4位含有取代基的2‑(2`‑羟基苯基)苯并唑化合物及其制法和用途的制作方法
本发明属于有机半导体光电技术领域,具体涉及一种4位含有取代基的2-(2'-羟基苯基)苯并唑化合物及其制备和应用。
背景技术:
1987年,美国的C.W.Tang等人发明以真空蒸镀法制成多层式结构的有机发光二极管(OLED),使得有机电致发光器件的研究受到各国科学家的高度重视。OLED技术的发展对光电显示领域产生了翻天覆地的变化。如今材料科学的发展和理论工艺的成熟对新型OLED的发展起着至关重要的作用,使得材料领域的研究人员对发光材料的探索产生了极大地兴趣。由于白光有机发光二极管(WOLED)具有效率高、亮度高、功耗低、视角广、响应速度快、主动发光、超薄超轻以及可柔性化等优异性能,并在显示和照明领域有广阔应用前景,受到学者和业界的广泛重视而成为研究热点。
1994年,日本山形大学的Kido教授等首先制备了世界上第一个WOLED器件,尽管当时器件的最大功率效率只有0.83lm/W,最大亮度只有3400cd/M2,但是还是在有机电致发光器件领域实现了突破性的进展。
2003年,WOLED器件效率达到了15lm/W并首次超过了白炽灯。2006年,美国圣地亚哥大学的Nobuhiro Ide研究小组利用全磷光材料制备出颜色可调控的堆叠式多层发光结构的WOLED,其器件效率达到了62.8lm/W。2008年,美国圣地亚哥大学的James Esler研究小组制备出最大功率效率为100 lm/W多发光层结构的WOLED,其工作电压为3.6V,外量子效率达到了20%。
多发光层WOLED的效率通常较高,但是结构和工艺条件相对复杂、电压较高,导致成本也相对较高,各发光层之间存在异质结,并且色坐标稳定性一般较差。如何合理地控制激子产生区域,激子复合区域,各发光层之间能量转移和激子扩散是获得高性能的保证。
目前,在WOLED领域,开发一种器件效率高、工艺简单,成本低,稳定性好的单层白光器件是实现白光有机发光二极管商业化的关键。
技术实现要素:
针对目前白光有机发光二极管中存在的器件制备工艺复杂、制作成本较高、重复性较差等技术问题,本发明提供了一种新的有机荧光材料,即4位含有取代基的2-(2'-羟基苯基)苯并唑化合物,及其制法和应用。所述的有机荧光材料是一系列具有激发态分子内质子转移化合物的四能级跃迁性质的2-(2'-羟基苯基)苯并唑,并连接多种含芳烃共轭结构单元的新材料。这类材料具有荧光量子产率高,斯托克位移大,合成简单,产率高等优点。通过混合商业化的蓝光材料组成二元互补色,可以制备工艺简单的单层白光器件。本发明采用的技术方案如下。
本发明提供了一种4位含有取代基的2-(2'-羟基苯基)苯并唑化合物。
本发明所述的4位含有取代基的2-(2'-羟基苯基)苯并唑化合物,其结构通式为:
式中:X为S、O或NH,Ar为含芳烃共轭结构单元。
所述含芳烃共轭结构单元选自以下取代基结构中的一种:
本发明还提供上述4位含有取代基的2-(2'-羟基苯基)苯并唑化合物的制备方法。
本发明所述的4位含有取代基的2-(2'-羟基苯基)苯并唑化合物的制备方法,其包括如下步骤:
(1)含芳烃共轭结构化合物的硼酸化:在氮气保护下,溴代芳烃溶于干燥的有机溶剂,温度低于-40℃条件下加入正丁基锂,搅拌2到4小时后,加入硼酸化合物,硼酸化,升温到常温后反应15-20小时。反应结束后,萃取有机相,将有机相合并干燥,得到产物。
(2)将步骤(1)的产物和2-(2'-4-溴苯酚)苯并唑,钯催化剂溶于有机溶剂,然后加入强碱水溶液,氮气保护下反应。反应结束后,萃取有机相,合并有机相并浓缩,分离提纯后,得到终产物,即为本发明的产物。
上述步骤(1)中,低温反应可使用干冰丙酮浴冷却;所述硼酸化合物为硼酸正三丁酯;使用水和二氯甲烷萃取有机相;所述有机溶剂为四氢呋喃。
上述步骤(2)中,所述钯催化剂为四(三苯基膦)钯、双(二亚苄基丙酮)钯、1,1-双(二苯膦基)二茂铁二氯,强碱为碳酸钾、氯化钾、氟化钾中的一种或几种,所述有机溶剂为四氢呋喃和甲苯的体积比为1:1的混合溶剂。反应温度为80-120℃,反应时间为24-60小时。所述的2-(2'-4-溴苯酚)苯并唑化合物的通式为:
其中,X为S、O或NH。
上述制备方法中,按摩尔比计,步骤(1)中,溴代芳烃和正丁基锂的比例为1:0.9~1.5,优选1:1.2;步骤(2)中,所述硼酸化溴代芳烃和2-(2'-4-溴苯酚)苯并唑的比例为1:0.5~1.5,优选1:1
所述溴代芳烃和有机溶剂的添加比例为1摩尔:2~8升。优选比例为1摩尔:4~6升。
本发明的再一目的在于提供基于4位含有取代基的2-(2'-羟基苯基)苯并唑化合物用于制备白光有机发光二极管的用途。
本发明所述的4位含有取代基的2-(2'-羟基苯基)苯并唑化合物可以作为发光材料,用于有机白光二极管器件中。通过混合商业化的蓝光材料组成二元互补色,可以制备工艺简单的单层白光器件。
本发明具有如下有益效果:1、本发明的4位含有取代基的2-(2'-羟基苯基)苯并唑化合物具有荧光量子产率高,斯托克位移大;2、上述化合物的制备方法合成简单,产率高,利于扩大生产规模;3、上述化合物可以作为发光材料,用于有机白光二极管器件中。
附图说明
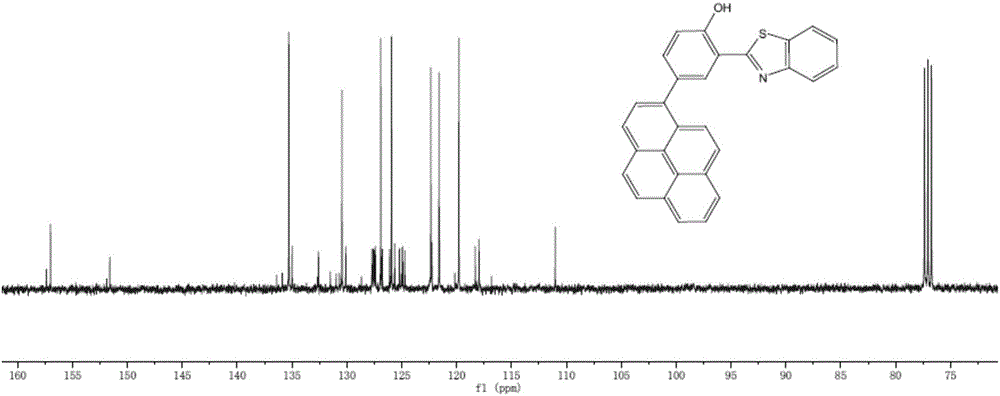
图1本发明实施例2化合物的碳谱图;
图2本发明实施例3化合物的碳谱图;
图3本发明实施例2化合物的荧光吸收-发射图;
图4本发明实施例3化合物的荧光吸收-发射图;
图5本发明实施例2化合物的氧化还原曲线。
具体实施方式
为了更好理解本发明专利的内容,下面通过具体的实例来进一步说明本发明的技术方案。但这些实施实例并不限制本发明。
该材料2-(2'-4-溴苯酚)苯并唑通过铃木反应连接多种芳烃共轭结构单元的给电子基团,该材料具有如下的通式:
式中:X为S、O或NH。
实施例1、
首先,2-(2'-4-溴苯酚)苯并噻唑的制备:
取二氨基酚(4.4g,20mmol,1equiv)和对氨基苯甲酸(2.5g,20mmol,1equiv)加入130ml多聚磷酸中,在N2的环境中加热到168℃,并回流6小时。待反应完全后,冷却后将产物溶于100ml冰水混合物中,向其中加入饱和的NaOH水溶液中和。待pH值调为6左右改加NaHCO3,边加边搅拌,到不再有CO2气泡产生。将中和后的溶液静置待溶液澄清后抽滤。把抽滤出来的固体加入饱和NaHCO3水溶液用乙酸乙酯萃取三次,合并有机层,用无水Na2SO4干燥旋蒸后得到绿色产物5g,产率为83%。(下述实施例中,2-(2'-4-溴苯酚)苯并噻唑的制备方法都与此相同,不再赘述)。
其次,2-(2'-苯并噻唑)-4-(9'-菲)苯酚的制备
(1)含芳烃共轭结构化合物的硼酸化:在氮气保护下,9-溴代菲溶于干燥的四氢呋喃,置于干冰丙酮浴(-78℃)中冷却,低温下加入正丁基锂,9-溴代菲和正丁基锂的添加比例为1:0.9搅拌2小时后,加入硼酸正三丁脂,硼酸化,继续反应0.5小时,撤去干冰丙酮浴,升温到常温后反应15小时。反应结束后,在冰水浴下淬灭反应,然后用水和二氯甲烷萃取有机相,将有机相合并干燥旋干,得到产物。
(2)取9-硼酸菲(0.18g,0.75mmol,1当量)和2-(2'-4-溴苯酚)苯并噻唑(0.12g,0.38mmol,0.5当量)混合溶于20ml四氢呋喃和甲苯的体积比为1:1的混合溶液中。加入催化剂Pd(pph3)4。避光通氮气,再加入10ml浓度为2mol/L的碳酸钾和氟化钾的水溶液。在80℃的条件下反应24小时。反应结束后,萃取有机相,合并有机相并浓缩,通过层析柱的方法进行分离提纯,得到黄色粉末状终产物。
实施例2、
2-(2'-苯并噻唑)-4-(9'-菲)苯酚的制备
(1)含芳烃共轭结构化合物的硼酸化:在氮气保护下,9-溴代菲溶于干燥的四氢呋喃,置于干冰丙酮浴中冷却至-78℃,低温下加入正丁基锂,9-溴代菲和正丁基锂的添加比例为1:1.2,搅拌3小时后,加入硼酸正三丁脂,硼酸化,继续反应0.5小时,撤去干冰丙酮浴,升温到常温后反应18小时。反应结束后,在冰水浴下淬灭反应,然后用水和二氯甲烷萃取有机相,将有机相合并干燥旋干,得到产物。
(2)取9-硼酸菲(0.18g,0.75mmol,1当量)和2-(2'-4-溴苯酚)苯并噻唑(0.24g,0.75mmol,1当量)混合溶于20ml四氢呋喃和甲苯的体积比为1:1的混合溶液中。加入催化剂Pd(pph3)4。避光通氮气,再加入10ml浓度为2mol/L的碳酸钾和氟化钾的水溶液。在90℃的条件下反应48小时。反应结束后,萃取有机相,合并有机相并浓缩,通过层析柱的方法进行分离提纯,得到黄色粉末状终产物0.31g,产率为82%。
对终产物进行核磁共振和氧化还原曲线检测,分别如图1和图5所示
实施例3、
2-(2'-苯并噻唑)-4-(1'-芘)苯酚的制备
将实施例2中的9-硼酸菲换成1-硼酸芘,具体步骤为:
(1)含芳烃共轭结构化合物的硼酸化:在氮气保护下,1-溴代芘溶于干燥的四氢呋喃,置于干冰丙酮浴中冷却至-78℃,低温下加入正丁基锂,1-溴代芘和正丁基锂的添加比例为1:1.2,搅拌3小时后,加入硼酸正三丁脂,硼酸化,继续反应0.5小时,撤去干冰丙酮浴,升温到常温后反应18小时。反应结束后,在冰水浴下淬灭反应,然后用水和二氯甲烷萃取有机相,将有机相合并干燥旋干,得到产物。
(2)取9-硼酸芘(0.6g,2.2mol,1当量)和2-(2'-4-溴苯酚)苯并噻唑(0.7g,2.2mmol,1当量)混合溶于30ml四氢呋喃和甲苯的体积比为1:1的混合溶液中。加入催化剂Pd(pph3)4。避光通氮气,再加入10ml浓度为2mol/L的碳酸钾和氟化钾的水溶液。在90℃的条件下反应48小时。反应结束后,萃取有机相,合并有机相并浓缩,通过层析柱的方法进行分离提纯,得到黄色粉末状终产物0.86g,产率为73%。
对终产物进行核磁共振检测,如图2所示。
实施例4、
2-(2'-苯并噻唑)-4-(1'-芘)苯酚的制备
(1)含芳烃共轭结构化合物的硼酸化:在氮气保护下,1-溴代芘溶于干燥的四氢呋喃,置于干冰丙酮浴中冷却至-78℃,低温下加入正丁基锂,1-溴代芘和正丁基锂的添加比例为1:1.5,搅拌4小时后,加入硼酸正三丁脂,硼酸化,继续反应0.5小时,撤去干冰丙酮浴,升温到常温后反应20小时。反应结束后,在冰水浴下淬灭反应,然后用水和二氯甲烷萃取有机相,将有机相合并干燥旋干,得到产物。
(2)取9-硼酸芘(0.6g,2.2mol,1当量)和2-(2'-4-溴苯酚)苯并噻唑(0.36g,3.3mmol,1.5当量)混合溶于30ml四氢呋喃和甲苯的体积比为1:1的混合溶液中。加入催化剂Pd(pph3)4。避光通氮气,再加入10ml浓度为2mol/L的碳酸钾和氟化钾的水溶液。在120℃的条件下反应60小时。反应结束后,萃取有机相,合并有机相并浓缩,通过层析柱的方法进行分离提纯,得到黄色粉末状终产物。
实施例5、
3-(2'-苯并噻唑)-4'-(1,2,2-三苯乙烯)-4'-[1,1'-联苯]的制备
(1)含芳烃共轭结构化合物的硼酸化:在氮气保护下,溴代四苯乙烯溶于干燥的四氢呋喃,置于干冰丙酮浴中冷却至-78℃,低温下加入正丁基锂,溴代四苯乙烯和正丁基锂的添加比例为1:1.2,搅拌3小时后,加入硼酸正三丁脂,硼酸化,继续反应0.5小时,撤去干冰丙酮浴,升温到常温后反应18小时。反应结束后,在冰水浴下淬灭反应,然后用水和二氯甲烷萃取有机相,将有机相合并干燥旋干,得到产物。
(2)取硼酸四苯乙烯(0.5g,1.9mol,1当量)和2-(2'-4-溴苯酚)苯并噻唑(0.65g,1.9mmol,1当量)混合溶于40ml四氢呋喃和甲苯的体积比为1:1的混合溶液中。加入催化剂Pd(pph3)4。避光通氮气,再加入10ml浓度为2mol/L的碳酸钾和氟化钾的水溶液。在90℃的条件下反应48小时。反应结束后,萃取有机相,合并有机相并浓缩,通过层析柱的方法进行分离提纯,得到淡黄色粉末状终产物。
实施例6、
本实施例是对实施例2和实施例3制备的化合物光谱的测定。
将实施例2和实施例3的化合物配成标准的1μM的四氢呋喃溶液。采用岛津-3150紫外可见光谱仪和RF-520XPC荧光光谱仪进行吸收光谱和发射光谱的测定。光致发光光谱是在紫外吸收的最大吸收出测得的。如图3和图4所示,由图可知,两个化合物的吸收和荧光重叠范围很小甚至不存在重叠,说明这两个材料有着斯托克位移大的优点。
实施例6、
本实施例是对实施例2和实施例3制备的化合物电化学的测定。
电化学循环伏安(CV)实验在一个Eco Chemie B.V.AUTOLABpotentiostat伏安分析仪上完成的,采用三电极体系,包括铂碳电极、Ag/Ag+为参比电极、铂丝为对电极。氧化过程采用二氯甲烷作为溶剂,还原过程采用四氢呋喃作为溶剂,六氟磷四丁基铵(Bu4N+PF6-)作为支持电解质,浓度为0.1M。所有电化学实验都是在常温下氮气气氛中进行的,电压扫描速度0.1V/S。使用二茂铁(FOC)作为基准,通过测量氧化和还原过程的开始电压可以计算材料的HOMO和LUMO能级。
从电化学测试结果图5可以看出,材料的氧化还原峰的位置,再通过公式:
HOMO=-[EOX–EFe/Fe++4.8]
LUMO=-[ERE–EFe/Fe++4.8]
计算可以得到实施例2材料的HOMO和LUMO能级分别为-5.44eV和-3.40eV。实施例3材料的HOMO和LUMO能级分别为-5.26eV和-3.44eV。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种二苯并噻吩的萃取方法与流程
- 取代的苯并噻吩基衍生物作为用于治疗II型糖尿病的GPR40激动剂的制造方法与工艺
- 2‑哌嗪‑1‑基‑4H‑1,3‑苯并噻嗪‑4‑酮衍生物及其用于治疗哺乳动物感染的用途的制造方法与工艺
- 一种甲硫基取代苯并[d]恶唑衍生物的制备方法与流程
- 包括一种或多种1,1‑二取代的烯烃化合物的乳液聚合物、乳液法及聚合物组合物的制造方法与工艺
- 包括一种或多种1,1‑二取代的烯烃化合物的聚合物及其聚合物组合物的制造方法与工艺
- 包括一种或多种1,1‑二取代的烯烃化合物的溶液聚合物、溶液聚合法及聚合物组合物的制造方法与工艺
- 一种异原子取代的苯并噻二唑基聚合物给体材料及其制备方法和应用与流程
- 5,7位取代的1H‑苯并[e][1,4]二氮杂‑2(3H)‑酮衍生物的制备方法与流程
- 具有两个二苯并呋喃或二苯并噻吩取代基的咔唑的制造方法与工艺
- 一种含硫的取代基取代的二噻吩衍生物及其共轭聚合物的制备方法及应用
- N2-糖基取代的1,2,3-三唑化合物及其合成方法与应用
- 4-(3-取代苯胺基)-6-取代基喹唑啉类化合物及制备和应用
- 被具有包含炔基取代基的苯基取代的除草活性环二酮化合物或其衍生物的制作方法
- 1-N-取代苄基-6-N’-取代基-2,3,6,9-四氢-1H-[1,4]噁嗪并[3,2-g]喹啉-9-酮-8-甲酸 ...的制作方法
- 具有取代苯基硫属原子代低级烷基和导入酯结构的苯基作为取代基的新型1,2-二氢喹啉 ...的制作方法
- 2
- 制备区域规则的聚-(3-取代的)噻吩、硒吩、噻唑和硒唑的方法
- 一种取代五并苯类红色有机电致发光材料及其制备方法
- 光子型间苯取代基全氟环戊烯类二芳烯光致变色材料的制备及其在双光子光存储中的应用的制作方法