一种雷迪帕韦的制备方法及制备雷迪帕韦的中间体与流程
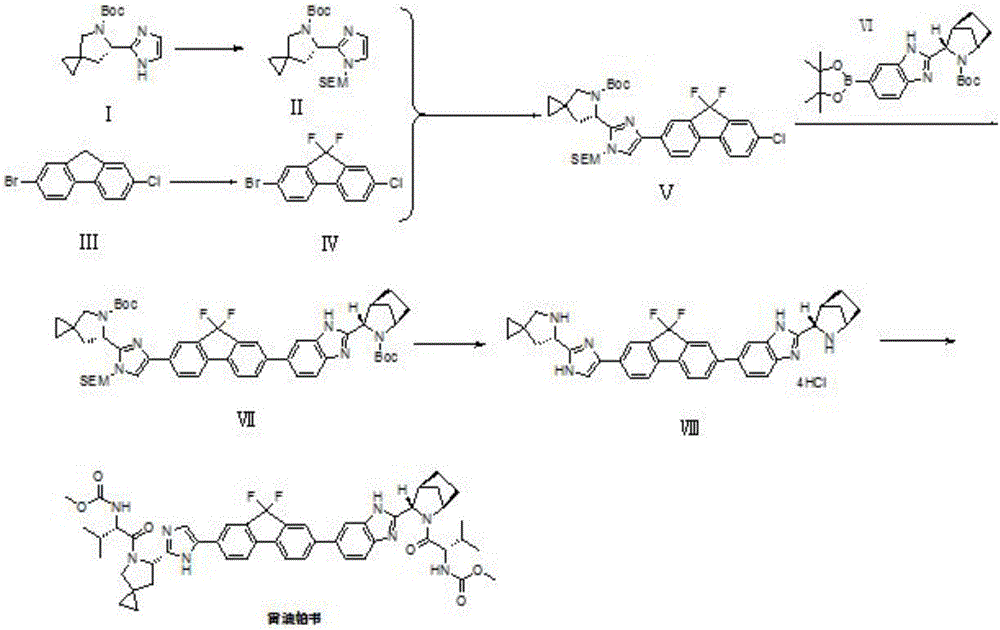
本发明涉及有机合成制药的技术领域,特别涉及一种雷迪帕韦的制备方法及制备雷迪帕韦的中间体。
背景技术:
丙型肝炎是由丙肝病毒感染引起的,丙肝病毒为直径约55~65nm球形颗粒,外有脂质囊膜和棘突结构,内有核心蛋白(core)和RNA组成的核衣壳。丙肝病毒的基因组含10个基因,表达产生10个结构蛋白(包含核心蛋白、包膜蛋白E1和E2、离子通道蛋白P7)和非结构蛋白(包含NS2、NS3、NS4A、NS4B、NS5A和NS5B)。其中NS5A是一种高度磷酸化的非结构蛋白,不具备酶催化活性,其磷酸化水平在HCV基因组的复制和翻译过程中都起着调节的作用。NS5A上丝氨酸残基产生两种磷酸化程度不同的蛋白,即基础磷酸化p56和高度磷酸化p58,均在HCV的生命周期中发挥重要作用。NS5A功能的重要性和多样性使它成为抗HCV的重要靶点,其抑制剂的研究开发已经取得了非常引人瞩目的成就。
Harvoni是索非布韦和雷迪帕韦的复方,临床研究显示,HCV患者接受这个药物治疗8周或12周,治愈率可达99%;复方中的雷迪帕韦是丙肝病毒NS5A抑制剂。
专利WO2013184702最早报道了雷迪帕韦,并且报道了由化合物XIII与化合物VI进行Suzuki偶联反应来合成雷迪帕韦的方法,合成路线如下式所示。其中涉及到使用昂贵的N-氟代双苯磺酰胺作为氟代试剂合成化合物X,化合物X再经过格式反应、酯化反应、环化反应、Suzuki偶合反应和脱保护反应共五步反应得到制备雷迪帕韦的关键中间体化合物VIII,该方法反应步骤多,制备过程过于复杂。
技术实现要素:
有鉴于此,本发明目的在于提供一种工艺简便、步骤少的雷迪帕韦的制备方法。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种雷迪帕韦关键中间体的制备方法,包括以下步骤:
(1)在碱性物质的作用下,将具有式I所示结构的化合物和2-(三甲基硅烷基)乙氧甲基氯在非质子有机溶剂中进行咪唑环保护反应,得到具有式Ⅱ所示结构的化合物;
(2)在有机胺盐的作用下,将2-溴-7-氯-9H-芴与N-氟代双苯磺酰胺在醚类溶剂中进行氟代反应,得到2-溴-7-氯-9,9-二氟-9H-芴;
(3)在碱性物质、钯催化剂和有机磷配体的作用下,将所述具有式Ⅱ所示结构的化合物和所述2-溴-7-氯-9,9-二氟-9H-芴在极性溶剂中进行碳-氢活化偶联反应,得到具有式Ⅴ所示结构的化合物;
(4)在碱性物质、钯催化剂和有机磷配体的作用下,将所述具有式Ⅴ所示结构的化合物和具有式Ⅵ所示结构的化合物在极性溶剂中进行进行Suzuki偶联反应,得到具有式Ⅶ所示结构的化合物;
(5)将所述具有式Ⅶ所示结构的化合物和酸性物质在非质子极性溶剂中进行脱保护基反应,得到雷迪帕韦关键中间体,具有式Ⅷ所示结构;
所述步骤(1)和步骤(2)之间没有时间顺序限制。
优选的,所述步骤(1)中的碱性物质为氢化钠、叔丁醇钾、叔丁醇钠、双(三甲基硅烷基)氨基锂、双(三甲基硅烷基)氨基钠和双(三甲基硅烷基)氨基钾中的一种或几种的混合物;
优选的,所述步骤(1)中的非质子有机溶剂为四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和N,N-二甲基甲酰胺中的一种或几种的混合物。
优选的,所述步骤(2)中的有机胺盐为双(三甲基硅烷基)氨基钾、双(三甲基硅烷基)氨基锂、双(三甲基硅烷基)氨基钠和二异丙基氨基锂中的一种或几种的混合物;
优选的,所述步骤(2)中的醚类溶剂为四氢呋喃、2-甲基四氢呋喃、异丙醚和甲基叔丁基醚中的一种或几种的混合物。
优选的,所述步骤(3)中的碱性物质为碳酸钾、叔丁醇钾、碳酸铯和醋酸铯中的一种或几种的混合物;
优选的,所述步骤(3)中的钯催化剂为Pd(OAc)2、Pd2(dba)3、Pd(PPh3)4和Pd(PPh3)2Cl2中的一种或几种的混合物;
优选的,所述步骤(3)中的有机磷配体为2-二环己基膦-2′,6′-二甲氧基-联苯、正丁基二(1-金刚烷基)膦和2-二环己基磷-2,4,6-三异丙基联苯中的一种或几种的混合物;
优选的,所述步骤(4)中的碱性物质为碳酸钾、碳酸钠、碳酸铯和碳酸氢钠中的一种或几种的混合物;
优选的,所述步骤(4)中的钯催化剂为Pd(OAc)2、Pd(dppf)Cl2、Pd2(dba)3和Pd(PPh3)4中的一种或几种的混合物。
优选的,所述步骤(4)中的有机磷配体为2-二环己基膦-2′,6′-二甲氧基-联苯、4,5-双二苯基膦-9,9-二甲基氧杂蒽和2-二环己基磷-2,4,6-三异丙基联苯中的一种或几种的混合物。
优选的,所述步骤(5)中的酸性物质为盐酸、氢溴酸、甲磺酸、对甲苯磺酸和三氟醋酸中的一种或几种的混合物。
优选的,所述步骤(4)为:当步骤(3)和步骤(4)中使用的碱和有机磷配体种类一致时,将所述步骤(3)得到的反应液中与具有式Ⅵ所示结构的化合物和钯催化剂混合,进行Suzuki偶联反应,得到具有式Ⅶ所示结构的化合物。
本发明提供了一种利用上述方案所述制备方法制备的关键中间体制备雷迪帕韦的方法,包括以下步骤:
在碱性物质和缩合剂的作用下,将所述雷迪帕韦关键中间体与Moc-L-缬氨酸在极性溶剂中进行缩合反应,得到雷迪帕韦。
本发明提供了用于制备雷迪帕韦的中间体化合物,具有式XV所示结构:
式XV中,R为H、
本发明提供了一种雷迪帕韦的制备方法,以具有式Ⅰ所示结构的化合物和2-溴-7-氯-9H-芴为起始物,分别制得具有式II所示结构的化合物和2-溴-7-氯-9,9-二氟-9H-芴,再经过碳-氢活化偶联反应、Suzuki偶联反应和脱保护基反应,即可得到合成雷迪帕韦的关键中间体—具有式Ⅷ所示结构的化合物,具有式Ⅷ所示结构的化合物再与Moc-L-缬氨酸进行缩合反应,即可得到雷迪帕韦。本发明提供的雷迪帕韦的制备方法简单、步骤少,关键化合物2-溴-7-氯-9,9-二氟-9H-芴(式Ⅳ)仅需经过碳-氢活化偶联反应、Suzuki偶联反应和去保护基反应,三步即可得到制备雷迪帕韦的关键中间体VIII,提高了氟化合物的利用率,降低了制备成本,实验结果表明,本发明的合成路线中Suzuki偶联反应的收率可以达到80%以上,去保护基反应的收率可以达到95%,氟化物的利用率可以达到50%以上。
附图说明
图1为本发明实施例合成雷迪帕韦的路线图。
具体实施方式
本发明提供了一种雷迪帕韦关键中间体的制备方法,包括以下步骤:
(1)在碱性物质的作用下,将具有式I所示结构的化合物和2-(三甲基硅烷基)乙氧甲基氯在非质子有机溶剂中进行咪唑环保护反应,得到具有式Ⅱ所示结构的化合物;
(2)在有机胺盐的作用下,将2-溴-7-氯-9H-芴与N-氟代双苯磺酰胺在醚类溶剂中进行氟代反应,得到2-溴-7-氯-9,9-二氟-9H-芴;
(3)在碱性物质、钯催化剂和有机磷配体的作用下,将所述具有式Ⅱ所示结构的化合物和所述2-溴-7-氯-9,9-二氟-9H-芴在极性溶剂中进行碳-氢活化偶联反应,得到具有式Ⅴ所示结构的化合物;
(4)在碱性物质、钯催化剂和有机磷配体的作用下,将所述具有式Ⅴ所示结构的化合物和具有式Ⅵ所示结构的化合物在极性溶剂中进行Suzuki偶联反应,得到具有式Ⅶ所示结构的化合物;
(5)将所述具有式Ⅶ所示结构的化合物和酸性物质在非质子极性溶剂中进行脱保护基反应,得到雷迪帕韦关键中间体,具有式Ⅷ所示结构;
所述步骤(1)和步骤(2)之间没有时间顺序限制。
本发明在碱性物质的作用下,将具有式I所示结构的化合物((S)-6-(1H-咪唑-2-基)-5-螺杂环[2.4]庚烷-5-羧酸叔丁酯)和2-(三甲基硅烷基)乙氧甲基氯在非质子有机溶剂中进行咪唑环保护反应,得到具有式Ⅱ所示结构的化合物((S)-6-[1-((2-(三甲基硅基)乙氧基)甲基-1H-咪唑-2-基]-5-螺杂环[2.4]庚烷-5- 羧酸叔丁酯)。在本发明中,所述咪唑环保护反应用的碱性物质优选氢化钠、叔丁醇钾、叔丁醇钠、双(三甲基硅烷基)氨基锂、双(三甲基硅烷基)氨基钠和双(三甲基硅烷基)氨基钾中的一种或几种的混合物;所述咪唑环保护反应用的非质子有机溶剂优选为四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚和N,N-二甲基甲酰胺中的一种或几种的混合物。
在本发明中,所述具有式I所示结构的化合物、碱性物质和2-(三甲基硅烷基)乙氧甲基氯的物质的量比优选为1:1~4:1~3,更优选为1:2~3:1.5~2.5;所述具有式I所示结构的化合物的质量与非质子有机溶剂的体积比优选为1g:15~25ml,更优选为1g:20ml。
在本发明中,所述咪唑环保护反应的温度优选为室温;所述咪唑环保护反应的时间优选为1.5~3h,更优选为2h。本发明优选在搅拌的条件下进行咪唑环保护反应;所述搅拌的速率优选为300~500r/min,更优选为350~450r/min。
在本发明的部分具体实施例中,可以将具有式I所示结构的化合物和非质子有机溶剂混合,得到具有式I所示结构的化合物溶液;在0~5℃条件下将碱性物质和具有式I所示结构的化合物溶液混合,搅拌0.5~1h,得到第一混合溶液;将2-(三甲基硅烷基)乙氧甲基氯滴加至第一混合溶液中,控制滴加过程中第一混合液的温度为0~5℃,得到第二混合溶液;将第二混合溶液自然升温至室温进行咪唑环保护反应,所述反应时间自升温至室温时开始计算。在本发明中,滴加过程中控制混合溶液的温度优选为0~5℃;所述滴加的速度优选为1~5滴/秒,更优选为2~3滴/秒。
所述咪唑环保护反应完成后,本发明优选将得到咪唑环保护反应液进行后处理,得到具有式Ⅱ所示结构的化合物。在本发明中,咪唑环保护反应液的后处理过程优选包括:将咪唑环保护反应淬灭,依次对得到的反应液进行萃取、洗涤、浓缩和纯化,得到式Ⅱ所示结构的化合物。
在本发明中,所述淬灭反应用淬灭剂优选为水;本发明优选使用薄层色谱法检测反应历程,薄层色谱法检测显示反应结束时,将反应淬灭。在本发明中,所述萃取用萃取剂优选为乙酸乙酯;所述萃取的次数优选为2~5次,更优选为3~4次;本发明将所述萃取得到有机相洗涤,所述洗涤的洗涤剂优选为饱和氯化钠溶液;所述洗涤的次数优选为1~3次。在本发明中,所述浓缩优选为减压浓缩;本发明优选将所述洗涤产物浓缩至干。在本发明中,所述纯化优选为硅胶柱纯化;所述硅胶柱纯化用展开剂优选为乙酸乙酯和石油醚的混合物;所述乙酸乙酯和石油醚的混合物中乙酸乙酯和石油醚的体积比优选为1:1~5,更优选为1:2~4。
本发明对式Ⅰ所示化合物的来源没有特殊要求,使用市售商品或使用本领域常规方法进行合成均可。
本发明在有机胺盐的作用下,将2-溴-7-氯-9H-芴(式Ⅲ)与N-氟代双苯磺酰胺在醚类溶剂中进行氟代反应,得到2-溴-7-氯-9,9-二氟-9H-芴(式Ⅳ)。在本发明中,所述氟代反应用有机胺盐优选为双(三甲基硅烷基)氨基钾、双(三甲基硅烷基)氨基锂、双(三甲基硅烷基)氨基钠和二异丙基氨基锂中的一种或几种的混合物;所述氟代反应用醚类溶剂优选为四氢呋喃、2-甲基四氢呋喃、异丙醚和甲基叔丁基醚中的一种或几种的混合物。
在本发明中,所述2-溴-7-氯-9H-芴、有机胺盐与N-氟代双苯磺酰胺的物质的量比优选为1:2.5~4.5:2~5,更优选为1:3.0~4.0:2.5~3.0;所述2-溴-7-氯-9H-芴的质量与醚类溶剂的体积比优选为1g:20~25ml,更优选为1g:21~23ml。
本发明对2-溴-7-氯-9H-芴的来源没有特殊要求,使用使用本领域常规方法进行合成或直接使用市售的商品均可。
在本发明中,所述氟代反应的温度优选为-60~-100℃,更优选为-70~-80℃;所述氟代反应的时间优选为1.5~3h,更优选为2h;本发明优选在惰性气体保护下进行氟代反应。
在本发明的部分具体实施例中,可以将具有式Ⅲ所示结构的化合物、N-氟代双苯磺酰胺和第一部分醚类溶剂混合,得到混合溶液;将碱性化合物溶解于第二部分醚类溶剂中,得到碱溶液;将所述碱溶液滴加到混合溶液中,进行氟代反应。在本发明中,滴加过程中控制混合溶液的温度为-70~-80℃;所述滴加的速度优选为1~5滴/秒,更优选为2~3滴/秒;所述第一部分醚类溶剂和第二部分醚类溶剂的总体积符合上述比例,在此不再赘述;所述第一部分醚类溶剂和第二部分醚类溶剂的体积比优选为1:0.9~1.2,更优选为1:1~1.1;本发明的反应时间自碱溶液滴加完毕开始计算。
所述氟代反应完成后,本发明优选将得到的氟代反应液进行后处理,得到2-溴-7-氯-9,9-二氟-9H-芴。在本发明中,所述后处理优选包括以下步骤:
将氟代反应液自然升温至-10~-20℃,向反应液中加入水后浓缩,得到浓缩物;
将所述浓缩物过滤,得到第一滤饼,将所述第一滤饼与甲醇混合,在30~50℃下搅拌0.5~1h,过滤得到第二滤饼;
将所述第二滤饼与甲苯混合,在80~95℃下搅拌1~1.5h,趁热过滤,滤液旋干得到第三滤饼;
将所述第三滤饼与异丙醇混合,在0~5℃下搅拌1~1.5h,过滤,得到第四滤饼;
将所述第四滤饼用异丙醇洗涤后干燥,得到2-溴-7-氯-9,9-二氟-9H-芴。
在本发明中,所述水的加入量优选为反应液体积的0.5~2.0倍,更优选为1.0~1.5倍;所述浓缩优选为减压浓缩;本发明优选将反应液浓缩至原体积的1/5~1/3;
在本发明中,所述甲醇的体积和第一滤饼的重量比优选为2~5ml:1g,更优选为3~4ml:1g;所述搅拌的转速优选为300~500r/min,更优选为350~450r/min;
在本发明中,所述甲苯的体积和第二滤饼的重量比优选为2~5ml:1g,更优选为3~4ml:1g;
在本发明中,所述异丙醇的体积和第三滤饼的重量比优选为4~7ml:1g,更优选为5~6ml:1g;
在本发明中,所述使用异丙醇洗涤的次数优选为2~5次,更优选为3~4次;所述干燥的温度优选为40~60℃,更优选为50℃,干燥至恒重即可;
在上述后处理过程中,所述各步骤中搅拌的转速独立的优选为300~500r/min,更优选为350~450r/min。
得到具有式Ⅱ所示结构的化合物和2-溴-7-氯-9,9-二氟-9H-芴后,本发明在碱性物质、钯催化剂和有机磷配体的作用下,将所述具有式Ⅱ所示结构的化合物和所述2-溴-7-氯-9,9-二氟-9H-芴在极性溶剂中进行碳-氢活化偶联反应,得到具有式Ⅴ所示结构的化合物((S)-6-(4-(7-氯-9,9-二氟-9H-芴-2-基)-1-[(2-(三甲基硅基)乙氧基)甲基-1H-咪唑-2-基]-5-螺杂环[2.4]庚烷-5-羧酸叔丁酯)。
在本发明中,所述碳-氢活化偶联反应用碱性物质优选为碳酸钾、叔丁醇钾、碳酸铯和醋酸铯中的一种或几种的混合物;所述碳-氢活化偶联反应用钯催化剂优选为Pd(OAc)2、Pd2(dba)3、Pd(PPh3)4和Pd(PPh3)2Cl2中的一种或几种的混合物;所述碳-氢活化偶联反应用有机磷配体优选为2-二环己基膦-2′,6′-二甲氧基-联苯(SPhos)、正丁基二(1-金刚烷基)膦(P(n-Bu)Ad2)和2-二环己基磷-2,4,6-三异丙基联苯(XPhos)中的一种或几种的混合物;所述碳-氢活化偶联反应用极性溶剂的极性值优选为4.5~7.5,更优选为5.0~7.0;所述极性溶剂具体的优选为二氧六环、N,N-二甲基乙酰胺、N-甲基吡咯烷酮和乙二醇二甲醚中的一种或几种的混合物。
在本发明中,所述具有式Ⅱ所示结构的化合物、2-溴-7-氯-9,9-二氟-9H-芴、碱性物质、钯催化剂和有机磷配体的物质的量比优选为1:1~1.5:2~3:0.005~0.1:0.01~0.2,和更优选为1:1.2~1.3:2.3~2.5:0.01~0.05:0.05~0.15;所述具有式Ⅱ所示结构的化合物的质量与极性溶剂的体积比优选为1:10~20ml,更优选为1:13~16ml。
在本发明中,所述碳-氢活化偶联反应的温度优选为110~130℃,更优选为115~125℃,最优选为120℃;所述碳-氢活化偶联反应的时间优选为11~13h,最优选为12h。
所述碳-氢活化偶联反应后,本发明优选将得到的碳-氢活化偶联反应液过滤,向得到的滤液中加水淬灭反应,再依次对所述淬灭后的滤液进行萃取、洗涤和浓缩,得到具有式Ⅴ所示结构的化合物的粗产品,粗产品可直接应用于下一步的制备。在本发明中,所述萃取用萃取剂优选为乙酸乙酯;所述萃取剂与滤液的体积比优选为1.5~3.5:1,更优选为2~3:1;所述萃取的次数优选为2~5次,更优选为3~4次。本发明优选将萃取得到的有机相合并,进行洗涤;所述洗涤的洗涤剂优选为饱和氯化钠溶液;所述洗涤的次数优选为1~3次。在本发明中,所述浓缩优选为减压浓缩;本发明对减压浓缩的具体操作方法没有特殊要求,使用本领域常规的减压浓缩方法即可,本发明优选浓缩至干,得到粗产品。
得到具有式Ⅴ所示结构的化合物后,本发明在碱性物质、钯催化剂和有机磷配体的作用下,将所述具有式Ⅴ所示结构的化合物和具有式Ⅵ所示结构的化合物((1R,3S,4S)-3-[6-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1H-苯并咪唑-2-基]-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯)在极性溶剂中进行Suzuki偶联反应,得到具有式Ⅶ所示结构的化合物((S)-6-(4-(7-(2-((1R,3S,4S)-2-(叔丁氧羰基)-2-氮杂双环[2.2.1]庚烷-3-基)-1H-苯并[d]咪唑-6-剂)-9,9-二氟-9H-芴-2-基)-1-[(2-(三甲基硅基)乙氧基)甲基-1H-咪唑-2-基]-5-螺杂环[2.4]庚烷-5-羧酸叔丁酯)。在本发明中,所述Suzuki偶联反应用碱性物质优选为碳酸钾、碳酸钠、碳酸铯和碳酸氢钠中的一种或几种的混合物;所述Suzuki偶联反应用钯催化剂不能为Pd(PPh3)2Cl2,其余和步骤(3)中使用的钯催化剂种类相同,且还可以为Pd(dppf)Cl2;所述Suzuki偶联反应用有机磷配体优选为2-二环己基膦-2′,6′-二甲氧基-联苯(SPhos)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(XantePhos)和2-二环己基磷-2,4,6-三异丙基联苯(XPhos)中的一种或几种的混合物;所述Suzuki偶联反应用极性溶剂的极性值优选为4.5~7.2,更优选为5~6.5;所述极性溶剂具体的优选为二氧六环、N,N-二甲基乙酰胺、N-甲基吡咯烷酮和乙二醇二甲醚中的一种或几种的混合物。
在本发明中,所述具有式Ⅴ所示结构的化合物、具有式Ⅵ所示结构的化合物、碱性物质、钯催化剂和有机磷配体的物质的量比优选为1:1~1.5:2~3:0.005~0.1:0.001~0.2,更优选为1:1.2~1.3:2.3~2.5:0.01~0.05:0.005~0.1;所述具有式Ⅴ所示结构的化合物的质量和极性溶剂的体积优选为1g:20~30ml,更优选为1g:25ml。
本发明对具有式Ⅵ所示结构的化合物的来源没有特殊要求,使用本领域方法进行合成或直接使用市售的商品均可。
在本发明中,所述Suzuki偶联反应的温度优选为100~110℃,更优选为103~105℃;所述Suzuki偶联反应的时间优选为6~8h,更优选为7h。
本发明还提供了一种通过“一锅法”实现碳-氢活化偶联反应和Suzuki偶联反应的方法,即当步骤(3)和步骤(4)中使用的碱性物质和有机磷配体的种类一致时,所述步骤(3)的反应完成后,将步骤(3)的反应液与具有式Ⅵ所示结构的化合物和钯催化剂混合进行Suzuki偶联反应;步骤(3)中得到的具有式Ⅴ所示结构的化合物无需进行分离和纯化处理;此处所用的钯催化剂与上述方案步骤(4)中的钯催化剂一致,在此不再赘述。
本发明提供的制备方法通过“一锅法”来实现具有式Ⅶ所示结构的化合物的合成,将步骤(3)和步骤(4)综合到一个反应器中,无需进行中间产物的分离和纯化,进一步简化了制备步骤,降低了生产成本。
所述Suzuki偶联反应完成后,本发明优选将反应淬灭,依次对反应液进行萃取、洗涤、浓缩和纯化,得到式Ⅶ所示结构的化合物。在本发明中,所述萃取、洗涤、浓缩和纯化的方法与分离咪唑环保护反应产物时的方法一致,在此不再赘述。
得到具有式Ⅶ所示结构的化合物后,本发明将所述具有式Ⅶ所示结构的化合物和酸性物质在非质子极性溶剂中进行脱保护基反应,得到雷迪帕韦关键中间体,具有式Ⅷ所示结构。在本发明中,所述酸性物质优选为盐酸、氢溴酸、甲磺酸、对甲苯磺酸和三氟醋酸中的一种或几种的混合物;所述盐酸优选为质量浓度为36~38%的浓盐酸;所述氢溴酸优选为质量浓度为47.5%的浓氢溴酸;所述甲磺酸优选为纯度大于98%的甲磺酸;所述对甲苯磺酸优选为纯度大于96%的对甲苯磺酸;所述三氟醋酸优选为纯度大于99%的浓三氟醋酸;所述脱保护基反应用非质子极性溶剂的极性值优选为4.5~7.2,更优选为5~6.5;所述非质子极性溶剂具体的优选为乙二醇二甲醚、丙酮和乙腈中的一种或几种的混合物。
在本发明中,所述具有式Ⅶ所示结构的化合物和酸性物质的摩尔比优选为1:4.0~6.0,更优选为1:4.3~5.0;所述具有式Ⅶ所示结构的化合物的质量和非质子极性溶剂的体积比优选为1g:15~25ml,更优选为1g:20ml。
在本发明中,所述脱保护基反应的温度优选为70~90℃,更优选为75~85℃,最优选为80℃;所述脱保护基反应的时间优选为5~7h,更优选为5.5~6.5h,最优选为6h;本发明优选在加热回流的条件下进行所述脱保护基反应。
在本发明的部分具体实施例中,可以将具有式Ⅶ所示结构的化合物和非质子极性溶剂混合,得到具有式Ⅶ所示结构的化合物的溶液;将酸性物质滴加至具有式Ⅶ所示结构的化合物的溶液中进行脱保护剂反应,得到具有式VIII所示结构的化合物。在本发明中,所述滴加速度优选为1~5滴/秒,更优选为2~3滴/秒;本发明的反应时间自酸性物质滴加完毕开始计算。
所述脱保护基反应完成后,本发明优选将脱保护基反应液进行后处理,得到雷迪帕韦关键中间体。在本发明中,所述脱保护基反应液后处理的方法优选具体为:将反应液过滤,得到滤饼;将所述滤饼用乙酸乙酯洗涤后浓缩去除残留溶剂。
本发明提供了一种利用上述方案所述制备方法制备的关键中间体制备雷迪帕韦的方法,包括以下步骤:
在碱性物质和缩合剂的作用下,将所述雷迪帕韦关键中间体与Moc-L-缬氨酸在极性溶剂中进行缩合反应,得到雷迪帕韦。
在本发明中,所述缩合反应用碱性物质优选为有机碱;更优选为N-甲基吗啉、三乙胺和二异丙基乙胺中的一种或几种的混合物;所述缩合剂优选为1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC.HCl)和1-羟基苯并三唑(HOBt)或2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU)中的一种或几种的混合物;所述缩合反应用极性溶剂的极性值优选为4.5~7.2,更优选为5.0~6.5;所述极性溶剂具体的优选为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮和二氯甲烷中的一种或几种的混合物。
在本发明中,所述雷迪帕韦关键中间体、缩合剂、碱性物质和Moc-L-缬氨酸的物质的量比优选为1:3~8:8~14:2~4,更优选为1:4~6:9~11:2.5~3.5;所述具有式Ⅷ所示结构的化合物的质量和极性溶剂的体积比优选为1g:8~12ml,更优选为1g:9~11ml,最优选为1g:10ml。
在本发明中,所述缩合反应的温度优选为0~40℃,更优选为10~30℃;所述缩合反应的时间优选为2~3h,更优选为2.5~2.8h。
在本发明的部分具体实施例中,可以将酸性物质、Moc-L-缬氨酸和极性溶剂混合并在0~40℃条件下搅拌0.5~1h,得到酸性物质和Moc-L-缬氨酸的混合溶液;将所述混合溶液温度降至0~5℃,向所述混合溶液中加入具有式Ⅷ所示结构的化合物,得到反应原液;将有机碱滴加至所述反应原液中,滴加完毕后将溶液温度升至2~30℃进行缩合反应,得到雷迪帕韦。在本发明中,所述滴加的速度优选为1~5滴/秒,更优选为2~3滴/秒;所述缩合反应的时间自升温至所需温度时开始计算;本发明步骤(6)中使用的有机碱均为液态物质,可以直接滴加,无需使用溶剂进行溶解。
所述缩合反应完成后,本发明优选将缩合反应产物分离,得到雷迪帕韦。在本发明中,所述分离缩合反应产物优选具体为:向反应液中加入乙酸乙酯和水,搅拌0.5~1h后静置分层,得到水相和第一有机相;将所述水相用乙酸乙酯萃取,得到第二有机相;将所述第二有机相和第一有机相合并后与吸水性物质混合,再搅拌1~1.5h后过滤,得到滤液;将所述滤液浓缩,得到浓缩物;将所述浓缩物与丙酮混合,在5~15℃下搅拌12~24h后过滤和干燥,得到雷迪帕韦粗产品;将所述雷迪帕韦粗产品与乙酸乙酯混合后过滤、浓缩至干,得到浓缩固体;将所述浓缩固体用丙酮溶解至澄清后,搅拌10~12h,析出固体后过滤和干燥,得到雷迪帕韦纯品。
在本发明中,所述反应液中加入的乙酸乙酯和反应液的体积比优选为1.8~2.2:1,更优选为2:1;所述反应液中加入的乙酸乙酯和水的体积比优选为2~3:1,更优选为2.3~2.5:1;
在本发明中,所述萃取用乙酸乙酯与水相的体积比优选为1.5~3.5:1,更优选为2~3:1;所述萃取的次数优选为2~3次;
在本发明中,所述吸水性物质优选为无水硫酸钠和/或硅胶;所述吸水性物质的加入量优选为0.05~0.2g/ml,更优选为0.1~0.15g/ml;
在本发明中,所述浓缩滤液的温度优选为70~100℃,更优选为80~90℃;所述浓缩滤液的时间优选为1~3h,更优选为2h;本发明优选将滤液浓缩至原体积的1/5~1/3,更优选为浓缩至原体积的1/4;
在本发明中,所述丙酮与浓缩物的体积比优选为4~5:1,更优选为4.5:1;所述雷迪帕韦粗产品的干燥温度优选为70~90℃,更优选为75~85℃,干燥时间优选为1~3h,更优选为1.5~2.5h;
在本发明中,所述雷迪帕韦粗产品与乙酸乙酯的质量比优选为1:3~3.5,更优选为1:3.1~3.3;
在本发明中,所述浓缩得到浓缩固体的步骤中的浓缩的温度优选为70~100℃,更优选为80~90℃,本发明此处对浓缩时间没有特殊要求,浓缩至干即可;
在本发明中,所述浓缩固体与丙酮的质量比优选为1:10~12,更优选为1:11;
在本发明中,所述雷迪帕韦粗纯品的干燥温度和时间与雷迪帕韦粗产品的干燥温度和时间一致,在此不再赘述。
本发明的制备过程中合成了用于制备雷迪帕韦的新的中间体化合物,具有式XV所示结构:
式XV中,R为H、该中间体化合物用于合成雷迪帕韦,能够减少合成步骤,提高合成效率。
下面结合实施例对本发明提供的雷迪帕韦的制备方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
(1)在250mL的四口瓶中加入5.0g具有式Ⅰ所示结构的化合物和100mL四氢呋喃,降温至0℃,加入质量分数为60%的分散于石蜡油的氢化钠912mg,温度控制在0℃;氢化钠加入结束后,在0℃下搅拌反应30分钟。滴加3.3g 2-(三甲基硅烷基)乙氧甲基氯,滴加过程温度控制在0℃,滴加结束后自然升温至室温,搅拌反应2小时;反应完成后取样,TLC(PE:EA=1:1)检测显示反应结束;
对产物进行分离提纯:缓慢滴加100mL水淬灭反应,用150mL乙酸乙酯萃取三次,合并有机相用40mL饱和食盐水洗涤一次,减压浓缩至干,浓缩物用快速硅胶柱纯化,展开剂为乙酸乙酯和石油醚混合物(PE:EA=10:1到PE:EA=3:1),收集含有产品的组分,浓缩得具有式Ⅱ所示结构的化合物5.2g,收率69.6%,纯度97%;
使用核磁共振对产物结构进行检测,可得1HNMR谱图数据为:1HNMR(400MHz CDCl3)δ6.99(s,1H),6.90(s,1H),5.78-5.73(m,0.5H),5.44–5.35(m,0.5H),5.20–5.13(m,1H),5.10–5.02(m,1H),3.80-3.64(m,1H),3.53–3.45(m,2H),3.33(d,1H,J=10Hz),2.33–2.23(m,1H),2.10-1.95(m,1H),1.40(s,4.5H),1.22(s,4.5H),1.00-0.83(m,2H),0.78–0.66(m,1H),0.64–0.58(m,2H),0.57–0.52(m,1H);
根据核磁共振结果可知,所得产物确实为具有式Ⅱ所示结构的化合物;
(2)在2.0L反应瓶中加入60g 2-溴-7-氯-9H-芴,186g N-氟代双苯磺酰胺和660mL四氢呋喃,在氮气保护下,反应液降温至-78℃,缓慢滴加690mL1.0mol/L的双(三甲基硅烷基)氨基锂的四氢呋喃溶液,滴加时温度控制在-70℃,滴加结束后,在-70℃下搅拌反应2小时;取样送LC-MS检测显示反应结束;
对产物进行分离提纯:将反应液自然升温至-20℃,滴加108mL水至澄清,减压浓缩至大量固体析出,分三批加入840mL水带蒸四氢呋喃,冷却至20℃,过滤;滤饼加入180g甲醇中,升温至40℃搅拌30分钟,降温至20℃搅拌30分钟,过滤;滤饼加入192g甲苯中,升温至90℃搅拌1小时;布氏漏斗中铺少量硅胶,趁热过滤;滤饼用热甲苯洗涤两次;减压浓缩大部分甲苯,滴加300g异丙醇,降温至5℃搅拌1小时,过滤,滤饼用异丙醇洗涤两次,滤饼在50℃下鼓风干燥得43.8g2-溴-7-氯-9,9-二氟-9H-芴;收率63.9%,纯度96.6%;
使用核磁共振对产物结构进行检测,可得1HNMR谱图数据为:1H NMR(400MHz,CDCl3),δ:7.80–7.70(m,1H),7.70–7.56(m,2H),7.46(q,J=8.4Hz,2H),7.50–7.36(m,1H);
根据核磁共振结果可知,所得产物确实为具有式Ⅳ所示结构的化合物;
(3)在氮气保护下,向250mL单口瓶中加入6.2g具有式Ⅱ所示结构的化合物,4.1g 2-溴-7-氯-9,9-二氟-9H-芴,3.6g碳酸钾,147mg Pd(OAc)2,550mg SPhos和90mL N,N-二甲基乙酰胺,加完后,反应体系升温至120℃搅拌反应12小时;取样TLC(PE:EA=3:1)检测显示反应结束;
将产物进行分离提纯:向反应液中加少量硅藻土,过滤除去黑色不溶物;向滤液中,加150mL水淬灭反应,用300mL乙酸乙酯萃取两次;合并有机相,用饱和食盐水洗涤两次,减压浓缩干;得到具有式Ⅴ所示结构的化合物的粗品,粗产品可直接用于下一步反应。
取样品,采用硅胶柱方法纯化,将得到的纯品进行质谱和核磁共振检测;所得数据为:MS=628[M++1];1HNMR(400MHz CDCl3)δ7.80–7.70(m,1H),7.63–7.54(m,3H),7.52–7.48(m,1H),7.45–7.42(m,1H),7.17–7.11(m,1H),5.70-5.60(m,0.5H),5.33–5.28(m,0.5H),5.20–5.13(m,2H),3.92-3.64(m,1H),3.53–3.45(m,2H),3.40–3.33(m,1H),2.12–2.07(m,1H),1.95-1.85(m,1H),1.40(s,4.5H),1.22(s,4.5H),0.97-0.92(m,2H),0.87–0.81(m,2H),0.68–0.62(m,1H),0.57–0.52(m,1H),0.00(s,9H);
根据检测结果可知,所得产物确实为具有式Ⅴ所示结构的化合物;
(4)在250mL单口瓶中加入4.0g具有式Ⅴ所示结构的化合物,3.4g具有式Ⅵ所示结构的化合物,1.8g碳酸钾,143mg Pd(dppf)Cl2,526mg SPhos,80mL 1.4-二氧六环和20mL水;反应体系用氮气置换三次,升温至100℃回流7小时;取样送LC-MS检测显示反应结束;
将产物分离提纯:加少量硅藻土和50mL水淬灭反应,加100mL乙酸乙酯萃取三次,合并有机相,用饱和食盐水洗涤两次,减压浓缩干;浓缩物用快速硅胶柱纯化,展开剂为乙酸乙酯和石油醚混合物(PE:EA=10:1到PE:EA=3:1),收集含有产品的组分,浓缩、干燥得具有式Ⅶ所示结构的化合物4.7g,纯度98%,收率为82%;
使用核磁共振对产物结构进行质谱和核磁共振检测,所得谱图数据为:MS=453[M/2+1];1HNMR(400MHz CDCl3)δ10.88–10.62(m,1H),7.90(s,1H),7.80–7.70(m,2H),7.63–7.54(m,2H),7.52–7.48(m,3H),7.18–7.10(m,1H),5.70-5.60(m,0.5H),5.33–5.28(m,0.5H),5.20–5.13(m,2H),4.58(s,1H),4.18(s,1H),3.82-3.64(m,1H),3.53–3.45(m,2H),3.40–3.33(m,1H),2.12–2.07(m,2H),1.78-1.70(m,3H),1.52(s,9H),1.45-1.38(m,4H),1.24(s,9H),0.97-0.92(m,2H),0.87–0.81(m,4H),0.68–0.62(m,2H),0.00(s,9H).
根据检测结果可知,所得产物确实为具有式Ⅶ所示结构的化合物。
(5)在250mL单口瓶中加入2.5g具有式Ⅶ所示结构的化合物和50mL DME,滴加1.3g浓盐酸,升温至80℃回流6小时;取样送LC-MS检测显示反应结束;
将产物分离提纯:将反应液过滤,滤饼用25mL乙酸乙酯洗涤两次,减压浓缩,除去残留的溶剂,得到雷迪帕韦关键中间体(具有式Ⅷ所示结构)1.87g,收率94%,纯度97%;
使用质谱和核磁共振对产物结构进行检测,可得谱图数据为:MS=288[M/2+1]。1HNMR(400MHz CDCl3)δ10.88–10.62(m,1H),10.40–10.32(m,2H),8.37(s,1H),8.29–8.24(m,1H),8.12–7.93(m,5H),7.83–7.70(m,3H),5.32-5.23(m,1H),4.84(s,1H),4.20(s,2H),3.69-3.63(m,1H),3.19–3.11(m,2H),2.90–2.80(m,1H),2.29–2.21(m,1H),2.15–2.05(m,1H),2.04–1.92(m,1H),1.72–1.62(m,3H),0.88–0.82(m,2H),0.75-0.68(m,2H).
根据检测结果可知,所得产物确实为具有式Ⅷ所示结构的化合物。
(6)向2L反应釜内加1000g DMF,87.8g的EDC.HCl,41.2g的HOBt,和80g MOC-L-valine,在25℃下搅拌30分钟,降温到0℃,加入100克具有式Ⅷ所示结构的化合物,在0℃下滴加100g的N-甲基吗啉,30分钟加完;加完后,升温到25℃反应2小时,送样检测HPLC,VIII的含量在0.3%以内为反应终点,否则继续反应到合格为止;
将产物分离提纯:反应结束后,将反应液转入5L反应瓶中,向反应液中加入2000g的乙酸乙酯和800g水,搅拌30分钟后,静置分层得有机相;水层用1440g的乙酸乙酯提取2次,将得到的有机相合并,用5%碳酸氢钠溶液2000g洗涤有机相一次,1000g的水洗涤有机相一次,1000g的饱和氯化钠溶液洗涤有机相一次;洗涤完成后向有机相中加入380g的元明粉和380g的硅胶,在25℃下搅拌1小时,过滤后浓缩滤液至无明显液滴(240克左右);向浓缩物中慢慢滴加1120g的丙酮(1小时加完),降温到10℃,搅拌24h,过滤后将产物在50℃下真空干燥至水分小于0.1%,得到雷迪帕韦粗品;粗品加入3倍重量的乙酸乙酯溶解后,过滤,在45℃下将滤液浓缩至干,加入11倍重量的丙酮溶解澄清后,搅拌10小时,慢慢析出固体,过滤得到产物雷迪帕韦,为白色粉末,收率为77.1%,纯度为99.5%。
使用质谱和核磁共振对产物结构进行检测,可得谱图数据为:ESI-MS m/z:889[M+H]+;1H NMR(400MHz,DMSO)δ12.19(s,1H),11.85(s,1H),8.08(s, 1H),7.96(s,2H),7.92–7.75(m,4H),7.70(s,1H),7.64–7.48(m,2H),7.30(d,J=8.3Hz,1H),7.21(d,J=8.6Hz,1H),5.20(dd,J=7.7,5.1Hz,1H),4.67(d,J=5.9Hz,1H),4.55(s,1H),4.17(t,J=7.9Hz,1H),4.01(t,J=8.2Hz,1H),3.82(d,J=9.7Hz,1H),3.72(d,J=9.7Hz,1H),3.55(d,J=2.5Hz,6H),2.67(s,1H),2.41(d,J=9.3Hz,1H),2.22(m,1H),2.10–1.69(m,7H),1.51(m,2H),0.95(dd,J=15.5,6.5Hz,6H),0.90–0.75(m,7H),0.70(m,1H),0.66–0.46(m,3H).13C NMR(101MHz,CDCl3)δ170.50,170.00,156.93,155.37,149.42,143.86,143.14,141.84,138.30,137.98,137.74,137.49,136.25,135.95,134.64,133.97,133.26,131.18,127.95,125.77,123.35,121.86,121.30,120.96,119.31,116.77,113.60,111.74,109.43,60.82,58.08,57.86,56.24,54.97,54.43,51.52,51.49,30.99,30.14,29.84,27.10,21.16,19.32,19.09,18.45,18.19,10.77,9.59.
根据检测结果可知,所得产物确实为雷迪帕韦。
对整个反应过程中N-氟代双苯磺酰胺的利用率进行计算,可得N-氟代双苯磺酰胺的利用率为59.4%。
实施例2
(1)在1L的四口瓶中加入20.0g具有式Ⅰ所示结构的化合物和400mL四氢呋喃,降温至5℃,加入10.2g叔丁醇钾,滴加13.2g 2-(三甲基硅烷基)乙氧甲基氯,滴加过程温度控制在5℃,滴加结束后自然升温至室温,搅拌反应4小时;反应完成后取样,TLC(PE:EA=1:1)检测显示反应结束;
按照实施例1中的方法对产物进行分离提纯,得到具有式Ⅱ所示结构的化合物23.3g,收率78%,纯度98%;
(2)在2.0L反应瓶中加入70g 2-溴-7-氯-9H-芴,190g N-氟代双苯磺酰胺和700mL四氢呋喃,在氮气保护下,反应液降温至-60℃,缓慢滴加700mL1.0mol/L的双(三甲基硅烷基)氨基锂的四氢呋喃溶液,滴加时温度控制在-60℃,滴加结束后,在-60℃下搅拌反应1.5小时;取样送LC-MS检测显示反应结束;
按照实施例1中的方法对产物进行分离提纯,得到得51.5g 2-溴-7-氯-9,9-二氟-9H-芴;收率65.2%,纯度98.6%;
(3)在氮气保护下,向500mL单口瓶中加入12.2g具有式Ⅱ所示结构的化合物,8.1g 2-溴-7-氯-9,9-二氟-9H-芴,16.7g碳酸铯,290mg Pd(pph3)4,1100mg SPhos和200mL N-甲基吡咯烷酮,加完后,反应体系升温至130℃搅拌反应11小时;取样TLC(PE:EA=3:1)检测显示反应结束;
按照实施例1中的方法将产物进行分离提纯,得到具有式Ⅴ所示结构的化合物的粗品,粗产品可直接用于下一步反应。
(4)在500mL单口瓶中加入8.0g具有式Ⅴ所示结构的化合物,6.5g具有式Ⅵ所示结构的化合物,2.1g碳酸氢钠,290mg Pd(dppf)Cl2,1050mgSPhos,200mL乙二醇二甲醚;反应体系用氮气置换三次,升温至110℃回流6小时;取样送LC-MS检测显示反应结束;
按照实施例1中的方法将产物分离提纯,得到具有式Ⅶ所示结构的化合物10.3g,收率89.5%,产物纯度98%;
(5)在250mL单口瓶中加入5g具有式Ⅶ所示结构的化合物和100mL丙酮,滴加3.8g氢溴酸,升温至70℃回流5小时;取样送LC-MS检测显示反应结束;
按照实施例1中的方法将产物分离提纯,得到雷迪帕韦关键中间体(具有式Ⅷ所示结构的氢溴酸盐)4.86g,收率98%,纯度98.5%;
(6)向2L反应釜内加1000g二氯甲烷,166g的HATU和165g MOC-L-valine,在20℃下搅拌30分钟,降温到0℃,加入200克具有式Ⅷ所示结构的化合物,在0℃下滴加200g的三乙胺,30分钟加完;加完后,升温到20℃反应3小时,送样检测HPLC,有式Ⅷ所示结构的化合物的含量在0.3%以内为反应终点,否则继续反应到合格为止;
按照实施例1中的方法将产物分离提纯,得到产物雷迪帕韦,为白色粉末,收率为80.1%,纯度为99.4%。
对整个反应过程中N-氟代双苯磺酰胺的利用率进行计算,可得N-氟代双苯磺酰胺的利用率为70.0%。
实施例3
(1)在2L的四口瓶中加入30.0g具有式Ⅰ所示结构的化合物和600mL四氢呋喃,降温至3℃,加入15g叔丁醇钠,滴加19.8g 2-(三甲基硅烷基)乙氧甲基氯,滴加过程温度控制在3℃,滴加结束后自然升温至室温,搅拌反应3小时;反应完成后取样,TLC(PE:EA=1:1)检测显示反应结束;
按照实施例1中的方法对产物进行分离提纯,得到具有式Ⅱ所示结构的化合物36.3g,收率81%,纯度98%;
(2)在2.0L反应瓶中加入50g 2-溴-7-氯-9H-芴,136g N-氟代双苯磺酰胺和500mL异丙醚,在氮气保护下,反应液降温至-65℃,缓慢滴加500mL1.0mol/L的双(三甲基硅烷基)氨基钾的异丙醚溶液,滴加时温度控制在-65℃,滴加结束后,在-65℃下搅拌反应2.5小时;取样送LC-MS检测显示反应结束;
按照实施例1中的方法对产物进行分离提纯,得到得37.4g2-溴-7-氯-9,9-二氟-9H-芴;收率66.2%,纯度98.9%;
(3)在1000mL单口瓶中加入45.4g具有式Ⅱ所示结构的化合物,30g2-溴-7-氯-9,9-二氟-9H-芴,26.3g碳酸钾,1.1g Pd(OAc)2,4.0g SPhos和600mL N,N-二甲基乙酰胺,氮气置换三次,升温至120℃回流反应5小时,取样送LC-MS检测显示反应结束;
缓慢降温至20℃,向反应混合液中加入50.3g具有式Ⅵ所示结构的化合物和3.5g Pd(dppf)Cl2,氮气置换三次,升温至120℃回流7小时,取样送LC-MS检测显示反应结束;
按照实施例1中的方法将产物分离提纯,得到具有式Ⅶ所示结构的化合物53.3g,收率62%,产物纯度98%;
(4)在500mL单口瓶中加入10g具有式Ⅶ所示结构的化合物和200mL丙酮,滴加7.6g氢溴酸,升温至75℃回流6小时;取样送LC-MS检测显示反应结束;
按照实施例1中的方法将产物分离提纯,得到雷迪帕韦关键中间体(具有式Ⅷ所示结构的氢溴酸盐)7.0g,收率98.5%,纯度98.9%;
(5)向2L反应釜内加1000g二氯甲烷,200g的HATU和200g MOC-L-valine,在20℃下搅拌30分钟,降温到0℃,加入250克具有式Ⅷ所示结构的化合物,在0℃下滴加250g的三乙胺,30分钟加完;加完后,升温到20℃反应3小时,送样检测HPLC,具有式Ⅷ所示结构的化合物的含量在0.3%以内为反应终点,否则继续反应到合格为止;
按照实施例1中的方法将产物分离提纯,得到产物雷迪帕韦253.3g,为白色粉末,收率为82.1%,纯度为99.5%。
对整个反应过程中N-氟代双苯磺酰胺的利用率进行计算,可得N-氟代双苯磺酰胺的利用率为50.0%。
由以上实施例可知,本发明提供的制备方法简单、步骤少,关键化合物2-溴-7-氯-9,9-二氟-9H-芴(式Ⅳ)仅需经过碳-氢活化偶联反应、Suzuki偶联反应和去保护基反应,三步即可得到制备雷迪帕韦的关键中间体VIII,且可以通过“一锅法”实现碳-氢活化反应和Suzuki偶联反应,提高了氟化合物的利用率,降低了制备成本。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!