来自样品背景的靶多核苷酸的纳米孔隙检测的制作方法
相关申请的交叉引用
本申请要求2016年2月2日提交的62/111,075号美国临时专利申请的优先权。其公开内容通过引用并入本文。
背景技术:
检测生物体基因组和其他生化机构的特定核酸序列或部分的核酸检测具有广泛的用途,其中包括病原体检测、遗传突变分析、疾病筛选和转基因筛选。核酸检测的一种方法是扩增一段dna以产生多个扩增子(即dna靶克隆),以获得有关核酸靶标的存在和/或数量的信息。这可以被用于测定,例如,有机体(例如病毒)是否被视为存在、基因是否突变或者基因是否被表达。常见的核酸检测方法依赖于扩增的dna的光学检测。当需要进行现场检测时,这可能会限制设备的便携性。此外,光学检测方法往往增加了仪器成本和复杂性。新兴技术通过使用电化学传感或纯电传感技术,简单得多的无化学检测方法,来改善这些问题。不幸的是,设备的高制造成本和低产量阻碍了这些技术在商业上的可行性。因此,所需要的是快速、简单和准确的,且不需要昂贵或耗时的光学或化学检测的用于靶多核苷酸序列的稳定检测的方法、装置和组合物。
技术实现要素:
本文所公开的各个方面可以满足上述一个或多个需求。本文所描述的系统和方法各自具有几个方面,其中没有单一的一个方面单独地负责其所需的特性。非是限制通过以下权利要求表达的本公开的范围,现在将简要讨论一些更突出的特征。在考虑此论述后,和特别是在阅读标题为“具体实施方式”的部分后,将了解本文所描述的样本特征是如何提供改进的系统和方法的。
在一些实施方式中,本文提供了用于电检测样品中目标分子或条件的存在或不存在的方法、组合物和装置。
在一些实施方式中,本文提供了一种用于检测疑似存在于样品中的靶多核苷酸序列的存在或不存在的方法,所述方法包括:提供一组引物,其中所述引物中至少之一可与包含所述靶多核苷酸序列的多核苷酸杂交,和其中所述引物中至少一个被修饰以包含能够特异性结合有效载荷分子的偶联位点;对所述样品进行扩增反应,其中所述样品包含所述引物组和用于扩增的试剂,使得由所述扩增反应产生的包含所述靶多核苷酸序列的扩增子将包含所述偶联位点;将所述有效载荷分子结合到所述偶联位点;将所述样品加载到包含纳米孔隙的装置中(其中所述纳米孔隙将所述装置的内部空间分隔成两个容积),并且将所述装置配置成使所述核酸通过一个或多个孔隙,其中所述装置包含用于每个孔隙的传感器,其被配置为识别穿过纳米孔隙的物体;以及通过使用来自所述传感器的数据测定与有效载荷分子结合的靶多核苷酸是否通过纳米孔隙来检测所述样品中所述靶多核苷酸序列的存在或不存在。
在一些实施方式中,在所述扩增之前将样品加载到所述装置中。在一些实施方式中,在所述扩增后将样品加载到所述装置中。在一些实施方式中,有效载荷分子在所述扩增后结合于所述扩增子的所述偶联位点。在一些实施方式中,有效载荷分子在所述扩增之前与所述引物的所述偶联位点结合。
在一些实施方式中,样品在所述扩增和所述纳米孔隙中的检测之间不经历纯化步骤。在一些实施方式中,将所述样品以至少1:20000、1:10000、1:5000、1:2000、1:1000、1:500、1:200、1:100、1:50、1:20、1:10、1:5、1:2、1:1.5、1:1.2、1:1.1或1:1.05的稀释被加载至所述纳米孔隙装置中。在一些实施方式中,所述样品未经稀释而被加载到所述纳米孔隙装置中。在一些实施方式中,样品包含非靶多核苷酸和扩增反应试剂。在一些实施方式中,纳米孔隙的直径为至少5nm、10nm、20nm、30nm、40nm或50nm。
在一些实施方式中,扩增反应选自于:聚合酶链式反应(pcr)、逆转录pcr、连接反应介导的pcr、环介导的扩增(lamp)、等温扩增、链置换扩增(sda)、多重置换扩增、链置换扩增、解旋酶依赖性扩增、切口酶扩增反应或重组聚合酶扩增。在一些实施方式中,在装置的内部空间中进行扩增反应。
在一些实施方式中,靶多核苷酸包含双链脱氧核糖核酸(dsdna)、单链dna(ssdna)、肽核酸(pna)、单链核糖核酸(ssrna)、dna/rna杂合体或双链核糖核酸(dsrna)。在一些实施方式中,靶多核苷酸是天然存在的多核苷酸。在一些实施方式中,靶多核苷酸是人工合成的多核苷酸。在一些实施方式中,靶多核苷酸是重组多核苷酸。
在一些实施方式中,有效载荷分子选自于:树状聚合物、双链dna、单链dna、dna适体、荧光团、蛋白质、抗体、多肽、纳米珠、纳米棒、纳米管、纳米颗粒、富勒烯、peg分子、脂质体或胆固醇-dna杂合体。
在一些实施方式中,有效载荷分子包含电荷。在一些实施方式中,带电荷的有效载荷分子选自于:肽、氨基酸、带电纳米颗粒、合成分子、核苷酸、多核苷酸、金属或离子。在一些实施方式中,与未结合的靶多核苷酸相比,当所述靶多核苷酸与所述带电的有效载荷分子结合时,检测靶多核苷酸的存在或不存在的灵敏度或特异性增加。
在一些实施方式中,与未结合的靶多核苷酸相比,当所述靶多核苷酸与所述有效载荷分子结合时,检测靶多核苷酸的存在或不存在的灵敏度或特异性增加。
在一些实施方式中,传感器包含被配置为在所述两个容积之间施加电压差,并且测量通过该分隔两个容积的纳米孔隙的电流以产生电流事件特征(currenteventsignature)的电极对。在一些实施方式中,当所述有效载荷结合的靶多核苷酸通过纳米孔隙时产生的电流事件特征与背景分子的电流事件特征按照平均深度、最大深度、持续时间、深度级数、深度和持续时间的面积(areaofdepthandduration)或噪音水平是可区分的。
在一些实施方式中,偶联位点和有效载荷分子通过共价键结合。在一些实施方式中,共价键通过点击化学(clickchemistry)形成。在一些实施方式中,点击化学是铜催化的。在一些实施方式中,点击化学是无铜的。在一些实施方式中,偶联位点和有效载荷分子通过非共价键结合。在一些实施方式中,该非共价键是氢键、离子键、范德华相互作用、疏水相互作用、极性键、阳离子-pi相互作用/平面堆叠相互作用或金属键。在一些实施方式中,偶联位点位于所述引物的3'或5'末端。在一些实施方式中,偶联位点位于所述扩增子的3'或5'端。
在一些实施方式中,偶联位点包含化学基团、反应性基团、小分子或肽。在一些实施方式中,该小分子包含生物素。在一些实施方式中,该反应性基团包含二苯并环辛基(dbco)或叠氮化物。在一些实施方式中,两个或更多个有效载荷分子与所述扩增子结合。在一些实施方式中,该装置至少包含两个串联的纳米孔隙,并且其中在转位过程中与所述有效载荷分子结合的所述扩增子同时处于所述至少两个纳米孔隙中。
本文还提供了用于检测疑似存在于样品中的靶多核苷酸序列的存在或不存在的方法,所述方法包括:提供一组引物,其中所述引物中的至少一个可与包含所述靶多核苷酸序列的多核苷酸杂交,并且其中所述引物中至少一个结合有效载荷分子;对所述样品进行扩增反应,其中所述样品包含所述引物组和用于扩增的试剂,使得其中由所述扩增反应产生的包含所述靶多核苷酸序列的扩增子将结合到所述有效载荷分子;将所述样品加载到包含纳米孔隙的装置中(其中所述纳米孔隙将所述装置的内部空间分隔成两个容积),并且将所述装置配置成使所述核酸通过一个或多个孔隙,其中所述装置包括用于每个孔隙的传感器,其被配置为识别穿过纳米孔隙的物体;以及通过使用来自所述传感器的数据测定与有效载荷分子结合的靶多核苷酸是否穿过纳米孔隙来检测所述样品中所述靶多核苷酸序列的存在或不存在。
在一些实施方式中,在所述扩增之前将样品加载到所述装置中。在一些实施方式中,在所述扩增后将样品加载到所述装置中。在一些实施方式中,样品在所述扩增和所述纳米孔隙中的检测之间不经历纯化步骤。
在一些实施方式中,所述样品以至少1:10000、1:1000、1:500、1:200、1:100、1:50、1:20、1:10、1:5、1:2、1:1.5、1:1.2、1:1.1或1:1.05的稀释被加载至所述纳米孔隙装置中。在一些实施方式中,样品不经稀释而被加载到所述纳米孔隙装置中。在一些实施方式中,样品包含非靶多核苷酸和扩增反应试剂。在一些实施方式中,纳米孔隙的直径为至少5nm、10nm、20nm、30nm、40nm或50nm。
在一些实施方式中,扩增反应选自于:聚合酶链式反应(pcr)、逆转录pcr、连接反应介导的pcr、环介导的扩增(lamp)、等温扩增、链置换扩增(sda)、多重置换扩增、链置换扩增、解旋酶依赖性扩增、切口酶扩增反应或重组聚合酶扩增。在一些实施方式中,在装置的内部空间中进行扩增反应。
在一些实施方式中,靶多核苷酸包含双链脱氧核糖核酸(dsdna)、单链dna(ssdna)、肽核酸(pna)、单链核糖核酸(ssrna)、dna/rna杂合体或双链核糖核酸(dsrna)。在一些实施方式中,靶多核苷酸是天然存在的多核苷酸。在一些实施方式中,靶多核苷酸是人工合成的多核苷酸。在一些实施方式中,靶多核苷酸是重组多核苷酸。
在一些实施方式中,有效载荷分子选自于:树状聚合物、双链dna、单链dna、dna适体、荧光团、蛋白质、抗体、多肽、纳米珠、纳米棒、纳米管、纳米颗粒、富勒烯、peg分子、脂质体或胆固醇-dna杂合体。
在一些实施方式中,有效载荷分子包含离子电荷。在一些实施方式中,带电的有效载荷分子选自于:肽、氨基酸、带电纳米颗粒、合成分子、核苷酸、多核苷酸、金属或离子。在一些实施方式中,与未结合的靶多核苷酸相比,当所述靶多核苷酸与所述带电的有效载荷分子结合时,检测靶多核苷酸的存在或不存在的灵敏度或特异性增加。
在一些实施方式中,与未结合的靶多核苷酸相比,当所述靶多核苷酸与所述有效载荷分子结合时,检测靶多核苷酸的存在或不存在的灵敏度或特异性增加。
在一些实施方式中,传感器包括被配置为在两个容积之间施加电压差,并且测量通过分隔两个容积的纳米孔隙的电流以产生电流事件特征的电极对。在一些实施方式中,当有效载荷结合的靶多核苷酸通过纳米孔隙时产生的电流事件特征与背景分子的电流事件特征按照平均深度、最大深度、持续时间、深度级数、深度和持续时间的面积或噪音水平是可区分的。
在一些实施方式中,引物和有效载荷分子通过共价键结合。在一些实施方式中,引物和有效载荷分子通过非共价键结合。在一些实施方式中,有效载荷分子结合在所述引物的3'或5'末端。
在一些实施方式中,两个或更多的有效载荷分子与所述引物结合。一些实施方式中,扩增子和有效载荷分子通过共价键结合。在一些实施方式中,扩增子和有效载荷分子通过非共价键结合。在一些实施方式中,两个或更多的有效载荷分子与所述扩增子结合。
在一些实施方式中,该装置包含至少两个串联的纳米孔隙,并且其中在转位过程中与所述有效载荷分子结合的所述扩增子同时处于所述至少两个纳米孔隙中。
本文还提供了用于检测疑似存在于样品中的靶多核苷酸序列的存在或不存在的方法,所述方法包括:提供一组引物,其中所述引物中的至少一个可与包含所述靶多核苷酸序列的多核苷酸杂交;对所述样品进行扩增反应,其中所述样品包含所述引物组和用于扩增的试剂,使得由所述扩增反应产生的包含所述靶多核苷酸序列的扩增子的长度为至少100个碱基对;将所述样品加载到包含纳米孔隙的装置中(其中所述纳米孔隙将所述装置的内部空间分隔成两个容积),并且将所述装置配置成使核酸通过一个或多个孔隙,其中所述装置包含用于每个孔隙的传感器,其被配置为识别穿过纳米孔隙的物体;以及通过使用来自所述传感器的数据测定与有效载荷分子结合的靶多核苷酸是否通过纳米孔隙来检测所述扩增样品中所述靶多核苷酸序列的存在或不存在,其中所述扩增样品未经纯化。
在一些实施方式中,在所述扩增之前将样品加载到所述装置中。在一些实施方式中,在所述扩增后将样品加载到所述装置中。在一些实施方式中,扩增子长度为至少200、500、1,000、2,000、5,000或10,000个碱基对。在一些实施方式中,样品在所述扩增和所述纳米孔隙中的检测之间不经历纯化步骤。
在一些实施方式中,所述样品以1:10000、1:1000、1:500、1:200、1:100、1:50、1:20、1:10、1:5、1:2、1:1.5、1:1.2、1:1.1或1:1.05的稀释被加载至所述纳米孔隙装置中。在一些实施方式中,样品未经稀释而将被加载到所述纳米孔隙装置中。在一些实施方式中,样品包含非靶多核苷酸和扩增反应试剂。
在一些实施方式中,纳米孔隙的直径为至少2nm、3nm、5nm、10nm、20nm、30nm、40nm或50nm。
在一些实施方式中,扩增反应选自于:聚合酶链式反应(pcr)、逆转录pcr、连接反应介导的pcr、环介导的扩增(lamp)、等温扩增、链置换扩增(sda)、多重置换扩增、链置换扩增、解旋酶依赖性扩增、切口酶扩增反应、重组聚合酶扩增、环介导的等温扩增(lamp)、自持序列复制、全基因组扩增或连接酶介导的pcr。在一些实施方式中,在该装置的内部空间中进行扩增反应。
在一些实施方式中,靶多核苷酸包含双链脱氧核糖核酸(dsdna)、单链dna(ssdna)、dna/rna杂合体、肽核酸(pna)、单链核糖核酸(ssrna)或双链核糖核酸(dsrna)。在一些实施方式中,靶多核苷酸是天然存在的多核苷酸。在一些实施方式中,靶多核苷酸是人工合成的多核苷酸。在一些实施方式中,靶多核苷酸是重组多核苷酸。
在一些实施方式中,传感器包含被配置为在所述两个容积之间施加电压差,并且测量通过分隔所述两个容积的纳米孔隙的电流以产生电流事件特征的电极对。在一些实施方式中,当靶多核苷酸通过纳米孔隙时产生的电流事件特征与背景分子的电流事件特征按照平均深度、最大深度、持续时间、深度级数、深度和持续时间的面积或噪音水平是可区分的。
在一些实施方式中,所述装置包含至少两个串联的纳米孔隙,并且其中在转位过程中所述扩增子同时处于所述至少两个纳米孔隙中。
本文还提供了一种试剂盒,其包含:包含纳米孔隙的装置,其中所述纳米孔隙将所述装置的内部空间分隔成两个容积,并且所述装置配置成使核酸通过一个或多个孔隙,其中所述装置包含用于每个孔隙的传感器,其被配置为识别通过纳米孔隙的物体;引物组,其中所述引物中的至少一个可与包含所述靶多核苷酸序列的多核苷酸杂交,并且其中所述引物中至少一个被修饰以包含能够特异性结合有效载荷分子的偶联位点;用于在扩增之前、期间或之后与所述偶联位点结合的有效载荷分子;用于检测样品中所述靶多核苷酸的存在或不存在的说明书。
本文还提供了一种试剂盒,其包含:包含纳米孔隙的装置,其中所述纳米孔隙将所述装置的内部空间分隔成两个容积,并且所述装置配置成使所酸通过一个或多个孔隙,其中所述装置包含用于每个孔隙的传感器,其被配置为识别通过纳米孔隙的物体;引物组,其中所述引物中的至少一个可与包含所述靶多核苷酸序列的多核苷酸杂交,并且其中所述引物中的至少一个与有效载荷分子结合;用于检测样品中所述靶多核苷酸的存在或不存在的说明书。
本文还提供了一种试剂盒,其包含:包含纳米孔隙的装置,其中所述纳米孔隙将所述装置的内部空间分隔成两个容积,并且所述装置配置成使核酸通过一个或多个孔隙,其中所述装置包含用于每个孔隙的传感器,其被配置为识别通过纳米孔隙的物体;引物组,其中所述引物中的至少一个可与包含所述靶多核苷酸序列的多核苷酸杂交,其中所述引物在扩增反应期间产生包含至少100、200、500、1,000、2,000、5,000或10,000个碱基对的所述靶多核苷酸序列的扩增子;以及用于检测样品中所述靶多核苷酸的存在或不存在的说明书。
本文还提供了用于定量样品中存在的靶多核苷酸序列的量的方法,其包括:提供包含已知量的对照多核苷酸的对照样品和包含未知量的靶多核苷酸的实验样品;扩增所述对照样品以产生包含所述对照多核苷酸的第一扩增子,和扩增所述实验样品以产生包含所述靶多核苷酸的第二扩增子;将所述对照样品和所述实验样品分别加载到包含纳米孔隙的装置中,其中所述纳米孔隙将装置的内部空间分隔成两个容积,并且该装置配置成使第一或第二扩增子通过一个或多个孔隙,其中该装置包含用于每个孔隙的传感器,其被配置为识别通过所述纳米孔隙的物体;以及将纳米孔隙中所述第一扩增子的捕获率与纳米孔隙中所述第二扩增子的捕获率进行比较,以定量所述实验样品中靶多核苷酸序列的量。
在一些实施方式中,在将所述对照样品或所述实验样品加载到所述装置中之后进行扩增。在一些实施方式中,在相同条件下扩增对照样品和所述实验样品。在一些实施方式中,对照多核苷酸和所述靶多核苷酸具有相同的长度或序列。在一些实施方式中,在相同条件下使用相同的纳米孔隙测定第一和第二扩增子的捕获率。在一些实施方式中,使用相似大小的纳米孔隙测定第一和第二扩增子的捕获率。
在一些实施方式中,通过汇总对于所述对照样品和所述实验样品随时间记录的一组传感器测量值,并比较两组数据以将捕获率转换为浓度而以数学方式产生对实验样品中靶多核苷酸浓度的估算。在一些实施方式中,在扩增反应内的每个循环之后,通过汇总对于所述对照样品和所述实验样品随时间记录的一组传感器测量值,并比较该两组数据以将捕获率转换为浓度而以数学方式产生对靶多核苷酸浓度的估算。
在一些实施方式中,所述方法还包括从所述传感器测量值的比较测定在扩增之前所述实验样品中的靶多核苷酸的量。
本文还提供了定量样品中存在的靶多核苷酸序列的量的方法,其包括:提供包含已知量的对照多核苷酸的对照样品和包含未知量的靶多核苷酸的实验样品;稀释所述对照样品以产生所述对照多核苷酸的至少两种不同已知浓度;以所述对照多核苷酸的所述至少两种不同的已知浓度将所述对照样品加载到包含纳米孔隙的装置中,其中所述纳米孔隙将所述装置的内部空间分隔成两个容积,并且所述装置配置为使所述对照多核苷酸通过一个或多个孔隙,其中所述装置包含用于每个孔隙的传感器,所述传感器被配置为识别通过所述纳米孔隙的物体;在所述至少两种已知浓度的每一种浓度下检测在所述纳米孔隙中对照多核苷酸的捕获率;扩增所述实验样品以产生包含所述靶多核苷酸的扩增子;将实验样品加载到包含纳米孔隙的所述装置中;检测所述纳米孔隙中靶多核苷酸的捕获率;以及将纳米孔隙中的所述第一扩增子的捕获率与纳米孔隙中所述第二扩增子的捕获率进行比较,以定量所述实验样品中靶多核苷酸序列的量。
在一些实施方式中,稀释是连续稀释。在一些实施方式中,所述方法还包括扩增所述对照多核苷酸。在一些实施方式中,在将所述对照样品加载到所述装置中之后进行稀释。在一些实施方式中,在将所述实验样品加载到所述装置之后进行扩增。
附图说明
从以下对本发明的特定实施方式的描述中,前述和其它对象、特征和优点将变得显而易见,如附图所示,其中相同的附图标记在所有不同的视图中指的是相同的部分。附图不一定按比例绘制,而是将重点放在说明本发明的各个实施方式的原理上。还提供了作为本公开的实施方式的数据图,其仅通过示例来说明特征,而非限制。
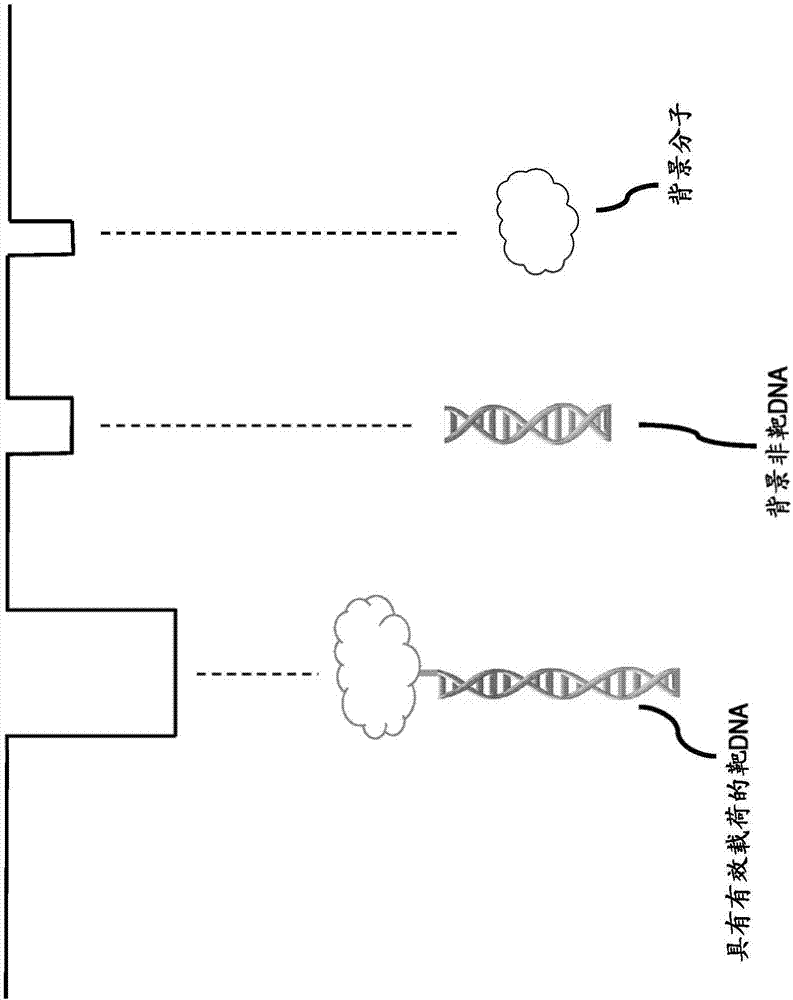
图1描绘了通过纳米孔隙的与有效载荷分子结合的包含靶多核苷酸序列的多核苷酸。
图2描绘了与非靶多核苷酸和一般的非多核苷酸背景分子相比,有效载荷结合的靶多核苷酸通过孔隙时的电流特征的差异。
图3描绘了用生物素化的引物扩增靶序列,然后用纳米孔隙检测由链霉亲和素结合的生物素化的扩增子的过程。
图4描绘了我们的整数(m)纳米孔隙(n)可观察(o)pcr(mnopcr)工作流程,其作为定量pcr扩增子的方法的部分。
图5描绘了在扩增或有效载荷连接之前或之后用于检测靶序列的工作流程,其不需要纯化。
图6描绘了(a)典型的纳米孔隙电流事件和(b)当用纳米孔隙检测3250bpdna时,纳米孔隙电流事件群体,以及将多核苷酸事件与背景事件区分。
图7显示了将470bpdna事件与背景噪声事件进行比较的数据,其显示两者不能被区分。
图8显示用无引物修饰、1生物素修饰的引物和2生物素修饰的引物扩增的470bpdna的琼脂糖凝胶(左图),随后是单链霉亲和素有效载荷连接后的琼脂糖凝胶(右图)。
图9比较470bpdna-[1生物素]-[1单链霉亲和素]和470bpdna-[2生物素]-[2单链霉亲和素]的纳米孔隙电流事件群体。
图10将470bpdna、470bpdna-[1生物素]-[1单链霉亲和素]和470bpdna-[2生物素]-[2单链霉亲和素]的阳性百分比检测标准与从背景建立的假阳性阈值(电噪声)事件进行了比较。
图11显示了在抗生物素抗体有效载荷连接后,(1)用无引物修饰扩增的470bpdna,(2)用1生物素修饰的引物扩增的dna和(3)用2生物素修饰的引物扩增的dna的琼脂糖凝胶。
图12对比了470bpdna(无有效载荷)与470bpdna-[2生物素]-[2抗生物素抗体]的纳米孔隙电流事件群体。
图13对比了以1:10稀释的pcr背景与相同pcr背景下扩增的1074bpdna的纳米孔隙电流事件群体。
图14对比了以1:10稀释的pcr背景、在相同pcr背景下扩增的1074bpdna、无有效载荷的扩增的500bpdna和扩增的500bpdna-[1生物素]-[1单链霉亲和素]的纳米孔隙电流事件群体。
图15显示了(1,4)470bpdna(无生物素引物修饰),(2)470bpdna-[1生物素]-[1单链霉亲和素]和(3)470bpdna-[2生物素]-[2单链霉亲和素]的琼脂糖凝胶,其中泳道1-3存在20x过量的单链霉亲和素,显示不存在非特异性的有效负荷连接。
图16对比了单独的pcr背景(1:60稀释)与pcr背景(1:60稀释)存在下的470bpdna(无有效载荷)、470bpdna-[1生物素]-[1单链霉亲和素]和470bpdna-[2生物素]-[2单链霉亲和素]的纳米孔隙电流事件群体。
图17针对从背景事件(pcr背景1:60稀释)确立的假阳性阈值,对比了470bpdna、470bpdna-[1生物素]-[1单链霉亲和素]和470bpdna-[2生物素]-[2单链霉亲和素]的阳性百分比检测标准。
图18对比了单独的全血背景(1:20000稀释)与全血背景(1:20000稀释)存在下的470bpdna(无有效载荷)、470bpdna-[1生物素]-[1单链霉亲和素]和470bpdna-[2生物素]-[2单链霉亲和素]的纳米孔隙电流事件群体。
图19针对从背景事件(全血背景1:20000稀释)确立的假阳性阈值,对比了470bpdna、470bpdna-[1生物素]-[1单链霉亲和素]和470bpdna-[2生物素]-[2单链霉亲和素]的阳性百分比检测标准。
图20显示了(1)362bpdna(无生物素引物修饰)、(2)362bpdna-[1生物素]-[1单链霉亲和素]和(3)362dna-[2生物素]-[2单链霉亲和素]的琼脂糖凝胶。
图21对比了362bp靶dna和470bp非靶dna(无有效载荷)的组合与362bp靶dna-[1生物素]-[1单链霉亲和素]的纳米孔隙电流事件群体。
图22针对从背景dna事件(470bp非靶dna,无连接的有效载荷)确立的假阳性阈值,对比了362bp靶dna-[1生物素]-[1单链霉亲和素]的阳性百分比检测标准。
图23描述了使用对照建立线性捕获率vs浓度趋势,并且随后通过将测量的捕获率映射到该线性趋势来估计靶多核苷酸的未知浓度。
图24描绘了从pcr反应产生的12、13和14循环扩增子对捕获率vs浓度趋势的浓度映射。
图25描绘了将从独立的pcr反应产生的不同组的20循环扩增子浓度映射到捕获率vs浓度趋势的重复性。
图26描绘了估计的浓度相对于pcr循环10、12、13、14、15、17、20、30、35、40的s形趋势。
图27对比了最小(1:1.17)稀释的pcr背景相对于在相同的pcr背景下扩增的1074bpdna的纳米孔隙电流事件群体。
图28描绘了1:5、1:2、1:43和1:1.17稀释下,阴性对照(不含起始模板的10个循环的pcr)对捕获率vs浓度趋势的浓度映射。
具体实施方式
在本申请的全文中,文本涉及本发明的装置、组合物、系统和方法的各种实施方式。所描述的各种实施方式旨在提供各种说明性实例,而不应该被解释为可选种类的描述。相反,应该注意的是,此处提供的各种实施方式的描述在范围上可能有重叠。此处讨论的实施方式仅仅是说明性的,并不是要限制本发明的范围。
还在本公开的全文中,各种出版物、专利和公开的专利说明书均通过明确的引文被引用。这些出版物、专利和公开专利说明书的公开内容通过引用由此作为整体并入本公开中。
如本文中使用的,术语“包含”意指所述系统、装置和方法包含所列举的成分或步骤,但不排除其它的成分或步骤。“基本上由……组成”在用于定义系统、装置和方法时意指排除对于组合有任何实质意义的其它成分或步骤。“由……组成”意指排除其它成分或步骤。通过这些过渡术语中各自限定的实施方式在本发明的范围内。
所有包括范围的数值表达,例如距离、大小、温度、时间、电压和浓度,是近似值,其按照0.1的增量变化(+)或(-)。需要明白的是,虽然并不总是所有数值表达前明确加有术语“大约”。也需要明白,尽管并不总是明确地表述,此处所描述的成分只是示例性的,并且此类成分的等价物是本领域中已知的。
如本文中使用的,“包含分隔内部空间的孔隙的装置”指的是具有在结构内包含开口的孔隙的装置,该结构将内部空间分隔成两个容积或腔室。该装置还可以具有多于一个纳米孔隙,并且在每对孔隙之间具有一个共同腔室。
本文所用的术语“多核苷酸”是指任何长度的核苷酸的聚合形式,是核糖核苷酸或脱氧核糖核苷酸。该术语仅指分子的一级结构。因此,该术语包括双链和单链的dna和rna。
如本文所用,术语“靶多核苷酸”是指包含目标序列(即靶多核苷酸序列或靶序列)的多核苷酸。靶多核苷酸可以包括与用于扩增靶多核苷酸的引物杂交的区域(例如,充分互补的序列)。这些区域可以是目标序列的一部分,位于目标序列侧翼,或者足够接近地在目标序列的更上游或下游以允许通过扩增反应扩增目标序列。在一些实施方式中,用于与引物杂交的这些区域位于通过扩增反应产生的扩增子的两个末端。本文描述了用于检测包含目标序列的靶多核苷酸的方法、装置和组合物。
本文所用的术语“引物”是指能够作为要复制的多核苷酸链(例如,靶多核苷酸)的合成的起始点的寡聚体。因此,在有利于杂交的条件下,引物将与包含靶序列的多核苷酸的互补区退火。在加入合适的反应物(例如,聚合酶、三磷酸核苷等)时,引物通过聚合剂延伸以形成靶多核苷酸的拷贝。引物可以是单链的,或者可以是部分或完全双链的。
如本文所用,术语“可杂交”是指能够杂交,即形成双链分子,例如rna:rna、rna:dna和/或dna:dna分子。为了扩增的目的,当其通过足以用作扩增反应的dna合成的起点的互补碱基配对相互作用能够与靶多核苷酸的链形成双链体时,则该引物可与靶多核苷酸杂交。
如本文所用,术语“结合”是指形成化学键,例如共价键、离子键或金属键。结合可以包括两个分子之间通过范德华力、疏水相互作用、阳离子-pi相互作用和/或平面堆叠相互作用的稳定缔合。结合包括两个分子通过点击化学的偶联。
如本文所用,术语“偶联位点”是指分子上用于将一个生物分子与另一个生物分子偶联的位点。在一些实施方式中,本文公开的引物所包含的偶联位点用于将引物本身或由引物产生的扩增子与有效载荷分子偶联以用于在纳米孔隙中检测有效载荷结合的扩增子。与偶联位点的结合机制可以包括任何稳定的结合相互作用,例如点击化学。
如本文所用的,术语“扩增”或“扩增反应”是指从靶多核苷酸序列产生多个包含该靶多核苷酸序列的克隆扩增子的反应。如本文所用的扩增反应试剂包括进行靶多核苷酸序列扩增所必需的任何分子。扩增反应试剂可以包括但不限于游离引物、dntp(脱氧核苷酸三磷酸酯、datp、dgtp、dctp、dttp)、聚合酶(例如taq或pfu)、盐(氯化镁、硫酸镁、硫酸铵、氯化钠、氯化钾)、bsa(牛血清白蛋白)稳定剂和洗涤剂(例如,tritonx-100)。扩增反应可以包括但不限于,例如,pcr、连接酶链反应(lcr)、转录介导的扩增(tma)、逆转录酶引发的pcr、dna或rna杂交技术、测序、等温扩增和环介导的等温扩增(lamp)。从靶多核苷酸序列产生扩增子的扩增技术是本领域技术人员所熟知的。
如本文所用,术语“骨架”或“聚合物骨架”是指在施加电压时转位穿过纳米孔隙的带负电或带正电荷的聚合物。在一些实施方式中,聚合物骨架包含可切割结构域或可切割接头。在一些实施方式中,聚合物骨架能够结合包含可切割接头的融合分子或其结合,且能够在施加电压时转位通过孔。在一些方面,聚合物骨架包含脱氧核糖核酸(dna)、核糖核酸(rna)、肽核酸(pna)、dna/rna杂合体或多肽。骨架也可以是化学合成的聚合物,而不是天然存在的或生物的分子。在优选的实施方式中,聚合物骨架是dsdna,以允许在转位通过纳米孔隙时给出更可预测的信号,并减少存在于ssdna或rna中的二级结构。在一些实施方式中,聚合物骨架包含可以位于骨架末端或在骨架的两个末端的融合分子结合位点。骨架和融合分子可以通过共价键、氢键、离子键、范德华力、疏水相互作用、阳离子-pi相互作用、平面堆叠相互作用或金属键连接。或者,可切割接头组分与骨架的直接共价系着可以连接骨架和融合分子。或者,融合体的接头组分可以通过直接共价系着将可切割接头连接到骨架上。在优选的实施方式中,融合分子包含骨架结合的结构域,其可以是dna、rna、pna、多肽、胆固醇/dna杂合体或dna/rna杂合体。
如本文所使用的,术语“有效载荷”或“有效载荷分子”是指与多核苷酸(例如引物)结合以增强在纳米孔隙中检测的选择性和/或敏感性的分子或化合物。在一些实施方式中,有效载荷分子可以是树状聚合物、双链dna、单链dna、dna适体、荧光团、蛋白质、多肽、纳米棒、纳米管、富勒烯、peg分子、脂质体或胆固醇-dna杂合体。在优选的实施方式中,多核苷酸和有效载荷通过共价键、氢键、离子键、范德华力、疏水相互作用、阳离子-pi相互作用、平面堆叠相互作用或金属键直接或间接连接。该有效载荷增加了靶多聚核苷酸或扩增子的大小,并且利于检测,其中当结合有效载荷的扩增子通过纳米孔隙时与背景分子相比具有明显不同的电流特征。
如本文所用,术语“背景”是指样品中可由纳米孔隙检测但不包含目标靶多核苷酸序列的分子。本发明的一个重要方面是从混合样品的背景中区分靶多核苷酸。
如本文所用,术语“纳米孔隙”是指具有的尺寸足够允许特定尺寸的聚合物通过的开口(孔洞或通道)。使用放大器,施加电压以驱动带负电的聚合物通过纳米孔隙,并且通过孔的电流检测分子是否通过它。
如本文所使用的,术语“传感器”是指从纳米孔隙装置收集信号的装置。在许多实施方式中,传感器包括放置在孔隙的两侧的一对电极,以在分子或其他实体,特别是聚合物骨架移动通过孔隙时,测量跨孔隙的离子电流。除了电极之外,附加的传感器,例如光学传感器可以检测纳米孔隙装置中的光学信号。其他传感器可用于检测如电流阻塞、电子隧穿电流、电荷诱导场效应、纳米孔隙通过时间、光学信号,光散射和等离子体共振等特性。
如本文所用,术语“电流测量”是指施加电压下随时间流过纳米孔隙的电流的一系列测量。电流被表示为用于量化事件的量度,并且通过电压(电导)标准化的电流也用于定量事件。
如本文所用,术语“开放通道”是指在噪声范围内通过纳米孔隙通道的电流的基线水平,其中电流不偏离由分析软件定义的值的阈值。
如本文所使用的,术语“事件”是指一组电流电阻测量值,该值从电流测量偏离开放通道值达到定义的阈值时开始,并且当电流返回到开放通道值的阈值之内时结束。
如本文所使用的,术语“电流阻抗特征”是指在检测到的事件中识别的电流测量值和/或谱的集合。在事件中也可能存在多个特征以增强分子类型之间的区分。
如本文所用,术语“捕获率”是指在纳米孔隙装置中随时间检测到的事件的数量。在一些实施方式中,捕获率可以具体指与特定的靶分子相关的事件的捕获率和/或转位率,例如有效载荷结合的扩增子的转位。如本文所述,与在类似的纳米孔隙条件下具有相似质/荷比的对照相比,捕获率可用于推断浓度。
概况
本文公开了通过产生在纳米孔隙装置中可检测的修饰的扩增子来检测靶多核苷酸的方法。本文公开的方法、组合物和装置允许dna分子在通过纳米孔隙时进行dna分子的纯电计数。在所提供的实例中证实了每次单个dna分子通过纳米孔隙时“有效负载连接的dna”提供确定的和稳定的信号。这允许从混合样品中进行准确和精确的dna定量的快速和简单的方法,从而允许使用不需要化学或光学检测的电检测方法在几分钟内从背景非靶分子计数并区分100s至1000s的靶分子。另外,鉴于便宜的硬件设备、低功率要求、小尺寸以及对广泛的纳米孔隙几何形状的耐受,制造和装置成本极低。由本文公开的本发明的几个实施方式提供的特征以及便于携带的小的纳米孔隙装置尺寸提供了一种新颖和有效的检测和诊断多核苷酸靶序列的方法。
背景区分
在一些方面,存在于样品中的靶多核苷酸可以是来自具有大量背景分子群体的原始的(甚至过滤的)天然液体(血液、唾液、尿液等)。这类背景分子,当在施加的正电压下充分带负电时,通过纳米孔隙。在一些情况下,这样的纳米孔隙事件可能看起来像靶多核苷酸。因此,这些背景分子可以产生假阳性,从而产生高错误检测率。添加足够的样品制备物以除去较大的分子将有助于这一点,但是产生假阳性事件的背景分子仍然存在,损害了纳米孔隙装置中靶多核苷酸检测的灵敏度和特异性。
为了提供背景分子和靶多核苷酸之间的区分,可以使用引物标记的方案。
具体地,将标记或标记序列(例如有效载荷分子)结合到可与靶多核苷酸的序列杂交的引物或探针。或者,引物或探针具有特异性结合选定标记或有效载荷分子的标记结合位点。在靶多核苷酸的扩增期间,将这些引物或探针并入扩增子中,从而将标记或有效载荷分子或标记/有效载荷分子结合位点并入包含靶多核苷酸的扩增子中。结合有效载荷分子的扩增子提供了独特的电流特征,其可用于识别含有转位通过纳米孔隙的包含靶序列的靶多核苷酸/扩增子产物的存在和/或身份。
在另一个实施方式中,单独的扩增子(未结合于标记)提供了足以区别于背景的区别性特征。这些实施方式通常需要较小的纳米孔隙和/或较长的扩增子来产生足以区别于背景的信号。
检测混合样品中的靶多核苷酸
可用固态纳米孔隙来电检测扩增子dna,和有效载荷连接在有效载荷仅与靶dna结合时有助于检测靶dna(图1)。
特别地,固态纳米孔隙可以用于电检测扩增子,如果其在穿过孔隙时提供充分大于开放通道电导以及在记录时同时存在的任何噪声或背景分子的阻抗信号(图2)。添大目标序列的“体积”使得能够在更大的孔隙(5-100nm)中进行纳米孔隙检测,因为体积将提供容易在背景噪声和分子之上测量的必要信号。没有这种体积的情况下,对于短于500bp的扩增子,大多数扩增子对于约5nm或更大的纳米孔隙也被错过了,阻碍了pcr产物的准确定量。例如,通过用修饰的引物进行扩增反应,引物中的修饰将被并入扩增子中,并且该修饰可被用于以必要的有效载荷“体积”标记扩增子,以使得能够在纳米孔隙中进行检测。在一个实施方式中,如图3所示,用生物素修饰引物。所得pcr扩增子与链霉亲和素有效载荷结合,并在纳米孔隙中检测其存在。
存在的体积增大(bulking)的实例包括生物素/链霉亲和素和表位/抗体,并且是两个示例性方法。或者,可以用化学反应性基团,例如炔烃修饰引物。这将使得可以通过使用点击化学用含有大体积叠氮化物的分子,例如叠氮基-聚乙二醇(peg)来修饰含有炔烃的扩增子。如果使用通过孔隙的光学检测,可以用荧光团修饰引物,因此可以通过荧光方法检测扩增子。总之,并入其合并依赖于扩增反应的分子的任何方法被并入本文的公开内容中。
结合反应具有如此高的亲和力,pcr扩增子可以被标记而无需首先将其纯化或从扩增缓冲液和组分中提取。标记后,使用纳米孔隙在合适的纳米孔隙记录缓冲液(例如1mlicl,10mmtris,1mmedta)中检测扩增子。与pcr反应混合物相关的或来自样品的背景与有效载荷结合的靶dna分子相比,具有明显不同的纳米孔隙电流阻抗特征(图2)。以这种方式,可以在背景存在下进行定量,消除了对样品或pcr反应混合物进行纯化的需要,极大地简化了工作流程。反过来,通过在测量之前仅需要稀释反应混合物,实施循环pcr反应和进行循环之间的扩增子产物的纳米孔隙测量的装置可以被便宜地包装和制造。另一方面,需要复杂的纯化步骤的装置在基础设施中将更加密集,并因此更昂贵和复杂。
图4显示了名为“mnopcr”(整数(m)纳米孔隙(n)可观察(o)pcr)的实时扩增子检测和定量的方法。随着mnopcr的进行,在每个循环之后产生的产物是有效载荷结合的,并然后在纳米孔隙中检测。实现这种实时检测方法是因为pcr扩增子在结合有助于检测的大体积的有效载荷之前不需要被纯化。该测量耐受不同稀释度(包括无稀释)的pcr反应混合物背景,不同稀释度(根据优化pcr反应的要求)的样品背景和有效载荷背景(未连接到靶多核苷酸的过量有效载荷)。
在反应进行时测量产物的能力提供了一种非常有效的方式来比较可能在模板/靶数目或浓度上不同的两个或更多个样品。此外,由于纳米孔隙检测和计数单分子,所以可以实现所得扩增子的量的准确定量,如实施例所示。
使用本文所述的方法,可以比较和对比靶核苷酸序列量不同的两个或更多个样品。这其中的两个非限制性情况是1)含有突变转录物和野生型序列的混合物的样品,以及2)转基因含量不同的样品。
对于情况1,通过用扩增靶序列(无论其是否含有突变)的引物组扩增样品,和然后将产生的产物量与用仅扩增突变序列的引物组产生的量进行比较,可以推断样品中突变体转录本的量,并比较样品之间的该值。例如,可以分辨出样品a含有20%的kras突变,而样品b含有30%的kras突变。
对于情况2,可以扩增存在于每个转录本(管家区域)中的dna的区段,并将其丰度与从仅靶向转基因的反应产生的转录本的量进行比较。可以使用纳米孔隙来测定材料的总量以定量管家扩增子,并且可以与产生的转基因扩增子的量比较。由于使用管家基因扩增子来测定样品的总浓度,因此可以比较开始浓度不同的两种不同样品的转基因丰度。
可以使用这些相同的概念来测定,例如来自口腔拭子的样品中是否存在细菌属,然后进一步描绘属于该属的物种。具体地,首先定量存在于所有细菌中的基因,例如,葡萄球菌,然后特异性地扩增和定量仅存在于抗生素抗性葡萄球菌的基因,如meca中。
图5描绘了使用所公开的方法来检测目标核酸序列的工作流程
对检测分配统计学显著性
在一些实施方式中,对随时间记录的传感器测量值的集进行汇总并应用数学工具以对疑似存在于样本中的靶多核苷酸的检测赋予数字置信度值,如前面章节中所详细描述的。
最近开发出基于纳米孔隙事件群体特征差异从背景(即其他分子类型)区分分子类型的定量方法(morin,t.j.等,“nanopore-basedtargetsequencedetection”2015年12月31日提交到ploson)。这种区分方法意味着可以在不同类型的其他背景分子的存在下检测特定分子类型,并且可以赋予检测的统计学显着性(例如,以99%置信度的试剂x的检测)。为将该方法应用到下面提供的实例中,我们这里首先总结该方法。
一般来说,在孔隙上方的腔室中有两类分子:类型1都是背景分子,而类型2是目标分子。在下面的实施例中,靶dna-有效载荷通常被认为是2型分子,其中非靶dna或游离的有效载荷、引物或来自样品或pcr反应混合物的分子被认为是背景(类型1)。基于来自实验的数据,我们确定存在于类型2事件的显著部分中的和存在于类型1事件的相对较小部分中的事件特征标准。特征标准可以取决于δg、持续时间、每个事件内的级别的数量和特征和/或从事件信号计算的任何其它数值或值的组合。
注意,可以手动或通过查表或以自动方式选择事件特征标准。例如,先前的实验可以建立用于一定范围的孔径和预期在给定测试中存在的其他条件的阳性和阴性对照的性能,且当对于给定的测试遇到相当的条件(即对于给定的扩增子长度和给定的有效载荷类型)时,可以使用从这些对照中确定的所选定标准(以查表的方式)。例如,基于恰好在样品前的对照运行,也可以实时地确定自动标准。如本申请中所公开的,自动标准选择对于实时定量pcr扩增子是合适和优选的方法。具体地,由于对照在分子大小/电荷和纳米孔隙事件特征/形状方面模拟扩增子,可以使用对照事件来建立捕获率vs浓度趋势,如所公开的,同时还提供用来使“2型事件边界”的计算自动化事件群体,该“2型事件边界”建立了在测量扩增的样本时,用于从背景中标示“扩增子”类型2事件的标准。可以通过围绕2d图中的点拟合曲线的任何方法来计算边界(例如,所述点是平均偏移对持续时间曲线图内的事件)。曲线拟合方法可以包括最小二乘法、线性或二次规划或者任何形式的数值优化,并通过分段多项式或样条的系数来对边界进行参数化。计算凸包可以提供边界。更高维的边界拟合途径也是可能的,例如,使用表征事件的3个属性(3d边界)。所得到的边界可以是多边形的或平滑的。用于用包含事件的特定百分比的边界来包装事件的子集(即,作为在包装用于自动标准确定的点时消减异常值的机制)的相关技术是计算被定义为包含总概率的z分数的最小区域的边界的z-分位数边界。例如,95%分位数边界是其中包含总概率的95%(数据的95%)的最小区域的边界。虽然概率密度未知,但可以使用标准技术用数据来估计。
一旦选择了标准(手动、通过查表或以自动方式),如果事件满足特征标准,则将该事件“标示”为类型2。我们将p定义为捕获事件是2型的概率。在没有2型分子的对照实验中p=0,和在具有2型分子的实验中p>0,但其值是未知的。我们定义假阳性概率q1=pr(标示的事件|1型事件)。在不具有2型分子的对照实验或实验组中(例如,使用非靶dna的20个循环之后的pcr反应混合物,并稀释到与纳米孔隙相邻的记录缓冲液中),从合理数目的捕获事件以良好准确性确定q1。在测定本体溶液中是否存在2型分子的检测实验中,捕获事件被标示的概率是p的函数,且可以近似计算为:
q(p)=(被标示事件数)/n
在该公式中,n是事件的总数。可以用qsd(p)=2.57*sqrt{q(p)*(1-q(p))/n}计算99%置信区间q(p)±qsd(p),其中sqrt{}为平方根函数。在实验过程中,随着事件数n的增加,q(p)的值收敛和不确定性界限减弱。作为记录时间的函数的q(p)±qsd(p)的曲线图示出了对于每种试剂类型它是如何演化的(图10、17、19和22)。在没有2型分子的对照实验中,观察到q(0)=q1。在2型分子已知以某种概率p*>0存在的对照实验中,可以在检测实验中使用计算值q(p*)来确定是否不存在2型分子,如下所定义的。
在检测实验中,当以下标准为真时,2型分子以99%的置信度存在:
q(p)-qsd(p)>q1(1)
如果上述标准是真的,我们得出结论p>0;如果其是非真的,我们不能说p>0。该框架在下面提供的实施例中被使用。
从测量的捕获率估算浓度
这里,我们讨论如何基于由对应于已知浓度的靶分子的序列的测量捕获率表示的线性关系来估计浓度。这是使用对照建立的。用c表示浓度,和r(c)表示对应于浓度c的真实/精确捕获率。我们预期r(c)与c成正比。
r(c)=ac
我们收集对应于已知浓度{c1,c2,...,cn}的序列的测量的捕获率。每个测量的捕获率是以下的形式
对于cj的测量的捕获率:rj±dj
我们解释对于cj的测量的捕获率如下:rj是来自正态分布n(acj,dj2)的随机样本,其中acj是精确的捕获率,和速率常数a是未知的。给定的数据集{rj±dj,j=1,2,...,n},a的后验分布具有正态分布:a~n(u,s2),其中u和s具有表达式
用于在捕获率对浓度曲线图中拟合对照的代表性实例示于图23-25,28中。
接下来,我们描述如何通过使用首先利用对照建立的捕获率度对浓度趋势(如上所述),然后测量未知浓度的靶分子的捕获率来估计未知物质的浓度。在注释方面,我们测量对应于未知浓度cx的捕获率,并且令:
对于cx的测量的捕获率:rx±dx
我们将cx记为
其中e1和e2是独立的标准正态随机变量。
ε1~n(0,1)和ε2~n(0,1)
假设斜率a以较小相对不确定度从其他数据点确定:s/u<<1。在这个假设下,我们可以近似cx。具体地,我们会将cx的估计值报告为:
其中u和s是使用下式从数据点计算的
用于通过将测量的捕获率与捕获率对浓度趋势(用对照建立)拟合来估计未知浓度的代表性实例示于图23-25,28中。
在所提供的实施例中,“实施例9:扩增子浓度的定量”通过举例说明如何实现本节中提出的方法。
组合物
在一些实施方式中,本文提供了与有效载荷分子结合的引物。在一些实施方式中,本文提供了包含有效载荷分子结合位点的引物。在任一实施方式中,引物可以产生结合或能够结合有效载荷分子以增强纳米孔隙中的检测的扩增子。
在一些实施方式中,有效载荷分子可以是树状聚合物、双链dna、单链dna、dna适体、荧光团、蛋白质、多肽、纳米棒、纳米管、富勒烯、peg分子、脂质体或胆固醇-dna杂合体。在优选的实施方式中,多核苷酸和有效载荷通过共价键、氢键、离子键、范德华力、疏水相互作用、阳离子-pi相互作用、平面堆叠相互作用或金属键直接或者间接连接。有效载荷增加了靶多核苷酸或扩增子的大小,并且使之便于检测,其中结合有效载荷的扩增子在通过纳米孔隙时具有与背景分子显著不同的电流特征。在一些实施方式中,有效载荷分子包含用于连接到引物的叠氮化物化学操作(chemicalhandle)。在一些实施方式中,引物结合于生物素分子。在一些实施方式中,有效载荷分子可以结合另一分子以影响该分子的体积,从而提高纳米孔隙中扩增子检测的灵敏度。在一些实施方式中,引物结合于用于与生物素分子结合的结合位点或包含该位点。在一些实施方式中,生物素进一步被链霉亲和素结合以增加有效载荷分子的大小来增强纳米孔隙在背景分子下的检测。增大的体积可以在包含靶序列的扩增子和背景分子之间产生更明显的特征差异。
在该实施方式中,有效载荷附接到引物或扩增子可以以各种方式实现。例如,引物可以是二苯并环辛炔(dbco)修饰的引物,从而用dbco化学基团有效地标记所有扩增子以用于通过无铜“点击”化学与叠氮化物标记的扩增子或引物偶联的目的。
在一些方面,引物包含引起或有助于有效载荷分子的识别和结合的化学修饰。例如,甲基化的dna序列可以被转录因子、dna甲基转移酶或者甲基化修复酶识别。在其它实施方式中,生物素可以被结合到抗生物素蛋白家族成员中,并被其识别。在此类实施方式中,生物素形成融合体结合结构域,和抗生物素蛋白或抗生物素蛋白家族成员是融合体上的聚合物骨架结合结构域。由于其结合互补性,引物/扩增子上的有效载荷分子结合结构域和有效载荷分子上的引物结合结构域可以颠倒,使得有效载荷结合结构域变为引物结合结构域,反之亦然。
能够特异性地识别核苷酸结合基序的分子,尤其是蛋白质,是本领域中已知的。例如,蛋白结构域如螺旋-转角-螺旋、锌指、亮氨酸拉链、翼状螺旋、翼状螺旋-转角-螺旋、螺旋-环-螺旋和hmg盒已知能够结合核苷酸序列。这些分子中的任何一个可以用作结合扩增子或引物的有效载荷分子。
在一些方面,有效载荷结合结构域可以是锁核酸(lna)、桥连核酸(bna)、所有类型的蛋白质核酸(例如bispnas、γ-pna)、转录激活因子样效应物核酸酶(talen)、成簇的规律间隔短回文重复序列(crispr)或适体(如dna、rna、蛋白质或其组合)。
在一些方面,有效载荷结合结构域是dna结合蛋白(例如锌指蛋白)、抗体片段(fab)、化学合成的粘合剂(例如pna、lna、talens或crispr)或合成聚合物骨架中的化学修饰(即反应性部分)(例如,硫醇盐、生物素、胺、羧酸酯)中的一个或多个。
纳米孔隙装置
所提供的纳米孔隙装置包括在将其内部空间分为两个容积的结构中形成开口的至少一个孔隙,以及至少一个配置为识别通过孔隙的物体(例如通过检测指示物体的参数的变化)的传感器。用于本文所描述的方法的纳米孔隙装置也在wo/2013/012881号pct公开中公开,其通过引用整体并入。
纳米孔隙装置中的孔隙是纳米级或微米级的。在一个方面,每个细孔的大小允许小或大分子或者微生物通过。在一个方面,每个孔隙直径至少约1纳米。可选地,每个细孔直径至少约2nm、3nm、4nm、5nm、6nm、7nm、8nm、9nm、10nm、11nm、12nm、13nm、14nm、15nm、16nm、17nm、18nm、19nm、20nm、25nm、30nm、35nm、40nm、45nm、50nm、60nm、70nm、80nm、90nm或100nm。
在一个方面,孔隙直径不超过约100nm。可选地,细孔直径不超过约95nm、90nm、85nm、80nm、75nm、70nm、65nm、60nm、55nm、50nm、45nm、40nm、35nm、30nm、25nm、20nm、15nm或10nm。
在一个方面,孔隙的直径在约1nm至约100nm之间,或者在约2nm至约80nm之间,或约3nm至约70nm之间,或约4nm至约60nm之间,或约5nm至约50nm之间,或约10nm至约40nm之间,或约15nm至约30nm之间。
在一些方面,纳米孔隙装置进一步包括用于将聚合物骨架移动跨过孔隙的手段和/或用来识别通过孔隙的物体的手段。进一步的细节在下面提供,以双孔隙装置为背景描述。
与单孔隙纳米孔隙装置相比,双孔隙装置可以更容易配置从而提供聚合物骨架跨孔隙运动的速度和方向的良好控制。
在一个实施方式中,纳米孔隙装置包括多个腔室,各腔室通过至少一个孔隙与相邻的腔室连通。在这些孔隙中,两个孔隙,即第一孔隙和第二孔隙,被布置为使得允许至少一部分靶多核苷酸移出第一孔隙并进入第二孔隙。此外,所述装置在每个孔隙处包括能够在运动过程中识别靶多核苷酸的传感器。在一个方面,该识别需要确定靶多核苷酸的单个成分。在另一个方面,该识别需要确定与靶多核苷酸结合的有效载荷分子。当采用单一传感器时,该单一传感器可包含两个放置在孔隙两端的用于测量跨孔隙的离子电流的电极。在另一个实施方式中,单一传感器包含电极以外的部件。
在一个方面,所述装置包括通过两个孔隙连接的三个腔室。具有三个以上腔室的装置可以很容易地设计以在三腔室装置任一侧或在三个腔室中任何两个腔室之间包括一个或多个额外的腔室。同样地,装置中可以包括连接腔室的超过两个孔隙。
在一个方面,两个相邻腔室之间可以有两个或更多个孔隙,以允许多个聚合物骨架同时从一个腔室移动到下一个腔室。这样的多孔隙设计可以提高装置中靶多核苷酸分析的通量。对于多路复用,一个腔室可以具有一种类型的靶多核苷酸,且另一个腔室可以具有另一种靶多核苷酸类型。
在一些方面,所述装置进一步包括用于将靶多核苷酸从一个腔室移动到另一个腔室的手段。在一个方面,该移动导致同时跨第一孔隙和第二孔隙两者加载靶多核苷酸(例如,包含靶序列的扩增产物或扩增子)。在另一个方面,所述手段进一步使靶多核苷酸能够在相同方向上通过两个孔隙移动。
例如,在三腔室两孔隙装置(“两孔隙”装置)中,每个腔室可以包含用于连接到电源的电极,从而可以在腔室之间跨各个孔隙施加单独的电压。
根据本发明的一个实施方式,提供了包括上腔室、中腔室和下腔室的装置,其中上腔室通过第一孔隙与中腔室连通,且中腔室通过第二孔隙与下腔室连通。这种装置可以具有此前在名称为双重孔隙装置(dual-poredevice)的美国公开no.2013-0233709中公开的任何尺寸或其他特征,该文献在此通过引用整体并入本文。
在一个方面,每个孔隙的直径为至少约1nm。或者,每个孔的直径为至少约2nm、3nm、4nm、5nm、6nm、7nm、8nm、9nm、10nm、11nm、12nm、13nm、14nm、15nm、16nm、17nm、18nm、19nm、20nm、25nm、30nm、35nm、40nm、45nm、50nm、60nm、70nm、80nm、90nm或者100nm。
在一个方面,每个孔隙的直径不超过约100nm。或者,孔隙的直径不超过约95nm、90nm、85nm、80nm、75nm、70nm、65nm、60nm、55nm、50nm、45nm、40nm、35nm、30nm、25nm、20nm、15nm或10nm。
在一个方面,孔隙的直径在约1nm至约100nm之间,或者约2nm至约80nm之间,或约3nm至约70nm之间,或约4nm至约60nm之间,或约5nm至约50nm之间,或约10nm至约40nm之间,或约15nm至约30nm之间。
在一些方面,孔隙基本上呈圆形。本文所用的“基本上圆形”是指至少约80或90%为圆柱体形式的形状。在一些实施方式中,孔隙形状为方形、矩形、三角形、椭圆形或六角形。
在一个方面,孔隙的深度介于约1nm至约10,000nm之间,或者在约2nm至约9,000nm之间,或在约3nm至约8,000nm之间,等。
在一些方面,纳米孔隙穿过膜延伸。例如,孔隙可以是插入脂质双层膜中的蛋白质通道,或者其也可以通过钻孔、刻蚀或以其他方式通过固态基质(如,二氧化硅、氮化硅、石墨烯或由这些或其他材料的组合形成的层)形成孔隙来工程化。纳米孔隙的大小设计为允许骨架:融合体:有效载荷,或该分子在酶活性之后的产物通过孔隙。在其它实施方式中,孔隙的临时阻塞对于鉴别分子类型可能是有利的。
在一些方面,纳米孔隙的长度或深度足够大,从而形成连接两个在其它方面分隔的容积的通道。在一些此类方面,每个孔隙的深度大于100nm、200nm、300nm、400nm、500nm、600nm、700nm、800nm或900nm。在一些方面,每个孔隙的深度不超过2000nm或1000nm。
在一个方面,孔隙以约10nm至约1000nm之间的距离分隔开。在一些方面,孔隙之间的距离大于1000nm、2000nm、3000nm、4000nm、5000nm、6000nm、7000nm、8000nm或9000nm。在一些方面,孔隙间隔不超过30000nm、20000nm或10000nm。在一个方面,距离为至少约10nm,或者至少约20nm、30nm、40nm、50nm、60nm、70nm、80nm、90nm、100nm、150nm、200nm、250nm或300nm。另一方面,该距离不超过约1000nm、900nm、800nm、700nm、600nm、500nm、400nm、300nm、250nm、200nm、150nm或100nm。
而在另一方面,孔隙之间的距离为约20nm至约800nm之间,约30nm至约700nm之间,约40nm至约500nm之间,或约50nm至约300nm之间。
两个孔隙可以以任何位置排列,只要它们允许腔室之间的流体连通并具有规定的大小和间距。在一个方面,孔隙布置为使得它们之间没有直接阻碍。仍然在一个方面,孔隙基本上是同轴的。
在一个方面,该装置在腔室中具有连接到一个或多个电源的电极。在一些方面,电源包含电压钳或膜片钳,其可以提供跨各孔隙的电压并独立地测量通过各孔隙的电流。在这个方面,电源和电极配置可以将中腔室设置成两个电源的共同地线。在一个方面,一个或多个电源配置为在上腔室(腔室a)和中腔室(腔室b)之间施加第一电压v1,和在中腔室和下腔室(腔室c)之间施加第二电压v2。
在一些方面,第一电压v1和第二电压v2是独立可调的。在一个方面,中腔室被调节为相对于两个电压的地电压。在一个方面,中腔室包含用于在各个孔隙和中腔室中的电极之间提供电导的介质。在一个方面,中腔室包含用于在各个孔隙和中腔室中的电极之间提供电阻的介质。保持相对于纳米孔隙电阻足够小的这种电阻有利于对跨孔隙的两个电压和电流解耦,这有助于电压的独立调节。
电压的调节可用于控制腔室内带电颗粒的运动。例如,当两个电压设置为极性相同时,适当带电的颗粒可以从上腔室顺序地移动到中腔室和到下腔室,或者反过来。在一些方面,当两个电压被设置成相反极性时,带电颗粒可以从上腔室或下腔室移动到中腔室并停留在那里。
装置中电压的调节可以特别地用于大分子如带电聚合物骨架的运动的控制,该大分子足够长以同时跨两个孔隙。在这个方面,分子移动的方向和速度可以通过电压的相对幅度和极性来控制,如下所述。
所述装置可以包含适合容纳液体样品(特别是生物样品)的材料和/或适合于纳米加工的材料。在一个方面,此类材料包括介电材料,例如但不限于硅、氮化硅、二氧化硅、石墨烯、碳纳米管、tio2、hfo2、al2o3或其它金属层,或这些材料的任何组合。在一些方面,例如,约0.3纳米厚的单片石墨烯膜可用作孔隙承载膜。
作为微流体装置且容纳双孔隙微流体芯片设施的装置可以通过多种手段和方法来制造。对于由两个平行的膜构成的微流体芯片,两个膜可以同时通过单波束钻孔以形成两个同心孔隙,尽管与任何适合的校准技术协同在膜的两侧使用不同的波束也是可能的。概括地说,外壳确保腔室a-c的密封分离。
在一个方面,所述装置包含微流体芯片(标记为“双孔隙芯片”),其由通过间隔体连接的两个平行的膜构成。每个膜包含通过膜中心用单波束钻孔形成的孔隙。进一步,所述装置优选具有用于芯片的
更具体地,孔隙承载膜可以用具有5-100纳米厚的硅、氮化硅或二氧化硅窗口的透射电子显微(tem)格栅制造。间隔体可采用绝缘体(如su-8、光刻胶、pecvd氧化物、ald氧化物、ald氧化铝)或蒸镀的金属材料(如银、金或铂),且占据在膜之间的腔室b的其它为水性的部分中的小空间而用于分隔膜。支持器置于由腔室b的最大体积部分构成的水性浴中。腔室a和c可以通过较大直径的通道(对于低接入电阻)达到,这导致膜密封。
聚焦的电子或离子束可以用来通过膜钻孔隙,从而使其自然对准。孔隙也可以通过对每层施加适当束聚焦来雕刻(收缩)达到较小尺寸。任何单一纳米孔隙钻孔方法也可以用来在两个膜中钻出孔隙对,考虑对于给定方法可能的钻孔深度和膜的厚度。预钻出微孔隙到规定的深度和然后通过膜的剩余部分钻出纳米孔隙对于进一步优化膜的厚度也是可能的。
通过在装置的孔隙处存在的电压,带电分子可以通过腔室之间的孔隙移动。移动的速度和方向可以通过电压的大小和极性控制。此外,由于两个电压可以各自独立地调节,所以带电分子的运动方向和速度可以在各腔室中精细地控制。
一个实例涉及靶多核苷酸,其长度大于包括两个孔隙的深度加两个孔隙之间的距离的综合距离。例如1000bp的dsdna长度约为340纳米,且显著大于间隔20纳米的两个10纳米深孔隙跨越的40纳米距离。第一步中,将多核苷酸加载到上腔室或下腔室中。由于其在ph约7.4的生理条件下带负电荷,多核苷酸可以跨被施加电压的孔隙移动。因此,第二步中,相同极性且大小相同或相近的两个电压施加于孔隙以顺序地跨两个孔隙移动多核苷酸。
大致在多核苷酸到达第二孔隙的时候,可以改变一个或两个电压。由于两个孔隙之间的距离选择为比多核苷酸的长度短,当多核苷酸到达第二孔隙时,它也在第一孔隙中。因此,在第一孔隙处的电压极性的迅速改变将产生将多核苷酸拉离第二孔隙的力。
假设所述两个孔隙具有相同的电压-力影响,且|v1|=|v2|+δv,则值δv>0(或<0)可以进行调节以在v1(或v2)方向上获得可调的运动。在实践中,虽然各个孔隙处电压诱导的力不会由于v1=v2而相同,校准实验可以确定对于给定的双孔隙芯片产生相等的拉力的适当偏置电压;且围绕该偏置电压的变化然后可以用于定向控制。
如果,在此时,第一孔隙处电压诱导力的大小小于第二孔隙处电压诱导力的大小,则多核苷酸会继续穿越两个孔隙移向第二孔隙,但速度较低。在这个方面,很容易理解,多核苷酸运动的速度和方向可以通过两个电压的极性和大小来控制。正如下面将进一步描述的,运动的这种精细控制有着广泛的应用。为了定量靶多核苷酸,双孔隙装置实施方式的效用是,在受控的递送和探测期间,可以重复地测量靶多核苷酸或有效载荷结合的靶多核苷酸以增加检测结果的置信度。
因此,在一个方面,提供了用于控制带电聚合物骨架通过纳米孔隙装置的运动的方法。所述方法包括(a)将包含靶多核苷酸(例如,靶多核苷酸扩增子)的样品加载到上述任一实施方式的装置的上腔室、中腔室或下腔室之一中,其中该装置被连接到用于在上腔室与中腔室之间提供第一电压和在中腔室与下腔室之间提供第二电压的一个或多个电源;(b)设置初始第一电压和初始第二电压以使靶多核苷酸在腔室之间移动,从而使聚合物骨架跨第一和第二孔隙两者定位;且(c)调节第一电压和第二电压以使两个电压都产生将带电靶多核苷酸拉离中腔室的力(电压竞争模式),其中两个电压在受控条件下大小不同,以使得靶多核苷酸骨架沿任一方向且以受控的方式跨两个孔隙移动。
在一个方面,将含靶多核苷酸的样品加载到上腔室中,且将初始第一电压设置为将靶多核苷酸从上腔室拉到中腔室,且将初始第二电压设置为将靶多核苷酸从中腔室拉到下腔室。同样地,样品可以被初始加载到下腔室中,且靶多核苷酸可以被拉到中腔室和上腔室。
在另一方面,将含有靶多核苷酸的样品加载到中腔室中;将初始第一电压设置成将带电聚合物骨架从中腔室拉到上腔室;且将初始第二电压设置成将靶多核苷酸从中腔室拉到下腔室。
在一个方面,在步骤(c)中第一电压和第二电压的实时或在线调节是采用专用的硬件和软件以高达数百兆赫的时钟频率通过主动控制或反馈控制进行的。第一电压或第二电压或两者的自动控制是基于第一或第二或两个离子电流测量值的反馈。
传感器
如上所述,在各个方面,纳米孔隙装置还包括一个或多个传感器用于完成靶多核苷酸的检测。
所述装置中使用的传感器可以是任何适合用于识别与有效载荷分子结合或未结合的靶多核苷酸扩增子的传感器。例如,传感器可以配置为通过测量与聚合物相关的电流、电压、ph值、光学特征或停留时间来识别靶多核苷酸。在其他方面,传感器可以被配置为识别靶多核苷酸的一个或多个单个组分或者与靶多核苷酸结合或连接的一种或多种组分。传感器可以由配置为检测可测量参数的变化的任何组件形成,其中此变化指示靶多核苷酸、靶多核苷酸的组分或优选与靶多核苷酸结合或连接的组分。在一个方面,传感器包括放置在孔隙两侧的一对电极以测量分子或其他实体(特别是靶多核苷酸)移动通过孔隙时的跨孔隙离子电流。在某些方面,在通过孔隙的靶多核苷酸片段结合于有效载荷分子时,跨孔隙离子电流发生可测量的变化。此类电流变化对应于,例如,存在的靶多核苷酸分子的存在、不存在和/或大小以可预测的、可测量的方式发生改变。
在优选的实施例中,传感器包括施加电压并用于测量跨纳米孔隙的电流的电极。分子转位通过纳米孔隙提供了电阻抗(z),其根据欧姆定律,v=iz,影响通过纳米孔隙的电流,其中v是施加的电压,i是通过纳米孔隙的电流,z是阻抗。相反,监测电导g=1/z以示意和定量纳米孔隙事件。当分子在电场中(例如,在施加的电压下)转位通过纳米孔隙时的结果是,当进一步分析电流信号时可能与通过纳米孔隙的分子相关的电流特征。
当使用来自电流特征的停留时间量度时,可以基于通过探测设备所需的时间长度,将组分的大小与特定组分相关联。
在一个实施方式中,在纳米孔隙装置中提供的传感器测量聚合物、聚合物的组分(或单元)或者结合或连接到聚合物的组分的光学特征。这种测量的一个实例包括通过红外(或紫外)光谱鉴定特定单元特有的吸收带。
在一些实施方式中,传感器是电传感器。在一些实施方式中,传感器检测荧光特征。孔隙出口处的辐射源可用于检测该特征。
实施例
本技术进一步参考以下实施例和实验进行定义。对于本领域技术人员明显的是许多改变可以在不背离本发明的范围的情况下实行。
实施例1:dna的纳米孔隙检测
固态纳米孔隙是在分隔两个液态体积的薄的固体膜中形成的纳米级开口。电压钳放大器在测量通过开放孔隙的离子电流的同时施加跨膜的电压v。与任何其他单分子传感器不同,纳米孔隙装置可以以非常低的成本封装成手持形式因子。当单一带电分子如双链dna(dsdna)通过电泳被捕获并被驱动通过孔隙时,测量的电流偏移和电导偏移深度(δg=δi/v)和持续时间被用于表征事件(图6a)。
在一些实施方式中,值δg(也标记为δg)被计算为平均电流偏移除以电压。在其他实施方式中,值δg(也标记为δg)被计算为最大电流偏移除以电压。通常,持续时间被计算为偏移宽度。
在实验期间记录许多事件之后,分析事件的分布以表征相应的分子。图6b显示在电压v=100mv(1mlicl)下通过27nm直径的纳米孔隙的0.1nm3.2kbdsdna的事件特性,产生在10分钟内记录的744个事件。这两个画圆圈的代表性事件显示:更宽的和更浅的事件对应于展开的dna穿过;以及更快的但更深的事件对应于折叠的dna通过。对于~1kb和更短的dsdna,dna仅在展开的状态下通过孔。
在纳米孔隙实验中电噪声尖峰产生假事件(电“背景噪声”)是常见的。这些假事件比3.2kb事件更快和更浅,因此容易区分。由于膜中的瞬时电容变化,背景事件每分钟1-2次通常用缓冲液观察到,产生快(<0.1ms)浅(<1.5ns)的事件。当使用较短的dna时,如以下实施例所述,背景电噪声假事件难以与真实dna事件分开,因此难以将真实事件与假事件分开。虽然小于5nm的纳米孔隙使得可能将dna真实事件与噪声假事件分离(briggs,kyle,haroldkwok和vincenttabard-cossa.“automatedfabricationof2-nmsolid-statenanoporesfornucleicacidanalysis.”small10,no.10(may28,2014):2077–86.doi:10.1002/smll.201303602),但当其他背景分子类型(例如,来自样品,或pcr反应混合物,或未结合有效载荷分子)即使在低丰度下存在时,这些孔隙可能堵塞。这是因为许多背景分子的大小等于或大于5nm(例如,作为有效载荷的单链霉亲和素蛋白为约5nm),和因此可以被捕获到孔隙中,但是不能通过为5nm或更小的孔隙。为了耐受不同类型的这种背景分子,我们优选直径至少为5nm的孔。因此,较短的扩增子(例如,500bp或更小)需要有效载荷附着以使其在(较大的)纳米孔隙中足够可观察,以进行定量。另一方面,大于500bp(例如,1kb)的扩增子可以用较大的孔(直径至少10nm和高达50nm)进行定量而无有效载荷附接,并且在不同类型的背景存在下,其中包括来自样品(口腔拭片、全血)和不同稀释度的pcr反应混合物。
实施例2:扩增子生成和有效载荷连接方法
以下使用有效载荷结合的dna的实施例使用以下扩增子长度:500bpdna、470bpdna(包含sry基因)和362bpdna(其中包含smcy基因)。
sry(性别决定区域y)基因位于y染色体上,且因此是男性特有的。缺乏sry基因(或更准确地说,其区域)是男性不育症(abusheikha,n.,a.lass和p.blinsden。“xxmaleswithoutsrygeneandwithinfertility:casereport.”humanreproduction16.4(2001):717-718)或其他健康状况(例如swyer综合征)的常见原因。因此,可以设计聚合酶链反应(pcr)分析以扩增不育男性中缺乏的sry基因的部分,从而提供对于男性和候选不育的检验。如果产生扩增子,该样品来自雄性,并含有sry基因的所述区域。
通过将来自可育男性的口腔拭子与50μl的pcr反应混合物混合实现sry基因的470碱基对区段的扩增,其中该pcr反应混合物含有1×terradirectbuffer(p/n639287),0.3μm的sry正向引物(gaatattcccgctctccgga),0.3μm的sry反向引物(gctggtgctccattcttgag)和1μl的pcrdirectpolymeraseterramix(p/n639287)。将该反应混合物进行6步pcr,其方案如下:1)98℃进行2分钟,2)98℃进行10秒,3)60℃进行15秒,4)68℃进行1分钟,5)重复步骤2-440个循环,6)保持在4℃。为了将生物素并入到分子的5'和/或3'末端,在反应混合物中使用5'末端生物素化的引物。还使用2μl的1:1000稀释的血滴作为起始样品材料进行相同的反应。为了测试非特异性扩增作为阴性对照,使用水或女性口腔拭子作为样品输入,预计不会产生产物。
smcy基因位于y染色体上,因此是男性特有的。在前列腺癌中观察到缺失smcy,并随着癌症的发展,频率越来越高(perinchery,geetha等人,“deletionofy-chromosomespecificgenesinhumanprostatecancer”。thejournalofurology163.4(2000):1339-1342)。
使用上述完全相同的方案,但使用smcy基因特异性引物(正向引物cctccagacctggacagaat,反向引物tgtggtctgtggaaggtgtca)完成smcy基因的扩增。这产生了362个碱基对的扩增子。
热循环后,通过加入相对于可用的生物素分子大约10倍过量的有效载荷,用单链霉亲和素有效载荷或抗生物素抗体有效载荷标记生物素化的dna(用于sry和smcy)。为了确定在扩增反应过程中产生多少生物素化的dna(对于sry和smcy),进行单独的实验,其中pcr产物从pcr反应混合物中纯化并用分光光度法定量。
通过用5μl单独的pcr反应产物运行2%琼脂糖凝胶来证明来自多个反应的扩增的一致性(图8左图)。随后,与单链霉亲和素蛋白(相对于生物素位点10x)进行温育以附接有效载荷(无、一或二个)。为了证实pcr产物通过1个(如果使用1个生物素修饰的引物)或2个(如果使用2个修饰的引物)单链霉亲和素蛋白有效地增大体积,对于血滴源材料运行2%的琼脂糖凝胶(图8,右图)。也通过运行凝胶来验证使用女性口腔拭子作为起始材料的阴性对照,其不产生可观察到的条带(未显示)。
从用于产生sry扩增子的口腔拭子源材料,试剂在150v下在5%聚丙烯酰胺凝胶中电泳80分钟。然后使用sybrgreendna特异性荧光染料的1x溶液(7.5μl样品+1.5μl染料)对dna进行染色并使用uv光成像。所示的所有凝胶中的“m”表示用于追踪dna的100bp尺寸标志物。图11显示按以下顺序运行的sry扩增子,泳道1)sry扩增子,泳道2)连接1个抗生物素抗体有效载荷的sry扩增子,3)连接2个抗生物素有效载荷的sry扩增子。泳道2和3中较小的下文条带分别表示无有效载荷和有一个有效载荷的sry。另一凝胶(图15)对比了不含引物修饰的sry(泳道1)与具有一个生物素化的引物的sry(泳道2)和具有两个生物素化的引物的sry(泳道3),其全部在20x单链霉亲和素存在下,显示了结合反应的特异性(泳道4为不存在单链霉亲和素的sry)。
从用于产生smcy扩增子的口腔拭子源材料,5μl(约100ngdna)pcr反应物在150v下在5%聚丙烯酰胺凝胶中电泳80分钟。图20显示按以下顺序运行的smcy扩增子,泳道1)smcy扩增子,泳道2)连接1个单链霉亲和素有效载荷的smcy扩增子,泳道3)连接2个单链霉亲和素有效载荷的smcy。
热循环和有效载荷附接(如果适用)后,将样品稀释到记录缓冲液中,导致在1mlicl,10mmtris-hcl,1mmedta,ph7.5中0.1nm至1nm的最终sry或smcy浓度。
实施例3:sry基因检测
没有连接的有效载荷的sry扩增子事件和电噪声背景假事件是难以区分的,使得该测定法非常容易受到假阳性的影响。特别地,图7显示了的单个分子通过孔隙(100mv)的最大偏移与持续时间的事件图,其将无有效载荷的sry扩增子(黑色)与电背景事件(红色)进行比较。虽然sry为1nm(高浓度),但大多数事件都遗漏了,且在此每分钟只产生3.5次检测的事件(69次事件,20分钟)。这与背景事件群体的事件率和分布重叠(47事件,75分钟)。由于无法检测大多数dna事件且无法与背景区分开,因此从捕获率推断浓度是不可能的。该实验使用15nm膜中的20nm孔隙。
另一方面,通过事件发生率的提高和更明确的事件分布(这不能由噪声事件产生),连接一个和两个单链霉亲和素蛋白有效载荷的sry扩增子(分别为sry-1ms和sry-2ms)可与电噪声背景假事件明显区分开。特别地,在凝胶检验有效载荷附接的sry分子(图8)之后,在图7所示的相同的纳米孔隙上依次测试该试剂,产生图9a中的最大偏移vs持续时间的事件分布图。最大偏移事件柱状图如图9b所示。sry-1ms复合体在45分钟内产生了1074个事件,其中大于0.072ms的事件百分比为41.5%(446)。sry-2ms复合体在80分钟内产生了937个事件,大于0.072ms的事件百分比等于43.9%(411)。相比之下,长于0.072ms的噪声假事件报告为28%。另外,没有有效载荷的sry事件只产生了4.4%的超过0.072ms的事件。
通过应用在“对检测分配统计学显著性”一节中建立的框架,我们可以将统计置信度用于检测1和2有效载荷结合的sry扩增子。具体来说,背景事件被认为是类型1,和dna-有效载荷事件被认为是类型2。一个示例性的标准是,如果它长于0.072ms,则将该事件标记为类型2。背景噪声假阳性事件可用于计算q1=0.28(28%)。
dna有效载荷结果可以用作模拟检测实验,并且通过应用数学框架的等式(1)来确定是否存在2型分子。对于sry-1ms作为2型分子,结果是q(p)-qsd(p)=0.415-0.037=0.38>0.28,这意味着我们可以说sry-1ms分子以99%的置信度存在。对于sry-2ms作为2型分子,结果是q(p)-qsd(p)=0.439-0.04=0.40>0.28,这意味着我们可以说sry-2ms分子以99%的置信度存在。另一方面,sry事件群体不符合等式(1)中的标准,因此我们不能以99%的置信度表示sry分子以高于背景存在。
图10中对于每种试剂类型(sry、sry-1ms、sry-2ms)显示了q(p)±qsd(p)作为记录时间的函数的曲线图。还将该趋势与从背景假阳性事件建立的假阳性阈值相比较。也观察到sry-1ms和sry-2ms在记录的前5分钟内以99%的置信度被检测到。
将事件标记为类型2的示例标准是是否事件持续时间大于0.072ms,其为两种有效载荷附接的sry分子类型都产生了阳性的检测结果。当改变阈值持续时间值时也保持该结果,表明结果不依赖于独特或窄的标准值范围。如果事件持续时间是0.02到0.1ms之间的任何值,则维持相同的检测结果。
另外,可以使用不同的标准,并且仍然保持该检测结果。具体地,如果最大δg>1ns,则考虑将事件标记为2型的标准。背景噪声假阳性事件可用于计算q1=0.23(23%)。用这个标准,对于sry-1ms作为2型分子,结果是q(p)-qsd(p)=0.333-0.037=0.29>0.23,这意味着我们可以说sry-1ms分子以99%置信度存在。对于sry-2ms作为2型分子,结果更明显,因为具有2个有效载荷的这些分子产生更多的较深的事件。具体地,q(p)-qsd(p)=0.581-0.041=0.54>0.23,这意味着我们可以说sry-2ms分子以99%的置信度存在。如前所述,sry事件群体不满足等式(1)中的标准(因为q(p)=0.13),因此我们不能以99%的置信度表示sry分子高于背景存在。
具有最大δg>1ns标准的结果表明,该标准也可用于检测包括sry-1ms分子的高于背景的sry-2ms分子。也就是说,如果试验首先使用单一ms有效载荷来检测1ms结合的靶标,并随后测试双重ms有效载荷产物,则可以使用最大δg>1ns标准来测定是否存在2ms结合的靶标。这提供了一种多路复用形式。对于该数据集,如果sry-1ms是背景的形式,则在等式(1)的应用中q1=0.333。由于q(p)-qsd(p)=0.581-0.041=0.54>0.333,我们可以说,sry-2ms分子以99%的置信度高于背景存在而不包括sry-1ms分子。
实施例4:具有较大有效载荷在较大纳米孔隙中的sry基因检测
以下数据证实了使用含有化学修饰(生物素)的引物“增大”扩增子和该“增大”有效载荷是抗体的方法。凝胶图像表明1和2有效载荷结合的sry分子的置信度(图11)。抗生物素抗体(bab)比单链霉亲和素(66kda)大大约3倍(150kda),从而提供了更大的有效载荷。利用更大的有效载荷,我们预计并且数据显示,该ab-结合的sry事件具有比观察到的ms结合的sry更深的阻抗偏移。例如,深度超过1.5ns的sry-2ms事件的百分比为16.0085%(150/937),且具有20nm的孔隙。相比之下,深度超过1.5ns的sry-2bab事件的百分比为60.023%(1042/1736),并且具有大2x的孔隙(直径45nm孔,30nm膜)。相反,没有有效载荷的sry在30分钟内仅产生41个事件,只有12.2%的事件深度超过1.5ns。图12显示了对于没有有效载荷的sry(黑色)和sry-2bab(粉红色),单个分子通过孔隙的最大δg对持续时间的事件图。因此,较大的有效载荷提供更深的事件特征,并且即使使用较大的纳米孔隙也可以进行检测(具有99%置信度)。
实施例5:区分扩增子与pcr背景
以下实施例证明,可以通过以下两种途径之一将pcr扩增子与由于pcr反应混合物导致的背景事件区分:通过使用长度至少为1000bp的扩增子长度(图13),或通过加入有效载荷(图14)。
图13显示了使用22nm膜中的25nm孔隙,对于在pcr背景存在下0.7nm的1074bp扩增子(蓝色)和单独pcr背景(黑色)的事件分布图。两种情况都使用pcr背景的1:10稀释。可以控制稀释以使得背景事件率与预期的扩增子率相比不会太大。由来自pcr的背景导致的更明显和分散的事件,靶序列的可检测事件发生率与背景相比成为判别因子。由于扩增子在此足够长,检测是可行的。
在15个循环的pcr(使用5.6骨架的808pg模板)后得到了0.7nm的1074bpdna,以1:10稀释到记录缓冲液中而无需纯化,并在10分钟内产生了1502个事件(2.5l/秒)。pcr背景在10分钟内产生了97个事件(0.171/秒)。使用水代替模板(相当于加入模板时使用的相同体积的水)的反应产生测量的pcr反应混合物,同样循环15次,然后以1:10稀释到记录缓冲液中。
pcr背景包括未杂交引物、dntp(脱氧核苷酸三磷酸,datp、dgtp、dctp、dttp)、聚合酶(例如taq或pfu)、盐(氯化镁、硫酸镁、硫酸铵、氯化钠、氯化钾)、bsa(牛血清白蛋白稳定剂)、洗涤剂(tritonx-100)以及根据所使用的扩增方案可能存在的其它元素。
图14从概念上示出较短的扩增子为了在pcr反应混合物背景之上可检测到,如何需要有效载荷连接。具体来说,没有有效载荷(绿色)和具有1ms有效载荷(单一引物在5'末端具有生物素修饰)(红色)的0.2nm500bp扩增子事件覆盖在图13的数据上。没有有效载荷的情况下,无有效载荷的dna的事件率与pcr背景相比不足以被感知(30分钟内29个事件),大多数dna通过传感器而未被检测到。另一方面,在有效载荷(500bp-ms)的情况下,事件率足以被用于检测(18分钟内238个事件),并且可以通过确定围绕由dna-有效载荷事件群体所包围的事件子集的检测标准以统计学置信度从背景进行区分。评估用于在pcr背景之上区分扩增子的统计学置信度是以下实施例的重点。
实施例6:pcr背景存在下的sry基因检测
图16和17显示可以在1:60稀释的pcr背景存在下,以99%的置信度检测sry-1ms和sry-2ms,而单独的sry不能从pcr背景中区分出来。使用15nm膜中的20nm孔隙(100mv)获得这些结果。图16显示了对于仅pcr背景(红色)、无有效载荷的sry(蓝色)、sry-1ms(黑色)和sry-2ms(青色),单个分子通过孔隙的平均δg对持续时间的事件图,所有的sry试剂在1:60稀释的pcr背景下进行测试。单独的pcr背景在40分钟内产生10个事件,与20分钟内73个事件的sry(无生物素,无有效载荷)相当。虽然sry事件率略高于背景,但事件群体重叠太多而无法以置信度检测sry。作为示例,pcr背景和sry分别具有10%和4%的长于0.072ms的事件。相反,0.5nm的有效载荷结合的sry事件具有显著更高的捕获率,因为这些分子由于有效载荷而变得更可观察。sry-1ms在34分钟内产生了1769个事件,和sry-2ms在仅6分钟内产生326个事件。我们还可以应用所呈现的数学框架来获得对这两种有效载荷结合的扩增子检测的99%的置信度,如下文所述。
视觉上,图16中的事件图表明,有效载荷结合的事件产生较高百分比的较深事件。由此我们可以基于事件深度选择标准。具体来说,考虑如果平均δg>2.7ns,则将事件标记为类型2的标准。pcr背景假阳性事件可用于计算q1=0.1(10%)。用这个标准,对于sry-1ms作为2型分子,结果是q(p)-qsd(p)=0.22-0.025=0.19>0.1,这意味着我们可以说sry-1ms分子以99%的置信度存在。对于sry-2ms作为2型分子,结果更明显,因为具有2个有效载荷的这些分子产生更多的较深的事件。具体地,q(p)-qsd(p)=0.34-0.067=0.27>0.1,这意味着我们可以说sry-2ms分子以99%的置信度存在。如前所述,sry事件群体不符合等式(1)中的标准(因为q(p)=0.066),因此我们不能以99%的置信度表示sry分子高于背景存在。
在图17中显示了以1:60稀释的pcr背景存在下对于每种试剂类型的作为记录时间的函数的q(p)±qsd(p)的图,其中由于记录时间的差异很大,水平轴上的时间标准化。还将该趋势与从pcr背景假阳性事件建立的假阳性阈值进行比较。在记录的前60秒内,sry-1ms和sry-2ms都以99%的置信度检测到。
实施例7:pcr和全血背景存在下的sry基因检测
图18和19示出了在同时存在两种形式的背景的情况下,可以以99%置信度检测sry-1ms和sry-2ms:第一种是模拟“样品”的形式(1:20000稀释的全血),和第二种是用于产生扩增子的pcr反应试剂的1:100稀释。如在上面考虑的其他情况中,单独的sry不能从这种形式的背景中区分开来。使用25nm膜中的25nm孔隙(100mv)获得这些结果。
首先测试1:1000稀释的全血,14分钟后导致孔隙堵塞。为了复原孔隙,灌注稀释液,和按照文献(beamish,eric,haroldkwok,vincenttabard-cossa和michelgodin“precisecontrolofthesizeandnoiseofsolid-statenanoporesusinghighelectricfields.”nanotechnology23,no.40(september14,2012):405301–8)中所建立的技术使用受控的电介质调理扩大孔隙。观察到如果需要小孔隙来检测短的扩增子(无有效载荷),则需要扩大孔径的这种堵塞事件将使测试终止,并且不产生结果。由于我们的有效负载附接方法即使在大孔隙中也会产生扩增子检测,因此需要扩大孔隙的堵塞事件(其可能会频繁出现,特别是用“复杂的”样品)不会导致测试失败;相反,较大的孔隙可以耐受甚至更多的背景(较大的孔隙更难堵塞),并且仍然可以获得测试结果。
图18显示了单个分子通过孔隙的最大δg对持续时间的事件图。记录的纪元(epoch)包括:仅缓冲液(12事件,30分钟);全血(wb)1:1000(154事件,14分钟)-未绘制;wb1:20000(157事件,16分钟)-绘制(青色);和dna(0.5nmsry无生物素,1:20000血稀释)(23分钟内293个事件)。无有效负载的sry具有对于wb而言更紧的持续时间和幅度分布。例如,wb集合有20%的事件高于0.072ms,而单独的sry只有8%(更快的事件)。在sry之后,测试1:20000wb中的0.5nm的sry-1ms,并且产生了事件数量的显著增加:在26分钟内1093个事件。随后是在1:20000wb中sry-2ms0.5nm,其在42分钟内产生1297个事件(在所有情况下,也存在1:100的pcr反应混合物)。如在先前的例子中,我们可以应用所呈现的数学框架对于两种有效载荷结合的扩增子获得检测的99%的置信度。
视觉上,图18中的事件图表明,有效载荷结合的事件产生较高百分比的较深事件,特别是对于更长的事件。据此,我们可以根据事件深度和用最短持续时间来选择标准。具体地,考虑如果最大δg>3ns和持续时间>24us,则将事件标记为类型2的标准。1:20000wb+1:100pcr背景假阳性事件可用于计算q1=0.096(9.6%)。按照这个标准,对于sry-1ms作为2型分子,结果是q(p)-qsd(p)=0.184>0.096,这意味着我们可以说sry-1ms分子以99%的置信度存在。对于sry-2ms作为2型分子,结果更明显,因为具有2种有效载荷的这些分子产生更多的较深的事件。具体地,q(p)-qsd(p)=0.348>0.096,这意味着我们可以说sry-2ms分子以99%的置信度存在。如前所述,无有效载荷的sry事件群体在背景存在的情况下不满足等式(1)中的标准(因为q(p)=0.024),因此我们不能以99%的置信度表示sry分子高于背景存在。
在图19中示出了在1:20000wb+1:100pcr背景存在下,对于每种试剂类型的作为记录时间的函数的q(p)±qsd(p)的图。还将该趋势与从背景假阳性事件建立的假阳性阈值相比较。在记录的前90秒内,sry-1ms和sry-2ms都以99%的置信度被检测到。
实施例8:非靶dna背景存在下的smcy基因检测
图21和22显示,可以在作为背景的模拟形式的丰富非靶dna存在下,以99%的置信度检测smcy-1ms。相比之下,单独的smcy不能从这种形式的背景中被区分开来。使用15nm膜中的21nm孔隙(100mv)获得这些结果。
在凝胶证实有效载荷附接的smcy扩增子之后(图20),在相同的纳米孔隙上依次测试以下试剂。作为背景的模拟形式,我们在相等浓度的sry(470bp)存在下以0.5nm总浓度(各0.25nm)测试smcy(362bp)。单独的smcy(未显示)不能与单独的sry区分开。特别地,较短的smcy扩增子具有与单独电背景相当的事件率,而sry具有约5倍于电噪声背景事件的事件率。因此,在sry+smcy中28分钟内记录的560个事件中的大多数可能归因于sry,尽管无法区分哪些事件可归因于任一扩增子。为了在这种形式的背景中检测smcy的存在,在一端使用生物素化的引物并附接ms。在0.5nmsmcy-1ms下,在20分钟内记录1879个事件。
图21显示单个分子通过孔隙的平均δg与持续时间的事件图。视觉上,事件图表明,有效载荷结合的事件产生更高比例的较深事件。据此,我们可以选择如果最大δg>4ns,将事件标记为2型的标准。sry+smcy背景假阳性事件可用于计算q1=0.116(11.6%)。用这个标准,对于sry-1ms作为2型分子,结果是q(p)-qsd(p)=0.287-0.026=0.26>0.116,这意味着我们可以说sry-1ms分子以99%的置信度存在。如前所述,无有效载荷的sry事件群体在背景的存在下不满足式(1)中的标准(未显示),且因此我们不能以99%的置信度表示smcy分子高于背景存在。
图22中对于smcy-1ms显示了与非靶dna(主要是sry)背景相比的作为记录时间的函数的q(p)±qsd(p)的图。在记录的最先2分钟内以99%的置信度检测到sry-1ms。
实施例9:扩增子浓度的定量
图23显示捕获率与浓度呈线性关系,并且可以通过使用对照来建立线性趋势并将测量的捕获率映射到线上以估计浓度来估算未知浓度。这意味着使用与未知物的捕获率动力学匹配的对照(即,使用相同的扩增子长度,并且具有相同的有效载荷(在使用的情况下))。虽然在孔隙大小之间存在捕获率的差异,但通过在同一孔隙上在未知物质之前运行对照,这种不确定性来源被消除。
使用15nm膜中的24nm孔隙,用无有效载荷的1074bp扩增子建立图23中的数据。通过将时间-捕获分布拟合为指数分布来测定每个已知和未知浓度的捕获率(这在纳米孔隙科学领域是众所周知的,例如在wang,hongyun,nicholashurt和williambdunbar“measurementandmodelingthekineticsofindividualdna-dnapolymerasecomplexesonananopore”。acsnano7,no.5(2013年5月28日):3876-86中)。
使用端点pcr产生对照以形成1074bp扩增子的储备(stock)。将其在二氧化硅上纯化并用分光光度法定量。为了产生每个对照,将储备溶液稀释至记录缓冲液中,并在记录实验之前再次测量。使用的标准浓度范围为0.075nm至1.1nm(0.075nm、0.15nm、0.8nm、1.1nm)。通过使用引物进行20个pcr循环来产生未知样品,从而从100pg长度为5600bp的起始材料产生1074扩增子。循环后,将反应物在记录缓冲液中1:50稀释,和进行纳米孔隙实验。与对照拟合并在将不确定度分配给估计值的同时估算未知值的方法在“从测量的捕获率估算浓度”一节中给出。对于实施例中的估计,在50x稀释度下的0.7nm对应于35nm,其接近于从对稀释前的样品的规格测量估计的51nm值。
实施例10:mnopcr的性能
图24和25呈现了我们的整数(m)纳米孔隙(n)可观察(o)pcr(mnopcr)方法的效能。对于两个数据集,首先使用纳米孔隙测量已知浓度的三种标准的捕获率,以建立未知样品可以映射的曲线。在图24中,在12、13和14个pcr循环之后运行未知浓度的样品,并估计相应的浓度。在图25中,通过比较使用共同的起始材料量和循环数的三个不同的pcr反应进行的捕获率来评估未知样品的测量精度。
图24显示了来自0.2、0.7和1.2nm标准品的标准曲线,且未知的12、13和14循环映射到曲线上,误差条表示测量值的99%置信度(+/-0.46nm)。在1mlicl,10mmtris,1mmedta,ph7.5中,在100mv下进行记录,并以10khz重新过滤。孔隙大小为22-25nm,膜厚为30nm。对于12、13和14个循环,未知物的所得浓度分别为1.32nm,2.42nm和4.56nm。与前面的例子一致,背景事件是总记录事件的<1%。标准品和未知物的事件图(未显示)显示紧密的重叠分组,如所预期的,所有记录的扩增子长度相同。图24中的图显示了与标准曲线相匹配的标准品(红点)和未知物(蓝色)(循环12、13和14)。根据每个标准品的直线的线性和测量误差,报告的浓度在5%以内为99%准确的。
随着反应进行,在每个循环(或循环的任何组合)之后进行测量使我们可以测定在哪个循环处扩增子是可检测的。当在反应进行时测量扩增子时,可以获得额外的信息,例如,反应处于对数阶段的时间、pcr反应的实际效率(即,由于理论倍增通常不能精确地实现)、终点检测以及两个或更多个不同靶序列数的样品之间的精确比较。这里,对数期从循环13开始,和每反应的效率为1.8倍(而不是理论值2.0)。检测在循环10时开始。
这些数据在估计浓度上产生5%的最大误差,这随之表明起始材料的1.1倍差异的区分应该是可能的。该实施例显示可以在起始模板浓度可能发生变化的两个或更多个样品之间进行准确的比较,其非常类似于qpcr,但是更准确,因为纳米孔隙计数单分子而不是来自分子的总体集合的荧光。
图25给出了评估定量精度的结果。具体地,使用相同的对数相循环数(20个循环)和相同的原料量,进行三次独立的pcr反应以产生材料。浓度估计是非常保守的,证明了精度。结果包括估计值:20循环a=54.3nm;20循环b=57.4nm;20循环c=58.1nm。测定精度=4%(散度(spread)/平均)。此外,当循环数变化时,在重复实验中观察到1-5%的精度性能。
实施例11:用mnopcr推断原料
该实施例中的汇总数据表明工作流程中用于在扩增之前计算样品中分子起始数的部分。使用对照,mnopcr方法可以确定反应效率,并确定反应何时进入对数阶段。图26显示了在循环10、12、13、14、15、17、20、30、35、40后测量反应后的s形曲线。具体地,进行pcr,并在循环10、12、13、14、15、17、20、30、35和40后取样。独立地在纳米孔隙中运行每个循环。作为对照,这可以用于建立捕获率与浓度线性趋势;作为未知物,将速率拟合于该线以通过浓度估计方法确定在指示的循环之后产生的产物的量。注意到,在测试一系列pcr循环产物之前,只需要测试两个对照(高浓度和低浓度)以建立捕获率vs浓度的趋势。对于该对照数据集,反应在循环15之后进入对数阶段,显示1.7倍的次最佳倍增,并在循环35之后达到终点。实际上,这可以作为对照曲线,与定量反应效率性能和未知曲线一起用来推断原料的量。
注意,通过扩增子被添加(或产生)的共同腔室的孔隙阵列,可以较早地检测较低的循环数,且具有更大的稀释度可容性,以便在曲线的较低浓度末端提供更好的分辨率和散度。这随之提高未知物的定量和降低推测误差。
实施例12:探索pcr反应混合物的最小稀释
图27和28探讨了在保持mnopcr方法的保真度的同时需要多小的稀释。图27显示单个分子通过孔隙的平均δg与持续时间的事件图。该图显示15个循环后的pcr反应混合物(红色)及阴性对照(水)以1:1.17稀释(85.7%pcr反应混合物)的事件,然后是相同的混合物,但具有产生的1074bp的扩增子(阳性对照,15个循环),其显示事件的大幅增加(黑色)。虽然1074bpdna在0.5nm下在10分钟内产生1856个事件,但是85.7%pcr产物在10分钟内仅产生39个事件,其使用25nm膜中的25nm孔隙。所用的所有阴性对照稀释液认为表现出相当的事件率和散度:1:5稀释(20%),1:2稀释度(50%),1:1.43稀释(70%),1:1.17稀释度(85.7%)。图28显示,当阴性对照pcr产物被最小稀释(1:1.17)时,它产生足够可感知事件以触发较不准确的浓度估计。当将样品稀释到>30%时,该效应最小化。
- 还没有人留言评论。精彩留言会获得点赞!