使用分裂循环扩增的小核酸定量的制作方法
相关申请的交叉引用
本申请要求于2015年2月17日提交的美国临时专利申请号62/117,381的优先权,该文的全部内容通过引用纳入本文用于所有目的。
发明背景
细胞中出现多种类型的非编码短rna。这类rna的示例包括但不限于mirna、snorna、pirna、或lncrna。其他类型的rna包括,例如,mrna。较短的rna可尤其存在扩增困难,因为序列不以对于使用标准引物和方法的杂交和扩增而言足够长的序列存在。
对短于30个核苷酸长的核酸靶标的常规pcr扩增和检测可能是困难的。pcr扩增需要用于扩增的引物在反应中使用的聚合酶范围内的温度下退火至靶dna或rna。由于pcr需要反应在各循环期间达到90℃或更高以使核酸物质的双链体解链,该聚合酶必须是热稳定的。这通过使用来自超嗜热菌的聚合酶来实现并且产生在以60℃为中心但范围为40℃至75℃的温度下有最佳的功能的酶。随着温度接近温度范围的较低和较高范围,聚合酶变得低效,导致理想的引物解链温度为50℃至65℃。这种要求导致引物的长度为约15至30个核苷酸。因此,对于dna结合染料检测,最小扩增子或靶标长度是约30-60个核苷酸,并且对于基于taqman探针的检测,最小扩增子或靶标长度是45-90个核苷酸。这些长度也取决于靶标的gc含量,使得,例如,at高度富集的靶标需要比gc富集的靶标长得多的扩增子长度。
技术实现要素:
在一些方面中,提供了对样品中靶dna模板的量进行定量的方法。在一些实施方式中,该方法包括:
a)形成多个混合物划分产物,其中所述混合物划分产物包含:
i)靶dna模板;
ii)热稳定的dna依赖性dna聚合酶;和
iii)正向和反向扩增引物,其中
所述扩增引物包含3’杂交区域,其与所述靶dna模板杂交并在所述dna依赖性dna聚合酶存在下引发模板引导的引物延伸;
所述正向或反向扩增引物还任选地包含不与所述靶dna模板互补的5’尾区域;并且
所述正向和反向扩增引物任选地具有超过所述靶dna模板长度的组合长度;并且
b)在适于通过聚合酶链式反应扩增靶dna模板的热循环条件下孵育所述混合物划分产物,其中所述热循环条件包括第一组温度循环和第二组温度循环,其中所述第二组温度循环包括比所述第一组温度循环的退火温度高至少5℃的退火温度;并且
c)检测所述混合物划分产物中扩增的靶dna模板的存在或不存在并确定其中所述靶dna模板存在时的划分产物的比例;从而对所述样品中靶dna模板的量进行定量。
在一些实施方式中,正向或反向扩增引物或两者的5’尾区域具有至少15%、20%、25%、30%、35%、40%、45%、或50%gc含量。在一些实施方式中,正向或反向扩增引物或两者的5’尾区域具有15%至80%、15%至75%、15%至70%、15%至65%、15%至60%、15%至50%、15%至45%、15%至40%、20%至80%、20%至75%、20%至70%、20%至65%、20%至60%、20%至50%、20%至45%、20%至40%、30%至80%、30%至75%、30%至70%、30%至65%、30%至60%、30%至50%、或30%至45%gc含量。
在一些实施方式中,划分产物中的靶dna模板的平均浓度是0.001-10个拷贝/划分产物。
在一些实施方式中,第一循环条件包括1-15、2-15、5-15、10-15、1-20、2-20、5-20、10-20、15-20、1-10、2-10或5-10个循环。
在一些实施方式中,热循环条件还包括第三组温度循环,其包括比第二组温度循环的退火温度高至少5℃(例如,高至少10℃、15℃或20℃,例如,5-25℃、5-20℃)的退火温度。
在一些实施方式中,第一循环条件包括1-20个以下循环(例如,1-15、2-15、5-15、10-15、2-20、5-20、10-20、15-20、1-10、2-10或5-10个循环):
i)变性步骤;
ii)组合的引物退火和延伸步骤;和
iii)任选地,在比第一引物退火和延伸步骤高的温度下进行的第二退火和延伸步骤。
在一些实施方式中,第二循环条件包括10-50个循环(例如,10、20、30、40或50个循环)。在一些实施方式中,第二循环条件包括10-50个以下循环(例如,10、20、30、40或50个循环):i)组合的引物退火和延伸步骤;和ii)变性步骤。
在一些实施方式中,第一循环条件包括低于55℃或50℃(例如,50℃至40℃,或55℃至40℃)的退火温度。
在一些实施方式中,第二循环条件包括至少60℃、55℃或50℃(例如,50℃至60℃、50℃至65℃、50℃至68℃、55℃至60℃、55℃至65℃或55℃至68℃)的退火温度。
在一些实施方式中,扩增引物与靶dna模板的相反链杂交,并且侧接具有低于55℃或50℃的退火温度的靶dna模板的区域。
在一些实施方式中,扩增引物杂交靶dna模板的相反链并且侧接长度为1-30(例如,1-10、8-12、8-20、10-25)个核苷酸的靶dna模板的区域。
在一些实施方式中,扩增引物杂交靶dna模板的相反链,使得引物的3’末端杂交至靶dna模板中的相邻核苷酸位置,即,零间插核苷酸。
在一些实施方式中,扩增引物杂交靶dna模板的相反链并且扩增引物的3’末端在与靶dna模板杂交时重叠。在一些实施方式中,重叠的长度是1、2或3个核苷酸。
在一些实施方式中,扩增引物的3’杂交区域的长度是至少3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个核苷酸。
在一些实施方式中,扩增引物的3’杂交区域的长度小于10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个核苷酸。
在一些实施方式中,靶dna模板的长度小于100、95、90、85、80、75、70、65、60、55、50、45、40或35个核苷酸。在一些实施方式中,靶dna模板的长度为100至15、100至25、100至35、90至15、90至25,或90至35个核苷酸。在一些实施方式中,靶dna模板包括与rna(例如,微小rna)互补的区域,其中与rna或微小rna互补的区域的长度小于50、35、30、25、22、20、18或15个核苷酸。在一些实施方式中,靶dna模板包括与rna(例如,微小rna)互补的区域,其中与rna或微小rna互补的区域的长度为50至15、50至18、50至20、50至22、50至25、50至30、50至35、35至15、35至18、35至20、35至22、35至25、或35至30个核苷酸。
在一些实施方式中,5’尾区域的长度是至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20个核苷酸。在一些实施方式中,5’尾区域的长度是1至20、2至20、3至20、4至20、5至20、1至15、2至15、3至15、4至15、5至15、1至10、2至10、3至10、4至10、或5至10个核苷酸。
在一些实施方式中,孵育包括这样的条件,所述条件使得正向或反向扩增引物的3’杂交区域不杂交至并引发来自第二dna模板的模板引导的延伸,所述第二dna模板在扩增引物杂交的区域中具有多态性。在一些实施方式中,正向或反向扩增引物的3’杂交区域包含区分核苷酸(discriminatorynucleotide),其中区分核苷酸与靶dna模板互补但不与具有多态性的第二dna模板互补,并且其中区分核苷酸处于引物的3’末端的终极(ultimate)位置,或者距离3’末端1、2、3、4或5个核苷酸。在一些实施方式中,正向或反向扩增引物的3’杂交区域包含区分核苷酸,其中该区分核苷酸与靶dna模板互补,但不与具有多态性的第二dna模板互补,并且其中该区分核苷酸位于距离引物的3’末端1、2、3、4、5、6或更多个核苷酸的位置处并且其中正向或反向扩增引物的3’杂交区域还包括不与靶dna模板或第二dna模板互补的核苷酸。
在一些实施方式中,正向或反向扩增引物的3’杂交区域包括与靶dna模板的均聚区域互补的至少1、2、3、4或5个核苷酸的均聚区域。在一些实施方式中,引物的均聚区域的长度是3至25个连续核苷酸。在一些实施方式中,均聚区域是不与靶dna模板互补的5’尾区域的3’。在一些实施方式中,引物的均聚区域是聚胸腺嘧啶区域。在一些实施方式中,引物的均聚区域是聚腺嘌呤区域。在一些实施方式中,该方法包括通过使靶dna模板与末端转移酶接触来向靶dna模板添加均聚区域。在一些实施方式中,靶dna模板的均聚区域是聚胸腺嘧啶区域。
在一些实施方式中,形成多个包含靶dna模板的混合物划分产物包括逆转录靶rna模板以形成靶dna模板。在一些实施方式中,逆转录靶rna模板包括使逆转录引物与靶rna模板杂交,其中所述靶rna模板在3’末端处多腺苷酸化,并且所述逆转录引物从3’到5’包含:
i)与靶rna模板核酸杂交的3’杂交区域;和以下之一:
ii)与靶rna模板的多腺苷酸化的3’末端的区域互补的均聚区域;或
iii)均聚的区域,除了具有与该区域中的其他核苷酸不同的一个或两个核苷酸,其中该区域除了所述的一个或两个核苷酸(其在任一侧上被均聚序列结合)以外,与靶rna模板的聚腺苷酸化的3’末端的区域互补。
在一些实施方式中,均聚区域是聚胸腺嘧啶区域。在一些实施方式中,均聚区域是聚腺嘌呤区域。在一些实施方式中,均聚区域是聚胞嘧啶区域。在一些实施方式中,均聚区域是聚鸟嘌呤区域。在一些实施方式中,均聚区域的长度是2-15个核苷酸。在一些实施方式中,逆转录靶rna模板包括将逆转录引物杂交至靶rna模板并生成cdna,其中靶rna模板在3’末端处聚腺苷酸化,并且cdna在cdna的5’末端处具有互补聚t序列,并且该方法还包括向该cdna的3’末端添加均聚区域,从而生成在5’和3’末端处有均聚序列的cdna。
还分开提供了生成和扩增cdna的方法。该方法也可任选地与上述或本文他处所述的方法组合。在一些实施方式中,该方法包括:
a.通过以下方式逆转录包含非聚a区域和聚a尾的靶rna:
i.将逆转录(rt)引物杂交至靶rna,其中rt引物从5’到3’包含:
x-(t)m-y-(t)n-z(seqidno:1)
其中x是任选的(即,可以不存在)、不与靶rna互补的1-10(例如,1-5或2-5)个核苷酸的5’尾核苷酸序列;
t是胸腺嘧啶;
y是单核苷酸,其是c、g或a;
z是与聚a尾相邻的非聚a区的部分互补的1-10(例如,1-5、2-5或2-10)个核苷酸的任选序列;
m是1-20(例如,1-10或2-10);
n是1-20(例如,1-10或2-10);
ii.通过用rna依赖性聚合酶延伸rt引物来逆转录靶rna以生成纳入rt引物的cdna;并且
b.通过以下方式扩增cdna:
i.杂交包含与-(t)m-y-(t)n-z互补的序列的扩增引物;并且
ii.用dna依赖性聚合酶延伸扩增引物。
在一些实施方式中,m=1-4或2-4,n=1-4或2-4,或者m和n独立地是1-4或2-4。在一些实施方式中,rt引物包括z(1-10(例如,1-5、2-5、或2-10)个与聚a尾相邻的非聚a区域的部分互补的核苷酸)并且所述扩增引物包括与-(t)m-y-(t)n互补的序列相邻的一个或多个3’核苷酸,且其与z互补。
在一些实施方式中,扩增包括聚合酶链式反应(例如,包括但不限于数字pcr反应)。
附图的简要说明
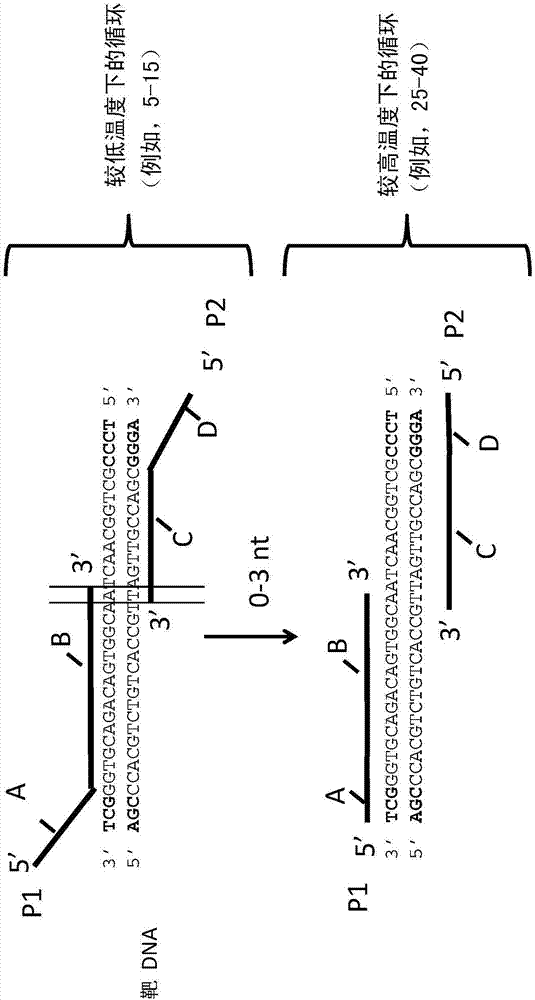
图1a显示了用于扩增靶dna的引物p1和p2的示例性实施例。p1具有不与靶dna互补的5’尾区域(a)和与靶dna互补的3’区域(b)。p2具有不与靶dna互补的5’尾区域和与靶dna的相反链互补的3’区域(seqidno:13-14)。靶dna显示为框。将理解靶dna可以单链或双链核酸开始,但在扩增之后,扩增子将是双链的。如图1a和1b所示,在较低的退火和延伸温度下进行第一组扩增循环,之后在较高的退火和延伸温度下进行第二组循环。
图2显示了示意性实施例,其中在具有靶dna和与靶dna相差1个核苷酸的非靶dna的样品上进行扩增(显示为具有“c”的靶dna和具有“t”的非靶dna)。如图1所示,靶dna显示为框并且可以是单链或双链的,所得的扩增子是双链的。非靶dna上的“x”表示该序列不被扩增,由于引物p1的3’区域与靶dna互补并且用于扩增的条件不允许非靶dna的明显扩增。
图3a、3b和3c示意性地显示了所述方法的实施方式,其中聚a序列存在于或被添加至rna分子,其随后经逆转录以生成靶dna。图3a显示了用引物p1的逆转录步骤,该引物包括与rna(seqidno:16)互补的3’区域(a),与rna的聚a序列互补的均聚聚t区域,并且可包括或不包括在引物的均聚区域(b)内某处的非均聚核苷酸。逆转录产生图3a中的底部分子。在许多实施方式中,rt引物不包括被区分的核苷酸。
图3b显示了使用cdna作为靶dna的第一组扩增循环,使用引物p1和p2来扩增靶标,引物的互补区在该组扩增期间退火。图3c显示了具有较高的退火温度和延伸温度的第二组扩增循环,引物的区域a和b将退火并扩增。
图4a、4b和4c示意性地显示了其他方面,其中均聚序列被添加到初始cdna3’末端(例如,通过末端转移酶)。在所示的实施方式中,聚t添加至图4a中的cdna(seqidno:18(聚-t)),虽然可添加其他核苷酸均聚物(例如,聚a、聚u、聚g、聚c)。图4b显示了使用cdna作为靶dna的第一组扩增循环,引物p1和p2用于扩增靶标,p2包括与cdna序列上的均聚物(seqidno:18(聚-t)和seqidno:19(聚-a))互补的均聚区段。图4c显示了具有较高退火温度和延伸温度的第二组扩增循环。
图5a、5b和5c示意性地显示了其他方面,其中通过使用非酶促反应来将均聚序列(seqidno:18(聚-t))添加至初始rna3’末端(例如,通过加尾的转录引物)。在所示的实施方式中,使用图5a中的加尾的逆转录引物将聚t添加至cdna,虽然可添加其他核苷酸和其他均聚物(例如,聚a、聚u、聚g、聚c)。图5b显示了使用cdna作为靶dna的第一组扩增循环,使用引物p1和p2来扩增靶标,p1包括与cdna序列上的均聚物互补的均聚区段。图5c显示了第二组扩增循环,其具有较高的退火温度和延伸温度(seqidno:18(聚-t)和seqidno:19(聚-a))。
图6显示了假拟使用针对靶rna的聚腺苷酸酶(polyadenylase)。seqidno如下所示:聚a反应:靶rna(顶部)=seqidno:20,靶rna(底部)=seqidno:21;rt反应:靶rna=seqidno:21,rt引物=seqidno:22,cdna产物=seqidno:23;pcr反应:引物(seqidno:24-25),pcr产物(seqidno:26-27)。
图7显示了第二假拟示例,其中使用加尾的引物。seqidno如下:rt反应:靶rna=seqidno:20,rt引物=seqidno:32,cdna产物=seqidno:23;pcr反应:引物(seqidno:24-25),pcr产物(seqidno:26-27)。
图8显示了涉及使用末端转移酶导入均聚序列的假拟示例。seqidno如下:rt反应:靶rna=seqidno:21,rt引物=seqidno:28,cdna产物=seqidno:29;末端转移酶反应:顶部序列=seqidno:30,末端转移酶=seqidno:31,pcr反应:引物(seqidno:24和33),pcr产物(seqidno:34-35)。
定义
术语“聚合酶”是指进行模板引导的多核苷酸合成的酶。该术语同时包括全长多肽和具有聚合酶活性的结构域。dna聚合酶是本领域技术人员熟知的,包括但不限于从激烈火球菌(pyrococcusfuriosus)、滨海嗜热球菌(thermococcuslitoralis)和海栖热袍菌(thermotogamaritime)分离或衍生的dna聚合酶或其修饰形式。它们包括dna依赖性聚合酶和rna依赖性聚合酶,如反转录酶。已知至少5个dna-依赖dna聚合酶家族,虽然大多数落入a、b和c家族。各家族之间几乎没有或没有序列相似性。大多数a家族聚合酶是可含有多重酶促功能(包括聚合酶、3′到5′核酸外切酶活性和5′到3′核酸外切酶活性)的单链蛋白质。b家族聚合酶通常有具有聚合酶和3′到5′核酸外切酶活性的单个催化结构域,以及辅助因子。c家族聚合酶通常是具有聚合和3′到5′核酸外切酶活性的多亚基蛋白质。在大肠杆菌中,已经发现了3种类型的dna聚合酶,dna聚合酶i(a家族)、dna聚合酶ii(b家族)和dna聚合酶iii(c家族)。在真核细胞中,核复制中涉及3种不同的b家族聚合酶,dna聚合酶α、δ和ε,并且a家族聚合酶,聚合酶γ用于线粒体dna复制。其它类型的dna聚合酶包括噬菌体聚合酶。相似地,rna聚合酶通常包括真核rna聚合酶i、ii和iii,和细菌rna聚合酶以及噬菌体和病毒聚合酶。rna聚合酶可以是dna依赖性和rna依赖性的。本文所述的聚合酶可能对于反应混合物、混合物划分产物,或混合物划分产物组中的靶核酸是异源的。本文所用术语“异源”是指在自然中未发现在一起的两种组分(例如,靶核酸和聚合物),例如,因为它们没有一起在相同野生型生物体中发现。
本文使用的“热稳定聚合酶”指使用dna或rna作为模板通过向核苷酸链中加入核苷酸单元以催化多核苷酸合成的酶且该酶在高于45℃的温度下具有最佳活性。
术语“核酸扩增”或“扩增反应”指用于倍增核酸靶序列拷贝的任何体外方法。这种方法包括但不限于聚合酶链式反应(pcr),dna连接酶链式反应(参见美国专利号4,683,195和4,683,202,《pcr方案:方法和应用指南》(pcrprotocols:aguidetomethodsandapplications)(innis等编,1990)),(lcr),qbetarna复制酶和基于rna转录(如tas和3sr)的扩增反应以及本领域技术人员已知的其它反应。
“扩增”指将溶液置于足以扩增多核苷酸的条件下的步骤。扩增反应的组分包括,例如,引物、多核苷酸模板、聚合酶、核苷酸等。术语扩增一般是指靶核酸的“指数型”增长。然而,本文所用的扩增也可指核酸的选择靶序列数量的线性增长,如由循环测序所得。
“聚合酶链式反应”或“pcr”是指靶双链dna的特定区段或子序列得以几何级数式扩增的一种方法。pcr是本领域技术人员所熟知的;参见例如,美国专利号4,683,195和4,683,202;和《pcr方案:方法和应用指南》,innis等编,1990。示例性pcr反应条件一般包括两步或三步循环。两步循环具有变性步骤,之后是杂交/延伸步骤。三步循环包括变性步骤,之后是杂交步骤,之后是独立的延伸步骤。pcr可以终点pcr(即仅在终点处监测)或定量pcr(“实时”监测)的方式进行。
“寡核苷酸引物”或“引物”指退火至靶核酸上的序列并且用作核酸合成的起始点的寡核苷酸序列。
术语“核酸”和“多核苷酸”在本文中可互换使用以表示脱氧核糖核苷酸或核糖核苷酸及其单链或双链形式的聚合物。该术语包括含有已知核苷酸类似物或修饰的主链残基或连接的核酸,其是合成的、天然产生的和非天然产生的,其与参比核酸具有相似结合性质,且以与参比核苷酸相似的方式代谢。这种类似物的示例包括但不限于:硫代磷酸(酯)、氨基磷酸(酯)、甲基膦酸(酯)、手性甲基膦酸(酯)、2-o-甲基核糖核苷酸和肽核酸(pna)。
术语“多肽”、“肽”和“蛋白质”在本文中可互换使用以表示氨基酸残基的聚合物。该术语适用于其中一个或多个氨基酸残基是相应天然产生氨基酸的人造化学模拟物的氨基酸聚合物,以及天然产生的氨基酸聚合物和非天然产生的氨基酸聚合物。
术语“氨基酸”指天然产生的和合成的氨基酸,以及作用方式类似于天然产生氨基酸的氨基酸类似物和氨基酸模拟物。天然产生的氨基酸是由遗传密码编码的氨基酸,以及随后修饰的氨基酸,如羟基脯氨酸、γ-羧基谷氨酸和o-磷酸丝氨酸。氨基酸类似物指与天然产生的氨基酸具有相同基本化学结构的化合物,即结合于氢、羧基、氨基和r基的α碳,如高丝氨酸、正亮氨酸、甲硫氨酸亚砜、甲硫氨酸甲基锍。这种类似物具有修饰的r基(如正亮氨酸)或修饰的肽主链,但保留了与天然产生的氨基酸相同的基本化学结构。氨基酸模拟物指结构不同于氨基酸的普通化学结构,但作用方式类似于天然产生氨基酸的化合物。
本文中氨基酸可按iupac-iub生物化学命名委员会推荐的通常已知的三字母符号或单字母符号表述。核苷酸同样可由其普遍接受的单字母代码表述。
术语“划分”或“划分的”指将样品分隔为多个部分,或“划分产物(partitions)”。划分产物可以是固体或流体。在一些实施方式中,划分产物是固体划分产物,例如微通道。在一些实施方式中,划分产物是流体划分产物,例如液滴。在一些实施方式中,流体划分产物(如液滴)是不互溶的流体(如水和油)的混合物。在一些实施方式中,流体划分产物(如液滴)是水性液滴,其被不互溶的运载体流体(如油)包围。
发明详述
i.引言
已经开发出用于扩增rna靶标的方法和反应混合物,所述rna靶标具体地是短rna靶标,诸如但不限于,微小rna。rna靶标被初始逆转录成靶dna。在其他实施方式中,可使用方法和反应混合物来扩增靶dna(例如,除cdna以外的dna)。该方法涉及使用第一组和第二组扩增循环(例如,pcr循环)来扩增靶dna,其中第二组循环包括在比第一组条件的退火温度更高的温度下的退火温度。另外,提供了正向和反向扩增引物,其中两个引物都具有与靶dna互补的3’区域,并且引物的至少一个(或两个)具有不与靶标互补的5’尾。在退火温度下进行第一组循环以允许基于引物的3’区域与靶dna的杂交的扩增。在第一组中的一定数量的循环(例如,5-10或5-15)之后,建立纳入引物的5’尾的扩增子,从而形成较长的扩增子,加尾的引物以较高tm与其杂交,允许第二组循环具有较高的退火和延伸温度,聚合酶在该温度下更好地发挥功能(接近或是最优)。
还提供了将聚腺苷酸化(聚a)rna逆转录成cdna并且使用逆转录引物扩增cdna的方法,该逆转录引物包含聚t序列,其含有除t以外的核苷酸的1个或2个间插核苷酸,由此生成具有包含1个或2个间插核苷酸的修饰的聚t序列的cdna。然后可使用与修饰的聚t序列互补的引物来扩增cdna。发明人已经发现该方法导致比使用其他相同引物但缺少间插核苷酸的方法更高的特异度和灵敏度。
可使用本文所述的方法检测并扩增的示例性靶rna,其包括但不限于mirna、snrna、snorna、pirna、或lncrna。微小rna(mirna),一般长度为18至25nt,是非蛋白质编码rna,其可抑制靶mrna的翻译(参见,例如,croce和calin,cell122(1):6-7(2005))。其他小rna包括小核质rna(snrna)和小核仁rna(snorna)。这些小rna可在例如,mrna剪接(u1,u2,和u4至u6snrna)、mrna和rrna加工(u7snrna;u3和u8snorna)、和通过2′羟基(盒c/dsnorna)甲基化或通过假尿苷形成(盒h/acasnorna)的rna修饰的位点选择中发挥功能。通过联合哺乳动物中piwi蛋白质来鉴定piwi-相互作用rna(pirna)。pirna的长度范围可以是26-30个核苷酸。也描述了长非编码rna(lncrna)。
本文提供了本发明的其他方面。
ii.反应组分
反应组分可包括包含靶dna以及本文所述的正向引物和反向引物的样品。
可如一种或多种聚合酶链式反应(pcr)扩增那样进行本文所述的方法。该方法可特别用于划分pcr方法,如数字pcr。因此,在一些实施方式中,制备了多种反应混合物,其各自具有低平均拷贝数的靶dna。例如,在一些实施方式中,平均靶dna拷贝数是0.001-10个拷贝/划分产物(例如,0.01、0.05、0.1、0.5、1、2、3、4、5、6、7、8、9或10个拷贝/划分产物)。划分产物的数量可如本领域中理解的那样变化,但范围可以是,例如,1000-1010个划分产物或更多。
反应混合物将包括至少2个寡核苷酸引物,在本文中仅将它们分为“正向”和“反向”引物。各引物将具有与靶dna杂交的3’区域。引物的3’区域的长度可以是,例如,3-20个核苷酸(例如,3、4、5、6、7、8、9、10、11、12、13、14、15或更多,例如,5-15、8-12、5-20等)。在一些实施方式中,3’区域与相应的靶dna序列完全互补。在其他实施方式中,3’区域可具有与靶dna序列的至多1个或2个错配。如上述和本文他处所述,该方法包括采用至少2个不同组的扩增循环,第二组循环具有比第一组更高的退火温度。可选择引物的3’区域以具有接近,例如,等于或低于第二退火温度的解链温度(tm)。例如,在一些实施方式中,一个或两个3’区域的tm与第二组循环的退火温度大致相同或比其低5、10、15、20、25、或30(例如,3-20、3-25)℃以内。
图1a-b显示了混合物的一些不同组分的示例。显示了引物p1和p2。引物区域b和c分别是引物p1和p2的3’区域。将理解引物p1与靶dna的一条链杂交并且引物p2与靶dna的相反链杂交。在其中初始靶dna是单链的情况中,第一循环后的扩增子将包括第二链。
正向引物、反向引物,或两者也可具有不与靶dna杂交的5’尾区域。该5’尾区域可发挥在初始轮次扩增后的数个扩增轮次中延长扩增子的功能。例如,初始轮次扩增将导致包含靶dna序列以及5’尾区域的扩增子。图1的顶部显示了一个实施方式,其中引物的3’区域与靶dna杂交而5’区域则不杂交。可选择5’尾区域的序列,使得引物的tm整体(3’区域和5’区域)具有等于或低于第二组扩增循环的延伸温度的tm。在一些实施方式中,引物的整体tm大约等于第二组循环的退火温度或比其低5、10、15、20、25或30℃。一般选择正向和反向引物的序列以避免引物二聚体形成或引物自杂交。在一些实施方式中,选择5’区域以具有大于30%、40%、50%、60%、70%、80%、85%、90%、或95%,例如,100%的gc含量。5’尾区域的长度可根据需要变化。例如,在一些实施方式中,5’尾区域的长度是1-20个核苷酸(例如,1、2、3、4、5、7、10个核苷酸等)。在一些实施方式中,5’尾部分(不计算引物的其余部分)具有等于或低于第一退火温度的tm。
在一些实施方式中,靶dna将与非靶dna序列高度相似。例如,在其中靶dna是来自待检测的微小rna的cdna,但是非靶微小rna高度相似的情况中,来自非靶微小rna的污染cdna可能会干扰检测。为了减少假阳性,在一些实施方式中,对一个或两个引物的3’区域进行选择,使得3’区域不完全与非靶dna互补(例如,至少1或2个核苷酸不互补)。在一些实施方式中,在本文中也称为“区分核苷酸”的非互补核苷酸(相对于非靶dna)位于引物的3’末端处或距离3’末端1、2、3、4或5个核苷酸。因为dna靶标可以非常短,在一些实施方式中,非互补核苷酸可位于距离引物的3’末端超过5个核苷酸。该方面示于图2,其中2个dna序列,一个含量靶c并且另一个dna序列在相同位置包含t。引物p1包含与靶dna完全互补的3’区域(即,具有g与靶标c互补),但具有相对于非靶dna的非互补核苷酸。然后可选择退火和延伸条件,使得引物与靶标杂交,但不与非靶标杂交。本文中的“不杂交”表示与非靶dna的量相比,靶dna扩增至少10倍,并且在一些实施方式中,至少100或1000倍。
在一些情况中,进一步选择在引物的3’区域中的另一个位置的核苷酸以与靶和非靶dna同时错配可能是有帮助的。这将降低引物针对靶标的tm,但是针对非靶dna的tm将甚至更低,因为3’区域将在2个位置处错配。通过将退火温度设为低于针对靶标的tm和高于针对非靶dna的tm,可扩增靶标,并且很少或没有非靶dna被扩增。
将靶dna上的正向和反向引物杂交的区域定位将与引物的3’区域部分地存在联系。因此,正向引物的3’区域将具有靶dna上的结合位点,并且反向引物的3’区域将具有靶dna上的结合位点(在双链靶dna的相反链上)。在一些实施方式中,2个结合位点将在靶dna中的结合位点之间具有1个或多个核苷酸(例如,1-25、1-20、8-12个核苷酸)。在其他实施方式中,结合位点将是相邻的(图1a-b中显示2个结合位点之间没有核苷酸,其中间隙是“0”)。在另一个实施方式中,2个引物的结合位点将是重叠的。例如,结合位点可重叠1、2、3、4或更多个(例如,1-3)核苷酸。该方面示于图1,其中重叠显示为“0-3”,表示在一些实施方式中,结合位点重叠3个核苷酸,在其他实施方式中,结合位点重叠2个核苷酸,在其他实施方式中,结合位点重叠1个核苷酸或是相邻的(“0”间隙)。
在一些实施方式中,靶dna将在5’末端、3’末端、或两者上包含均聚序列。均聚序列是至少2、3、4、5、6、7、8、9、10或更多个(例如,3-10、3-20、3-50)相邻的相同核苷酸的序列。示例性的均聚序列包括聚a、聚t、聚g、聚c或聚u序列。在其中靶dna具有一个或多个均聚序列的实施方式中,针对靶标扩增的相应引物可具有互补的均聚序列。例如,在图3a-c、4a-c和5a-c中显示了示例性的方面。引物中的均聚序列不需要与靶dna中的均聚序列长度相同。例如,引物均聚序列可短于靶均聚序列。在一些实施方式中,1个或2个引物将具有与靶dna中的均聚序列互补的均聚序列并且引物中的一个或多个将还包含不与靶dna互补的5’尾区域。
可从任何生物样品生成靶dna。本发明的优势是能够扩增短序列,并因此在一些实施方式中,靶dna的长度小于90、80、70、60、50、40、35或30个核苷酸,例如,10-30、20-30、20-40、20-50或10-90个核苷酸。在许多实施方式中,dna是来自样品中存在的rna的cdna。样品可以是,例如,含有短rna的任何混合物。在许多实施方式中,样品衍生自生物流体、细胞或组织。该样品可以是粗的或纯化的。在一些情况中,该样品是来自一个或多个细胞的rna的制备物。在一些实施方式中,细胞是动物细胞,包括但不限于人或非人哺乳动物细胞。非人哺乳动物细胞包括但不限于灵长类细胞、小鼠细胞、大鼠细胞、猪细胞和牛细胞。在一些实施方式中,细胞是植物或真菌(包括但不限于酵母)细胞。细胞可以是,例如,培养的原代细胞、永生化培养细胞或者可来自活检或组织样品的细胞,任选地经培养和刺激以在试验之前分裂。在透化和/或dna修饰步骤之前和/或期间,培养的细胞可处于悬浮或粘附状态。细胞可来自动物组织、活检等。例如,细胞可来自肿瘤活检物。
在一些实施方式中,样品包括仅具有降解的,或者由于核酸降解而难以扩增的可扩增短区域(例如,其中靶区域具有少于50、40、30、25、或20个连续可扩增核苷酸)的rna或dna靶标。例如,由于固定,福尔马林固定的样品可仅具有核酸的短序列。在其他实施方式中,在阅读了本文所述方法的扩增比一般可在pcr中扩增的序列短的序列的能力之后,可通过该方法扩增已经暴露于降解核酸的化学或温度条件的以往核酸样品。
在其他实施方式中,可使用上述的反应混合物的相同基本组分来扩增任何cdna或其他dna分子,无论较长或较短。当区分相差单核苷酸的靶dna与不同的非靶dna时,对这些方面是特别感兴趣的。在这些方面中,一个引物的3’区域(例如,也具有如上所述的5’尾序列的一个引物)较短(例如,3-18、5-19、8-22、3-8、5-10个核苷酸),3’区域中的核苷酸之一与靶dna中的与非靶dna不同的核苷酸互补。通过选择较短的3’区域,靶dna的与非靶dna相比的tm差异将被增强,并因此使得较短互补区域能够区分靶和非靶dna。另外,该方法不易于在延伸期间发生dna聚合酶的误读或错误率,因为该方法使用非杂交的3’核苷酸来防止延伸步骤从非靶模板上开始。在第一组循环之后,在针对靶dna的该较低tm下,所得的扩增子将纳入5’尾序列并因此可如本文他处所述使用在较高tm下的第二组扩增循环。因为该方面可用于较长的靶序列,可设计引物以适应标记的探针(例如,taqman或分子信标探针),如果需要。在其他实施方式中,可使用本文他处所述的插入染料来检测扩增产物。
iii.方法
提供了用于采用至少2个不同组的扩增循环进行扩增的本文所述的方法,其中第二组扩增循环具有高于第一组循环的退火温度。在一些实施方式中,第二组扩增循环具有比第一组扩增循环高至少1、2、3、4、5、6、7、8、9或10(例如,5-15)℃的退火温度。在一些实施方式中,第一组循环采用低于,例如,55、50、48、或45℃的退火温度。在一些实施方式中,第二组循环采用高于,例如,50、55、或60℃(例如,50-75℃)的退火温度。
在一些实施方式中,第一组循环中的循环数量将小于第二组中的循环数量。这部分是因为第一组循环生成包含纳入的引物的初始扩增子,之后第二组循环继续以更高的效率扩增放大的扩增子。在一些实施方式中,第一组循环将具有3-20个循环,例如,5-15个循环。在一些实施方式中,第二组循环将具有至少15个循环,例如,15-45、15-40、20-40、25-35个循环。
扩增“循环”是指一系列的温度变化,其支持双链dna变性,引物退火至靶dna,和通过dna聚合酶延伸引物。一般而言,使用3-步或2-步循环。3-步循环包括分开的变性步骤(例如,90-98℃),分开的退火步骤(例如,50-65℃),和分开的延伸步骤(例如,65-75℃)。在2-步循环中,上述的变性步骤之后是组合的退火/延伸步骤(例如,50-65℃)。虽然本文是指具有比第一组更高的退火温度的第二组循环,应理解在2-步循环中,第二组循环的延伸步骤也将必然高于第一组循环中的情况。在3-步循环中,与第一组循环中的延伸步骤相比,延伸步骤可以,但不必然在第二组循环中较高。
如上所述,所述方法的一个用途是用于检测和定量小rna,包括但不限于mirna、snorna、pirna、或lncrna。如此,在上述步骤之前,可进行逆转录反应以生成cdna。可根据需要使用逆转录酶来进行逆转录以生成cdna。可使用各种逆转录酶中的任意酶。示例性的反转录酶包括但不限于鼠白血病病毒(mlv)反转录酶、禽类成髓细胞血症病毒(amv)反转录酶、呼吸道合胞病毒(rsv)反转录酶、犬感染性贫血病毒(eiav)反转录酶、劳氏-相关病毒-2(rav2)反转录酶、superscriptii反转录酶、superscripti反转录酶、thermoscript反转录酶和mmlvrna酶h-反转录酶。在其他实施方式中,可使用具有rna聚合酶功能的dna聚合酶。例如,在锰存在下,dna聚合酶tth和z05可具有逆转录酶的功能。逆转录酶的浓度可变化并且可凭经验确定并取决于使用的特定逆转录酶。
在一些实施方式中,来自感兴趣的样品的rna含有聚a尾。然而,许多小rna不一定具有聚a尾。在一些实施方式中,聚a尾可在生成cdna之前添加到rna。例如,可用聚(a)聚合酶和atp孵育rna以使rna聚腺苷酸化。该步骤之后可以是本文所述的逆转录。
一旦可获得靶dna,即可添加均聚3’序列,例如,通过连接或将dna与末端转移酶和单核苷酸(例如,dttp、datp、dgtp、dctp、dutp)孵育。所得的dna分子将包含原始靶dna序列和3’均聚物。在一些实施方式中,使用一种或多种本文所述的方法将均聚序列添加至靶dna的两个末端(例如,rna的聚腺苷酸化并且可用末端转移酶将聚t序列添加至dna)。在其中均聚序列已经添加至rna的实施方式中,所得的用于扩增的模板将更长,使得用于扩增的引物设计更简单,并且能够在扩增中使用更高温度的退火。同样,聚腺苷酸化或其他延长cdna的方法可通过提供足够的序列以将pcr引物的3’末端置于引物的3’末端的1、2、3、4或5个核苷酸内的待区分的核苷酸上来允许靶标区分。
还提供了对具有聚a尾的rna进行逆转录的方法。该方法可与上述的分裂循环方法(例如,用于任何逆转录目的)分开进行或可与本文所述的分裂循环方法结合使用。在一些实施方式中,rna将天然包含聚a尾(例如,mrna)。在其他方面中,可使用聚a聚合向rna添加聚a尾。参见,例如,cao,g.j.和sarkar,n.proc.natl.acad.sci.usa.89,10380-10384(1992)。
一旦提供含聚a的rna,可使用逆转录(rt)引物来逆转录rna,其中rt引物从5’到3’包含:
x-(t)m-y-(t)n-z(seqidno:1)
其中x是1-10个核苷酸的任选的5’尾核苷酸序列,其不与靶rna互补;
t是胸腺嘧啶;
y是单核苷酸,其是c、g或a;
z是与聚a尾相邻的非聚a区的部分互补的1-10个核苷酸的任选序列;
m是1-10(例如,2-10、2-5);并且
n是1-10(例如,2-10、2-5)。
上述引物没有与rna的聚a序列互补的单个均聚聚t序列,并且相反具有由一个或两个除t以外的核苷酸分隔(在两侧上有聚t)的聚t序列。因此,当rt引物与聚arna杂交时,rt引物的聚t部分将与rna的聚a部分杂交,但是除t以外的一个或两个核苷酸将不与聚a部分互补,从而将形成“凸起”和非杂交的区域。因为rt条件经选择以允许杂交,不过,引物将引发rt反应以生成cdna。值得注意的是,所得的cdna将在其5’末端处含有与一个或两个核苷酸互补的互补序列。这将允许cdna在后续扩增中特异性高得多的扩增,该后续扩增使用与-(t)m-y-(t)n序列互补的引物。例如,如果rt引物是5'tttttctttttacgc3’(即,m=5,y=c,n=5;(seqidno:2)),pcr引物可以是:5’gcgtaaaaagaaaaa3’(seqidno:3)。
在一些实施方式中,rt引物将包括一个或多个3’核苷酸,其特异性杂交至靶rna的非聚a部分的3’末端,从而使rt引物更有特异性。例如,在上述示例中,“acgc”是与靶rna的最后4个非聚a核苷酸互补的序列(并且因此pcr引物具有互补“gcgt”序列)。
可根据需要检测本文所述的扩增反应。在一些实施方式中,使用在双链dna存在下生成信号的标记物。在一些实施方式中,可以使用插入双链dna时产生信号的插入试剂。示例性的试剂包括sybrgreentm、sybrgoldtm和evagreentm。由于这些试剂不是模板特异性的,推定基于模板特异性扩增产生信号。如果需要,这可以通过监测温度导致的信号变化得到证实,因为模板序列的熔点通常比例如引物-二聚体等高很多。
优选通过数字pcr来进行对靶dna的检测和定量。在一般的方面中,通过将稀释样品划分成多个离散的划分产物使得多个离散的测试位点中的大部分包含低数量的初始靶dna拷贝来进行数字pcr。然后分析并定量扩增产物,导致表示对应于靶dna存在或不存在的感兴趣的基因组区域的存在或不存在。然后可对靶dna(并且因此靶rna)的拷贝数进行定量以估计样品中的靶dna的拷贝数。划分产物的数量可根据应用变化并且可以实现统计学显著的水平。
包含靶dna(或rna,如果逆转录在划分产物中进行)的分离的样品可被划分成多个划分产物。划分产物可包括多种类型的划分产物中的任一种,包括固体划分产物(如孔或管)和流体划分产物(如油相内的水性液滴)。在一些实施方式中,这些划分产物是液滴。在一些实施方式中,这些划分产物是微通道。划分样品的方法和组合物描述于,例如,公开的专利申请wo2010/036352、us2010/0173394、us2011/0092373、us2014/0170736和us2011/0092376,其全部内容各自通过引用纳入本文。
在一些实施方式中,将样品划分成多个液滴。在一些实施方式中,液滴包含乳液组合物,即不互溶的流体(如水和油)的混合物。在一些实施方式中,液滴是水性液滴,其被不互溶的运载体流体(如油)包围。在一些实施方式中,液滴是油性液滴,其被不互溶的运载体流体(如水性溶液)包围。在一些实施方式中,本文所述液滴是相对稳定的并在两种或更多种液滴之间具有最小聚结。在一些实施方式中,由样品生成的液滴中少于0.0001%、0.0005%、0.001%、0.005%、0.01%、0.05%、0.1%、0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%或10%与其他液滴聚结。这些乳液还可具有有限的絮凝,一种分散相从薄片中悬浮液产生的过程。
在一些实施方式中,使油相流过包含待检测标记物的水性溶液,从而形成液滴。在一些实施方式中,包含待检测标记物的水性样品包括缓冲溶液和检测标记物的试剂。用于油相的油可经合成或天然存在。在一些实施方式中,所述油包括碳和/或硅。在一些实施方式中,所述油包括氢和/或氟。示例性的油包括但不限于硅油、矿物油、氟碳油、植物油或其组合。
该油相可包含氟化基础油,其可通过与氟化表面活性剂(如全氟聚醚)联用而进一步稳定。在一些实施方式中,该基础油包括以下一种或多种:hfe7500、fc-40、fc-43、fc-70或另一种常见氟化油。在一些实施方式中,该油相包含阴离子含氟表面活性剂。在一些实施方式中,该阴离子含氟表面活性剂是ammoniumkrytox(krytox-as)、krytoxfsh的铵盐或krytoxfsh的吗啉代衍生物。krytox-as的浓度可以是约0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%、2.0%、3.0%或4.0%(w/w)。在一些实施方式中,krytox-as的浓度是约1.8%。在一些实施方式中,krytox-as的浓度是约1.62%。krytoxfsh的吗啉基衍生物的浓度可以是约0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%、2.0%、3.0%或4.0%(w/w)。在一些实施方式中,krytoxfsh的吗啉代衍生物的浓度是约1.8%。在一些实施方式中,krytoxfsh的吗啉代衍生物的浓度是约1.62%。
在一些实施方式中,该油相还包含用于调节油性质(如蒸气压、粘性或表面张力)的添加剂。非限制性示例包括全氟辛醇和1h,1h,2h,2h-全氟癸醇。在一些实施方式中,1h,1h,2h,2h-全氟癸醇以约0.05%、0.06%、0.07%、0.08%、0.09%、0.1%、0.2%、0.3%、0.4%、0.5%、0.6%、0.7%、0.8%、0.9%、1.0%、1.25%、1.50%、1.75%、2.0%、2.25%、2.5%、2.75%或3.0%(w/w)的浓度添加。在一些实施方式中,1h,1h,2h,2h-全氟癸醇以约0.18%(w/w)的浓度添加。
在一些实施方式中,该乳液配制为生成具有类液界面膜的高度单分散液滴,其可通过加热转化为具有类固界面膜的微胶囊;这类微胶囊可作为生物反应器以通过一段时间的孵育保持其含量。转化为微胶囊形式可在一经加热后即发生。例如,这类转化可发生于大于约40°、50°、60°、70°、80°、90°或95℃的温度下。加热过程期间,可使用流体或矿物油覆盖物来阻止蒸发。可在加热前除去或不除去过量的连续相油。这些生物相容性胶囊可在大范围的热和机械处理下抗聚结和/或絮凝。
在一些实施方式中,将样品分配成至少500个划分产物(如液滴)、至少1000个划分产物、至少2000个划分产物、至少3000个划分产物、至少4000个划分产物、至少5000个划分产物、至少6000个划分产物、至少7000个划分产物、至少8000个划分产物、至少10,000个划分产物、至少15,000个划分产物、至少20,000个划分产物、至少30,000个划分产物、至少40,000个划分产物、至少50,000个划分产物、至少60,000个划分产物、至少70,000个划分产物、至少80,000个划分产物、至少90,000个划分产物、至少100,000个划分产物、至少200,000个划分产物、至少300,000个划分产物、至少400,000个划分产物、至少500,000个划分产物、至少600,000个划分产物、至少700,000个划分产物、至少800,000个划分产物、至少900,000个划分产物、至少1,000,000个划分产物、至少2,000,000个划分产物、至少3,000,000个划分产物、至少4,000,000个划分产物、至少5,000,000个划分产物、至少10,000,000个划分产物、至少20,000,000个划分产物、至少30,000,000个划分产物、至少40,000,000个划分产物、至少50,000,000个划分产物、至少60,000,000个划分产物、至少70,000,000个划分产物、至少80,000,000个划分产物、至少90,000,000个划分产物、至少100,000,000个划分产物、至少150,000,000个划分产物或至少200,000,000个划分产物。
在一些实施方式中,所述样品划分为足量划分产物,使得至少大部分划分产物含有不超过1、2、3、4、5、6、7、8、9、10、15、20、30、40、50、60、70、80、90、100、200、300、400、或500个拷贝的标记物。在一些实施方式中,大部分划分产物含有不超过1、2、3、4、5、6、7、8、9、10、15、20、30、40、50、60、70、80、90、100、200、300、400、或500个拷贝的一种或多种待测标记物。在一些实施方式中,各划分产物存在平均不超过1、2、3、4、5、6、7、8、9、10、15、20、30、40、50、60、70、80、90、100、200、300、400、或500个拷贝的一种或多种标记物。
在一些实施方式中,所述样品划分为足量划分产物,从而平均至少一个划分产物缺少所述标记物的拷贝。在一些实施方式中,所述样品划分为足量划分产物,从而平均至少5、10、15、20、25、30、35、40、50、60、70、80、90、100、125、150、175、200、250、300、350、400、450或500个划分产物缺少所述标记物的拷贝。在一些实施方式中,所述样品划分为足量划分产物,使得平均至少5、10、15、20、25、30、35、40、50、60、70、80、90、100、125、150、175、200、250、300、350、400、450或500个划分产物缺少所述标记物的拷贝并且使得平均至少5、10、15、20、25、30、35、40、50、60、70、80、90、100、125、150、175、200、250、300、350、400、450或500个划分产物具有所述标记物的至少一个拷贝。
在一些实施方式中,生成的液滴在形状和/或尺寸方面基本均匀。例如,在一些实施方式中,这些液滴在平均直径方面基本均匀。在一些实施方式中,生成的液滴的平均直径为约0.001微米、约0.005微米、约0.01微米、约0.05微米、约0.1微米、约0.5微米、约1微米、约5微米、约10微米、约20微米、约30微米、约40微米、约50微米、约60微米、约70微米、约80微米、约90微米、约100微米、约150微米、约200微米、约300微米、约400微米、约500微米、约600微米、约700微米、约800微米、约900微米或约1000微米。在一些实施方式中,生成的液滴的平均直径为小于约1000微米、小于约900微米、小于约800微米、小于约700微米、小于约600微米、小于约500微米、小于约400微米、小于约300微米、小于约200微米、小于约100微米、小于约50微米,或小于约25微米。在一些实施方式中,生成的液滴在形状和/或尺寸方面是不均匀的。
在一些实施方式中,生成的液滴在体积上基本均匀。例如,在一些实施方式中,生成的液滴的平均体积为约0.001nl、约0.005nl、约0.01nl、约0.02nl、约0.03nl、约0.04nl、约0.05nl、约0.06nl、约0.07nl、约0.08nl、约0.09nl、约0.1nl、约0.2nl、约0.3nl、约0.4nl、约0.5nl、约0.6nl、约0.7nl、约0.8nl、约0.9nl、约1nl、约1.5nl、约2nl、约2.5nl、约3nl、约3.5nl、约4nl、约4.5nl、约5nl、约5.5nl、约6nl、约6.5nl、约7nl、约7.5nl、约8nl、约8.5nl、约9nl、约9.5nl、约10nl、约11nl、约12nl、约13nl、约14nl、约15nl、约16nl、约17nl、约18nl、约19nl、约20nl、约25nl、约30nl、约35nl、约40nl、约45nl或约50nl。
数字毒气试验,例如,数字分析可用于通过划分样品和反应组分并且然后用引物和如本文所述用至少两组扩增循环扩增反应来检测并定量样品中的rna或dna。通常,数字分析的过程涉及:针对待检测标记物的存在,确定样品的各划分产物是阳性还是阴性。为了进行定量,检查各划分产物中可检测信号的存在或不存在。如果在划分产物中检测到信号,则该划分产物对抗原存在为“阳性”。如果在划分产物中没有检测到信号,则该划分产物是“阴性”。
在一些实施方式中,能够检测单个信号或多个信号的检测器被用于分析各划分产物中是否存在信号。例如,在一些实施方式中,使用双色读取器(荧光检测器)。阳性计数划分产物的分数可用于测定待测量靶dna或rna的绝对浓度。
一旦确定各样品划分产物的二态“是-否”结果,即可使用基于泊松统计的算法分析各划分产物的数据以定量样品中的靶标的量。定量靶标的浓度或含量的统计学方法描述于例如wo2010/036352,其通过引用全文纳入本文。
可用于本发明的dna聚合酶可以是能够复制dna分子的任何聚合酶。示例性的dna聚合酶是热稳定聚合酶,尤其适用于pcr。热稳定的聚合酶可以从多种嗜热细菌分离得到,例如水生栖热菌(thermusaquaticus(taq))、布鲁克栖热菌(thermusbrockianus(tbr))、黄栖热菌(thermusflavus(tfl))、红栖热菌(thermusruber(tru))、嗜热栖热菌(thermusthermophilus(tth))、滨海热球菌(thermococcuslitoralis(tli))以及热菌属的其他菌种、嗜酸热原体(thermoplasmaacidophilum(tac))、那不勒斯栖热袍菌(thermotoganeapolitana(tne))、海栖热袍菌(thermotogamaritima(tma))、以及热袍菌属(thermotoga)的其他菌种、激烈火球菌(pyrococcusfuriosus(pfu))、沃氏火球菌(pyrococcuswoesei(pwo)、以及其他火球菌属(pyrococcus)的其他菌种、嗜热脂肪芽孢杆菌(bacillussterothermophilus(bst))、嗜酸热硫化叶菌(sulfolobusacidocaldarius(sac))、硫磺矿硫化叶菌(sulfolobussolfataricus(sso))、隐蔽热网菌(pyrodictiumoccultum(poc))、阿比热网菌(pyrodictiumabyssi(pab))、和嗜热自养甲烷杆菌(methanobacteriumthermoautotrophicum(mth))、以及它们的突变体、变体或衍生物。
在一些实施方式中,聚合酶是包含聚合酶结构域和dna结合结构域的杂交聚合酶。已知这些杂交聚合酶具有提高的处理能力。参见例如,美国专利申请公开号2006/005174;2004/0219558;2004/0214194;2004/0191825;2004/0081963;2004/0002076;2003/0162173;2003/0148330;2003/0138830和美国专利号6,627,424和7,445,898,它们以其全部内容通过引用纳入本文用于所有目的,尤其是关于聚合酶、杂交/嵌合聚合酶以及用于制备和使用这些聚合酶的方法的全部启示。在一个方面中,杂交聚合物缺乏3’-5’外切核酸酶活性。在一个实施方式中,这些杂交聚合酶在聚合酶结构域中包括提供外切核酸酶缺陷的双重点突变。在一个具体的实施方式中,杂交聚合酶在聚合酶结构域中可包括双重点突变d141a/e143a。
在一些实施方式中,杂交聚合酶的dna结合结构域来自热稳定的生物体并且在较高的温度下(例如在45℃以上)提供增强的引物退火。例如,sso7d和sac7d分别是来自超嗜热古细菌硫磺矿硫化叶菌(sulfolobussolfataricus)和嗜酸热硫化叶菌(sulfolobusacidocaldarius)的小(约7kdmw)、碱性染色体蛋白(例如,参见choli等,biochimicaetbiophysicaacta950:193-203,1988;baumann等,structuralbiol.1:808-819,1994;和gao等,naturestruc.biol.5:782-786,1998)。这些蛋白以不依赖序列的方式结合dna,在一些情况下,一旦结合,使得dna的tm升高最多达40℃(mcafee等,biochemistry34:10063-10077,1995)。这些蛋白质及其同系物常常用作改善的聚合酶融合蛋白中的序列非特异性dna结合结构域。sso7d、sac7d、sac7e及相关序列(在这里称为“sso7序列”或“sso7结合域”)是本领域已知的(例如,参见登录号(p39476(sso7d);p13123(sac7d);和p13125(sac7e)))。这些序列通常具有至少75%或更大,80%,85%,90%,或95%或更大的氨基酸序列相同性。例如,sso7蛋白通常具有至少75%的sso7d序列相同性。
在其他实施方式中,有用的杂交聚合酶描述于例如美国专利申请公开号2006/005174;2004/0219558;2004/0214194;2004/0191825;2004/0081963;2004/0002076;2003/0162173;2003/0148330;2003/0138830;pct公开号wo2012/138417;以及美国专利号6,627,424和7,445,898,它们各自以其全部内容通过引用纳入本文用于所有目的,尤其是关于聚合酶、杂交/嵌合聚合酶以及用于制备和使用这些聚合酶的方法的全部启示。生成杂交蛋白的方法和杂交聚合酶蛋白的示例公开于wo2004011605,其通过引用全文纳入本文用于所有目的,并且具体用于与生成杂交蛋白相关的所有技术启示。
可使用在重组遗传学领域中使用的常规条件来进行许多上述步骤(例如,逆转录、扩增等)。公开本发明所用一般方法的基础文本包括sambrook和russell,molecularcloning,alaboratorymanual(《分子克隆,实验室手册》)(第3版,2001);kriegler,genetransferandexpression:alaboratorymanual(《基因转移和表达:实验室手册》)(1990);以及currentprotocolsinmolecularbiology(《新编分子生物学实验指南》)(ausubel等编,1994-1999))。
实施例
图6-8提供了各方面的示例性方案。图6显示了针对靶rna的聚腺苷酸酶(“聚a反应”)的假拟应用。然后用rt引物来逆转录聚腺苷酸化的rna。rt引物包括在均聚聚t序列中间的非-t核苷酸(在该实施例中是g),其与rna上添加的聚a序列互补,从而产生具有包含非-t核苷酸的5’聚t序列的cdna。随后使用包含聚t序列部分的引物并具有间插的非-t核苷酸(例如,pcr引物2带下划线的序列)的引物来进行pcr反应。2个使用的pcr引物包括加粗显示的5’尾序列。
图7显示了第二假拟示例,其中使用加尾的引物。图的rt反应部分显示了包含与靶rna互补的3’部分的rt引物和不与靶rna互补的带下划线的5’尾。在逆转录之后,产生了所示的“cdna产物”。然后在pcr反应(例如,本文所述的分裂循环扩增)中使用引物1和引物2来扩增cdna产物,引物1具有加粗的5’非互补尾和与cdna产物互补的3’部分(正常字体),引物2包含5’非互补尾、含有由rt引物5’尾引入的cdna的5’尾的至少部分,和与所得的双链cdna互补的3’区域。图8显示了涉及使用末端转移酶导入均聚序列的假拟示例。在rt反应中,用与靶rna互补的特异性选择性rt引物来逆转录靶rna。虽然在该示例中不使用5’尾,可以使用这样的尾。随后,在末端转移酶中使用cdna以向cdna的5’末端添加均聚序列(如图所述,聚t)。然后在pcr反应(例如,本文所述的分裂循环扩增)中使用引物1和引物2来扩增cdna产物,引物1具有加粗的5’非互补尾和与crna互补的3’区域(常规字体),引物2在cdna链的框架内并包含5’非互补尾部分、包含均聚序列的至少部分的带下划线的序列,和3’区域。
在工作实施例中,合成dna(22个核苷酸加15个腺嘌呤核苷酸的均聚物)和含有所述的加尾的5’区域的引物(整合dna技术公司(integrateddnatechnologies),爱荷华州克拉威尔)被添加至evagreenddpcrsupermixcat#186-4034(生物辐射实验室公司(bio-radlaboratories),加利福尼亚州赫尔克里斯)。通过将所有组分添加至1个50μl反应中来制备反应混合物并且将20μl移取至2个分开的孔中用于使用qx200ddpcr系统的液滴生成(生物辐射实验室公司,加利福尼亚州赫尔克里斯)。将来自这些孔的液滴反应各自添加至分开的板,其中一个是使用标准热循环方案的热循环:95℃持续5分钟,40个循环的:95℃持续30秒并且50℃持续1分钟,4℃持续5分钟,90℃持续5分钟,4℃保持,并且另一个是使用分裂循环的热循环方案的热循环:95℃持续5分钟,10个循环的:95℃持续30秒和40℃持续1分钟,30个循环的:95℃持续30秒和50℃持续1分钟,4℃持续5分钟,90℃持续5分钟,4℃保持。下表显示了在分裂循环的孔中每微升拷贝数的浓度与标准的单一温度热循环方案相比高25%。其他重复实验显示了一致性地观察到更高效的扩增,如当分裂循环热循环方案与具有5’尾区域的试验联用时较少的中幅(mid-amplitude)阳性和高25-50%的每微升拷贝数(示于图1)。
以下提供了比对。人微小rnalet7家族序列和加粗并下划线表示的与let-7a-5p不同的序列的比对(分别是seqidno:4-12)。
已经使用以下组合来实现了这9个高度相似的微小rna的区分:用逆转录引物的分裂循环的热循环,该逆转录引物具有被单个非均聚物核苷酸间插的均聚物区域,位于引物的3’末端的1、2、3或4个内的区分核苷酸,和一些情况中添加的错配以进一步使引物与不正确靶标的结合去稳定。试验的结果如下所示:
自如上显示了9个不同的试验的各家族成员微小rna各自的检测和扩增的rna百分比。沿左纵轴显示了试验名称并且沿顶部横轴显示了添加的合成微小rna模板。该图表显示了扩增并测量的样品的百分比。
购买了所有9个hsa-mir-let-7的合成微小rna(各22个核苷酸)、逆转录引物和5’加尾的试验引物(爱荷华州克拉威尔的集成dna技术公司(integrateddnatechnologies))。合成微小rna具有使用聚腺苷酸酶(马萨诸塞州伊普斯维奇的新英格兰生物实验室公司)添加到微小rna中的聚腺嘌呤的均聚物区域。该聚腺苷酸化的合成rna然后使用iscriptselectcdna合成试剂盒目录号170-8896(加利福尼亚州赫尔克里斯的生物辐射实验室公司(bio-radlaboratories))和逆转录引物添加到基因特异性逆转录反应中,该逆转录引物具有与聚腺苷酸化的区域部分互补的均聚物和如图3a中所示的合成微小rna。在逆转录反应中生成的cdna与图3b和3c中所述的5’加尾的引物一起被直接添加到evagreenddpcrsupermix目录号186-4034(加利福尼亚州赫尔克里斯的生物辐射实验室公司)中。通过用9个靶标中的每一个单独测试各区分试验来评价9个接近相同的微小rna靶标与组图a中所示的let-7家族的交叉反应性或脱靶扩增。通过将非区分试验(图1)检测到的靶标的浓度设为100%来计算交叉反应性百分比。通过将当向各试验中单独添加9个靶标中的每一个时扩增的靶标的浓度除以100%靶标扩增的浓度来计算交叉反应性。所有测试的试验都能够以小于4%的交叉反应性区分所有9个靶标(小于1%交叉反应性,除了三个,1.6、1.7和3.5%)(b)。一些微小rna方法需要牺牲效率以获得低交叉反应性,但用本文所述的方法,我们证明我们能够以100%的效率获得高度特异性(低交叉反应性)。
本文引用的所有文件(例如,专利、专利申请、书籍、期刊论文或其它公开物)通过引用全文纳入本文以用于所有目的,就好像将各篇单独的文件特定且单独地通过引用全文纳入本文用于所有目的一样。对于通过引用纳入的此类文件与本说明书中所含公开内容相矛盾的内容,均意于以本说明书为准和/或本说明书优先于任何相矛盾的材料。
可获得本发明的多种修改形式和变化形式而不背离本发明的精神和范围,这对于本领域技术人员而言是显见的。本文所述的的具体实施方式仅起示例作用,且不意于构成任何方式的限制。
序列表
<110>生物辐射实验室股份有限公司(bio-radlaboratories,inc.)
d·玛尔(maar,dianna)
s·库珀(cooper,samantha)
w·杨(wei,yang)
<120>使用分裂循环扩增的小核酸定量
<130>1002066
<150>us62/117,381
<151>2015-02-17
<160>35
<170>patentinversion3.5
<210>1
<211>61
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸引物
<220>
<221>misc_feature
<222>(1)..(10)
<223>n是a,c,g或t;n可以存在/缺失
<220>
<221>misc_feature
<222>(12)..(30)
<223>n是t;可以存在或缺失
<220>
<221>misc_feature
<222>(33)..(51)
<223>n是t;可以存在或缺失
<220>
<221>misc_feature
<222>(52)..(61)
<223>n是a,c,g或t;n可以存在/缺失(例如,1-5,2-5,或
2-10)
<220>
<221>misc_feature
<222>(52)..(61)
<223>序列与聚a尾相邻的非聚a区域的部分互补
<400>1
nnnnnnnnnntnnnnnnnnnnnnnnnnnnnytnnnnnnnnnnnnnnnnnnnnnnnnnnnn60
n61
<210>2
<211>15
<212>dna
<213>人工序列
<220>
<223>合成rt引物序列
<400>2
tttttctttttacgc15
<210>3
<211>15
<212>dna
<213>人工序列
<220>
<223>与rt引物互补的合成pcr引物
<400>3
gcgtaaaaagaaaaa15
<210>4
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7a-5p比对序列
<400>4
aactatacaacctactacctca22
<210>5
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7b-5p比对序列
<400>5
aaccacacaacctactacctca22
<210>6
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7c-5p比对序列
<400>6
aaccatacaacctactacctca22
<210>7
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7d-5p比对序列
<400>7
aactatgcaacctactacctct22
<210>8
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7e-5p比对序列
<400>8
aactatacaacctcctacctca22
<210>9
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7f-5p比对序列
<400>9
aactatacaatctactacctca22
<210>10
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7g-5p比对序列
<400>10
aactgtacaaactactacctca22
<210>11
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-let-7i-5p比对序列
<400>11
aacagcacaaactactacctca22
<210>12
<211>22
<212>dna
<213>人工序列
<220>
<223>合成hsa-mir-98-5p比对序列
<400>12
aacaatacaacttactacctca22
<210>13
<211>34
<212>dna
<213>人工序列
<220>
<223>合成靶dna序列5'至3'-互补
<400>13
tcccgctggcaactaacggtgacagacgtgggct34
<210>14
<211>34
<212>dna
<213>人工序列
<220>
<223>合成靶序列5'至3'
<400>14
agcccacgtctgtcaccgttagttgccagcggga34
<210>15
<211>16
<212>dna
<213>人工序列
<220>
<223>具有非均聚核苷酸的合成聚-t序列
<220>
<221>misc_feature
<222>(1)..(5)
<223>n是a,c,g,或t
<400>15
nnnnnttttttwtttt16
<210>16
<211>16
<212>dna
<213>人工序列
<220>
<223>合成聚-a序列
<400>16
aaaaaaaaaaaaaaaa16
<210>17
<211>11
<212>dna
<213>人工序列
<220>
<223>具有非均聚核苷酸的合成聚-a序列
<400>17
aaaawaaaaaa11
<210>18
<211>16
<212>dna
<213>人工序列
<220>
<223>合成聚-t序列
<220>
<221>misc_feature
<222>(12)..(16)
<223>n是t;可以完全存在(n=5)或完全缺失(n=0)
<400>18
tttttttttttnnnnn16
<210>19
<211>11
<212>dna
<213>人工序列
<220>
<223>合成聚-a序列
<400>19
aaaaaaaaaaa11
<210>20
<211>20
<212>dna
<213>人工序列
<220>
<223>合成序列-靶rna
<400>20
ggtgcagacagtggcaatca20
<210>21
<211>39
<212>dna
<213>人工序列
<220>
<223>合成序列-靶rna
<400>21
ggctgcagacagtggcaatcaaaaaaaaaaaaaaaaaaa39
<210>22
<211>19
<212>dna
<213>人工序列
<220>
<223>合成rt引物
<400>22
ttttttttttgtttttgat19
<210>23
<211>35
<212>dna
<213>人工序列
<220>
<223>合成cdna序列
<400>23
ttttttttttgtttttgattgccactgtctgcacc35
<210>24
<211>14
<212>dna
<213>人工序列
<220>
<223>合成pcr引物1
<400>24
tcgggtgcagacag14
<210>25
<211>17
<212>dna
<213>人工序列
<220>
<223>合成pcr引物2
<400>25
aggttgtttttgattgc17
<210>26
<211>33
<212>dna
<213>人工序列
<220>
<223>合成pcr产物
<400>26
aggttgtttttgattgccactgtctgcacccga33
<210>27
<211>33
<212>dna
<213>人工序列
<220>
<223>合成pcr产物(反向互补)
<400>27
tcgggtgcagacagtggcaatcaaaaacaacct33
<210>28
<211>10
<212>dna
<213>人工序列
<220>
<223>合成rt引物
<400>28
tgattgccac10
<210>29
<211>20
<212>dna
<213>人工序列
<220>
<223>合成cdna序列
<400>29
tgattgccactgtctgcacc20
<210>30
<211>21
<212>dna
<213>人工序列
<220>
<223>合成寡核苷酸序列
<400>30
tgattgccactgtctgcagta21
<210>31
<211>38
<212>dna
<213>人工序列
<220>
<223>合成末端转移酶序列
<400>31
tttttttttttttttttttgattgccactgtctgcacc38
<210>32
<211>30
<212>dna
<213>人工序列
<220>
<223>合成rt引物
<400>32
tttttttttttttttgtttttgattgccac30
<210>33
<211>21
<212>dna
<213>人工序列
<220>
<223>合成pcr引物2
<400>33
aggttttttttttttgattgc21
<210>34
<211>36
<212>dna
<213>人工序列
<220>
<223>合成pcr产物
<400>34
aggtttttttttttgattgccactgtctgcacccga36
<210>35
<211>36
<212>dna
<213>人工序列
<220>
<223>合成pcr产物(反向互补)
<400>35
tcgggtgcagacagtggcaatcaaaaaaaaaaacct36
- 还没有人留言评论。精彩留言会获得点赞!