲苯7g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应48小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大、分散系数在1.7至1.8之间。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数114.4326001.71333.3383001.72542.8489001.701057.7508001.792471.8546001.7648100583001.78实施例12化合物III调控MMA单体在THF中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.095g,MMA15g,及THF35g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应10小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大、分散系数在2.0。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数113.8234001.94222.3301001.96332.1305002.10436.9338001.95545.1298002.14647.4294002.16756.8321002.02864.9347001.88984.9316002.0310100.0366001.77实施例13化合物III调控MMA单体在有ABVN引发时THF中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.095g,偶氮二异庚腈0.037g,MMA15g,及THF35g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应8小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大、分散系数在2.0。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数118.6252001.941.528.1252002.07235.9284001.95346.5290002.02462.5302002.04565.4317002.01684.7273002.35788.9300002.198100.0324002.16实施例14化合物III调控St单体在有ABVN引发时THF中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.095g,偶氮二异庚腈0.037g,St15g,及THF35g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应48小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大、分散系数在1.8至3.0。各时段取样测定转化率,分子量和多分散系数结果如下:实施例1520%St单体浓度时,化合物III调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.064g,偶氮二异庚腈0.025g,St10g,及甲苯40g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应50小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到25%,分散系数在1.8至2.4。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数1.45.9166001.824.511.2175002.10916.8225002.0713.420.2242002.1224.524.3275002.363425.6293002.405025.7299002.35实施例1630%St单体浓度时,化合物III调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.095g,偶氮二异庚腈0.037g,St15g,及甲苯35g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应72小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到32%,分散系数在1.8至2.5。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数16.4170001.77312.7188001.98515.6217002.02719.8230002.191021.4257002.222424.1279002.523426.7293002.314829.0266002.585829.6277002.477232.1276002.47实施例1740%St单体浓度时,化合物III调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.128g,偶氮二异庚腈0.050g,St20g,及甲苯30g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应48小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到38%,分散系数在1.8至2.4。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数13.2322001.77411.8336001.86716.5314002.001631.4354002.122435.4402002.163036.1377002.323838.5383002.324838.6354002.22实施例1850%St单体浓度时,化合物III调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.160g,偶氮二异庚腈0.063g,St25g,及甲苯25g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应48小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到47%,分散系数在1.7至2.4。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数14.0361001.89412.2393001.73718.1329001.961634.1347002.002439.2406002.093042.1425002.313844.2422002.384847.1437002.44实施例1975℃下,化合物III调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物III0.095g,偶氮二异庚腈0.037g,St15g,及甲苯35g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在75℃下反应72小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到32%,分散系数在1.9至2.7。各时段取样测定转化率,分子量和多分散系数结果如下:实施例2065℃下,化合物IV调控MMA单体在有ABVN引发时THF中的溶液聚合反应。在25ml单口瓶中,加入化合物IV0.0199g,偶氮二异庚腈0.0075g,MMA3g,及THF7g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应48小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到78%,分散系数在1.7至1.8。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数212.74219513001.77417.57449552001.78830.23153568001.791046.51984631001.792451.00428669001.774878.76701710001.78实施例2165℃下,化合物IV调控St单体在有ABVN引发时THF中的溶液聚合反应。在25ml单口瓶中,加入化合物IV0.0199g,偶氮二异庚腈0.0075g,St3g,及THF7g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应48小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到47%,分散系数在1.7至2.0。各时段取样测定转化率,分子量和多分散系数结果如下:实施例22化合物V调控MMA单体在THF中的溶液聚合反应。在100ml单口瓶中,加入化合物V0.117g,MMA15g,及THF35g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应9小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大、分散系数在1.5到2.0。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数114.6323001.98232.0361001.89440.9510001.86541.6531001.79657.4571001.74764.5484001.65881.4492001.53988.4608001.49实施例2320%St单体浓度时,化合物V调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物V0.078g,偶氮二异庚腈0.025g,St10g,及甲苯40g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应72小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到36%,分散系数在1.5至2.2。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数11.20173001.53713.6144001.911019.8152002.003428.9179002.194528.7184002.135829.8176002.157236.7182002.13实施例2450%St单体浓度时,化合物V调控St单体在有ABVN引发时甲苯中的溶液聚合反应。在100ml单口瓶中,加入化合物V0.195g,偶氮二异庚腈0.062g,St25g,及甲苯25g。密封后经过液氮冷冻抽排三次使溶液处于氮气保护之下,在65℃下反应72小时。定时用注射器抽取样品称量后立即加入石油醚沉淀剂。样品经过真空干燥后,通过差重法测得单体的转化率随反应时间呈线性增长;通过GPC观测到数均分子量随单体转化率的增加而增大,转化率最终达到51%,分散系数在1.4至1.8。各时段取样测定转化率,分子量和多分散系数结果如下:时间(小时)转化率(%)分子量多分散系数14.8248001.62721.9239001.851024.6225001.772439.8245001.643439.4242001.624542.4233001.565846.4257001.477251.3301001.43当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1