多肽的制作方法
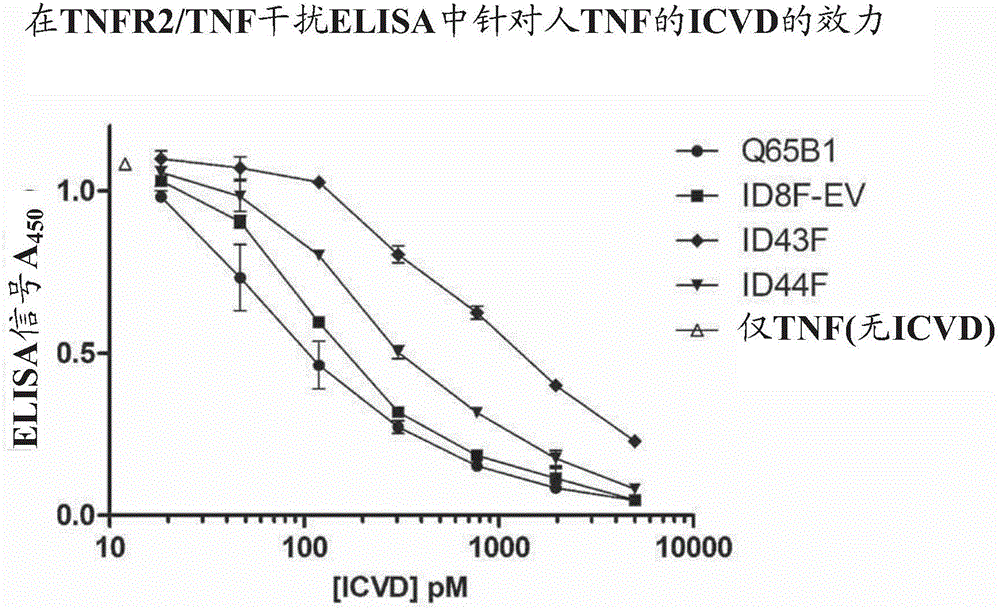
发明领域本发明涉及包含能够以高亲和力结合靶标的区域的多肽,特别是包含免疫球蛋白链可变结构域(icvd)的那些,以及包含所述多肽的构建体和包含此类多肽和构建体的药物组合物。本发明的多肽、构建体和药物组合物都适合于口服施用。本发明还涉及增加包含免疫球蛋白链可变结构域的多肽的肠稳定性的方法,制备包含免疫球蛋白链可变结构域的多肽的方法,以及利用该多肽、包含此类多肽的构建体、编码此类多肽的核酸、包含编码此类多肽的核酸的cdna和载体、表达或能够表达此类多肽的宿主细胞、包含此类多肽的药物组合物的方法,以及此类多肽的用途。发明背景药物研究开发越来越关注于生物药物,如治疗性多肽,包括抗体。通常,治疗性多肽通过全身途径直接或间接地施用给循环。然而,理想地,许多治疗性多肽经由口服途径递送。口服递送治疗性多肽可以提供以下优点:(a)直接靶向胃肠道(git)以局部治疗胃肠道疾病(jones和martino2015critrevbiotechnol20:1-15),(b)不良免疫反应的风险可以由于git的天然免疫耐受性质而降低,确保反复摄取治疗性多肽材料的长期安全性,(c)没有制造可注射治疗性多肽的严格监管要求,可以降低生产成本,(d)可以实现更高水平的患者接受度和长期顺应性(shaji和patoleindianjpharmsci200870(3):269-277)。然而,许多治疗性多肽在肠道中是不稳定的,并因此从口服施用获得的有益效果通常是有限的(bruno等人,2013therdeliv4(11):1443-1467)。因此,用于常规小分子药物的口服剂型已被用于口服多肽递送。目前正在研究的各种策略包括制剂介载体、酶抑制剂的使用、吸收促进剂以及粘膜粘附聚合物(shaji和patole,同上)。还采用了涉及对治疗性多肽本身的修饰的替代策略,例如引入(额外的)半胱氨酸桥。hussack等人2011plosone6(11):e28218描述了将额外的半胱氨酸桥引入抗tcdavhhs中。这些额外的半胱氨酸桥对增加蛋白水解稳定性的有效性高度依赖于所涉及的特异性蛋白酶,并且在某些情况下,这些额外的半胱氨酸桥对重组生产水平是不利的。类似地,kim等人mab6:219-235改造了具有二硫键桥的人vl结构域,具有混合结果。理论上,可以考虑取代被认为负责治疗性多肽的低肠稳定性的治疗性多肽中的特异氨基酸,以增强肠道中的稳定性。然而,在免疫球蛋白链可变结构域的上下文中,氨基酸序列中的单个取代可能不利地影响结合能力。这特别与负责结合靶标抗原的免疫球蛋白链可变结构域的互补决定区(cdr)相关。例如,特别关于vhh的cdr3,已知“...因为cdr3氨基酸或者与抗原直接接触或者保持并影响直接接触抗原的cdr3氨基酸的构象,负责降低稳定性的cdr3氨基酸不能在没有严重的亲和力丧失下被取代”(muyldermansannurevbiochem201382:775-797)。这种观点通过例如以下发现得到加强:对靶向c.jejuni鞭毛的vhh的取代(特别是cdr2中的r对g取代)导致vhh的结合能力的大幅度降低(接近对照)(hussack等人,2014proteinengineering,design&selection27(6):191-198)。因此,对于具有增加的肠稳定性的多肽以及用于增加这些多肽的肠稳定性的方法,长期存在需要。在至少一些实施方案中,与现有技术的物质相比,本发明的多肽可具有一个或多个以下优点:(i)口服施用适应性增加;(ii)口服施用后局部递送至肠道的适应性增加;(iii)肠道稳定性增加,同时基本保持结合亲和力和/或效力;(iv)在肠道模型中,例如标准胰蛋白酶肠道模型,标准小鼠小肠上清液肠道模型或标准人粪便上清液肠道模型中,稳定性增加,同时保持结合亲和力和/或效力;(v)在蛋白酶存在下增加的稳定性,例如(a)在小肠和/或大肠中可见的蛋白酶和/或ibd炎性蛋白酶,例如胰蛋白酶、糜蛋白酶、mmp、组织蛋白酶、肠肽酶、宿主炎性蛋白酶的存在下和/或(b)在通过在小肠和/或大肠中发现的微生物细胞的裂解而主动分泌和/或释放的、来自肠道共生微生物群落和/或病原菌的蛋白酶存在下;(vi)当在异源宿主(如酵母,例如属于曲霉属(aspergillus)、酵母属(saccharomyces)、克鲁维酵母属(kluyveromyces)、汉逊酵母属(hansenula)或毕赤酵母属(pichia)的酵母)中表达时(由于增加的对酵母蛋白酶的抗性)而增加的稳定性;(vii)不良免疫反应的风险降低;(viii)生产成本降低;(ix)肠道感染或自身免疫和/或炎性疾病的治疗和/或预防改善;(x)患者接受度和长期顺应性提高;(xi)在重组生产过程中产量提高;(xii)生物活性和/或生物分布改善;(xiii)所需剂量减少;(xiv)对用于药物的适用性和改进的性能;(xv)对用于功能性食品的适用性和改进性能。技术实现要素:本发明人已经生产了出人意料地有利的适合口服施用的、包含免疫球蛋白链可变结构域的多肽。这些多肽由于其增加的肠稳定性(即肠道中的稳定性增加)而特别有利。可以预期,这些多肽在预防或治疗胃肠道疾病(例如自身免疫性和/或炎性疾病如炎症性肠病),或预防或治疗来自肠道定居病原微生物的感染中具有特别的用途。还提供了增加包含免疫球蛋白链可变结构域的多肽的肠稳定性的方法和制备具有增加的稳定性的包含免疫球蛋白链可变结构域的多肽的方法。因此,本发明提供了包含免疫球蛋白链可变结构域的多肽,其包含三个互补决定区(cdr1-cdr3)和四个框架区,其中:(a)cdr1、cdr2和/或cdr3中的至少一个赖氨酸残基已被至少一个组氨酸残基取代和/或(b)cdr1、cdr2和/或cdr3中的至少一个精氨酸残基已被至少一个组氨酸残基取代;其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。还提供了增加包含免疫球蛋白链可变结构域的多肽的肠稳定性的方法,其中免疫球蛋白链可变结构域包含三个互补决定区(cdr1-cdr3)和四个框架区,其中该方法包括以下步骤:(a)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个赖氨酸残基,和/或(b)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个精氨酸残基。还提供了制备包含免疫球蛋白链可变结构域的多肽的方法,其中所述免疫球蛋白链可变结构域包含三个互补决定区(cdr1-cdr3)和四个框架区,其中所述方法包括以下步骤:(a)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中至少有一个赖氨酸残基,和/或(b)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个精氨酸残基,其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。还提供了包含能够以高亲和力结合靶标的区域的多肽,其中:(a)该区域中至少一个赖氨酸残基已被至少一个组氨酸残基取代,和/或(b)该区域中的至少一个精氨酸残基已被至少一个组氨酸残基取代;其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。附图说明图1-vero细胞上的tcda剂量-反应曲线实施例图2a-在tnfr2/tnf干扰elisa中抗tnficvdq65b1、id8f-ev、id43f和id44f(实验1)对人tnf的效力图2b-在tnfr2/tnf干扰elisa中抗tnficvdq65b1和id8f-ev(实验2)对人tnf的效力图3a-孵育6小时后小鼠小肠上清液中抗tnficvdq65b1、id8f-ev、id43f和id44f的稳定性图3b-孵育16小时后人粪便和小鼠上清液中抗tnficvdq65b1和id8f-ev的稳定性图4-在tnfr2/tnf干扰elisa中icvdid32f和id34f对人tnf的效力图5a-孵育16小时后小鼠小肠上清液中抗tnficvdid32f和id34f的稳定性图5b-孵育16小时后人粪便上清液池4中的抗tnficvdid32f和id34f的稳定性图6a-在vero细胞细胞毒性测定中通过id45b-id50b的tcdb027中和图6b-通过western印迹分析,孵育30分钟后人粪便上清液池4中的抗tcdbicvdid45b-id50b的稳定性图7-在vero细胞细胞毒性测定中通过id2b、id20b、id21b和id22b的tcdb027中和图8a-id2b胰蛋白酶测定-染色的聚丙烯酰胺凝胶图8b-id20b和id21b胰蛋白酶测定-染色的聚丙烯酰胺凝胶图8c-id22b胰蛋白酶测定-染色的聚丙烯酰胺凝胶图9-孵育1小时后人粪便上清液中抗tcdbicvdid2b和id21b的稳定性图10a-在vero细胞细胞毒性测定中通过027id1b、id24b、id25b和id27b的tcdb中和图10b-1小时后人体粪便上清池2中抗tcdbicvdid1b,id24b,id25b和id27b的稳定性图11a-id1b胰蛋白酶测定-染色的聚丙烯酰胺凝胶图11b-id24b和25b胰蛋白酶测定-染色的聚丙烯酰胺凝胶图11c-id27b胰蛋白酶测定-染色的聚丙烯酰胺凝胶图12a-在vero细胞细胞毒性测定中通过双头构建体id41b和id43b的tcdb017中和图12b-孵育4小时后在艰难梭菌阴性人粪便上清液池2中抗tcdb双头构建体id41b和id43b的稳定性(三次重复elisa)图12c-孵育4小时后在艰难梭菌阴性人粪便上清液池3中的抗tcdb双头构建体id41b和id43b的稳定性(三次重复elisa)图12d-孵育4小时后在艰难梭菌阴性人粪便上清液池4中的抗tcdb双头构建体id41b和id43b的稳定性(三次重复elisa)图13a-在vero细胞细胞毒性测定中通过id17a和id29a的tcda087中和图13b-孵育1小时后在人体粪便上清液中抗tcda双头构建体id17a和id29a的稳定性序列说明seqidno:1-抗tnf-αicvdq65b1的多肽序列seqidno:2-抗tnf-αicvdid8f-ev(id32f)的多肽序列seqidno:3-抗tnf-αicvdid43f的多肽序列seqidno:4-抗tnf-αicvdid44f的多肽序列seqidno:5-抗tnf-αicvdid34f的多肽序列seqidno:6-抗tcdbicvdb10f1的多肽序列seqidno:7-抗tcdbicvdq31b1的多肽序列seqidno:8-抗tcdbicvdid1b的多肽序列seqidno:9-抗tcdbicvdid2b的多肽序列seqidno:10-抗tcdbicvdid20b的多肽序列seqidno:11-抗tcdbicvdid21b的多肽序列seqidno:12-抗tcdbicvdid22b的多肽序列seqidno:13-抗tcdbicvdid24b的多肽序列seqidno:14-抗tcdbicvdid25b的多肽序列seqidno:15-抗tcdbicvdid27b的多肽序列seqidno:16-抗tcdb构建体id41b的多肽序列seqidno:17-抗tcdb构建体id43b的多肽序列seqidno:18-抗tcdbicvdid45b的多肽序列seqidno:19-抗tcdbicvdid46b的多肽序列seqidno:20-抗tcdbicvdid47b的多肽序列seqidno:21-抗tcdbicvdid48b的多肽序列seqidno:22-抗tcdbicvdid49b的多肽序列seqidno:23-抗tcdbicvdid50b的多肽序列seqidno:24-抗tcda构建体id17a的多肽序列seqidno:25-抗tcda构建体id29a的多肽序列seqidno:26-实施例cdraseqidno:27-实施例cdra的第一个三分之一seqidno:28-实施例cdra的第二个的三分之一seqidno:29-实施例cdra的第三个三分之一seqidno:30-实施例cdrbseqidno:31-实施例cdrb的第二个的三分之一seqidno:32-抗il-6ricvd7f6的多肽序列seqidno:33-抗il-6ricvdid-3v的多肽序列seqidno:34-抗il-6ricvd5g9的多肽序列seqidno:35-抗il-6ricvdid-54v的多肽序列发明的详细说明包括免疫球蛋白链可变结构域(icvd)例如vh和vhh的多肽、抗原结合多肽、抗体及抗体片段多肽是由结合在一条链中的多个氨基酸残基组成的有机聚合物。如本文所用,“多肽”与“蛋白质”和“肽”可互换使用。多肽当它们含有一个或多个形成抗原结合位点的氨基酸残基区段时,被认为是抗原结合的,能够以亲和力结合靶标抗原上的表位(适当地表示为kd值,ka值,kon率和/或koff率,如本文进一步描述的)。抗原结合多肽包括多肽,例如抗体,修饰为包括额外结合区域的抗体和抗原结合片段。多肽可以包含能够以高亲和力结合靶标的区域(适当地表示为kd值,ka值,kon率和/或koff率,如本文进一步描述的)。此类多肽包括darpins(binz等人,journalofmolecularbiology332(2):489-503),affimerstm,fynomertm,centyrins,nanofitins和环肽。常规抗体或免疫球蛋白(ig)是包含四条多肽链的蛋白质:两条重(h)链和两条轻链(l)链。每条链分为恒定区和可变结构域。重链可变结构域在本文中缩写为vhc,并且轻(l)链可变结构域在本文中缩写为vlc。这些结构域,与其相关的结构域和由其衍生的结构域在本文中称为免疫球蛋白链可变结构域。vhc和vlc结构域可以进一步细分为被称为“互补决定区”(“cdr”)的高变区域,散布有更保守的区域(称为“框架区”)(“fr”)。已经精确定义了框架区和互补决定区(kabat等人,1991sequencesofproteinsofimmunologicalinterest,第五版,u.s.departmentofhealthandhumanservices,nih出版号91-3242,其通过整体引用并入本文)。在常规抗体中,每个vhc和vlc由以下顺序从氨基末端至羧基末端排列的三个cdr和四个fr组成:fr1,cdr1,fr2,cdr2,fr3,cdr3,fr4。两个重免疫球蛋白链和两个轻免疫球蛋白链的常规抗体四聚体是由通过例如,二硫键,互相连接的重和轻的免疫球蛋白链,以及相连的重链相似物形成的。重链恒定区包括三个结构域ch1,ch2和ch3。轻链恒定区由一个结构域cl组成。重链的可变结构域和轻链的可变结构域是与抗原相互作用的结合结构域。抗体的恒定区域通常介导抗体与宿主组织或因子(包括免疫系统的各种细胞(例如效应细胞))和经典补体系统的第一组分(c1q)的结合。术语抗体包括iga、igg、ige、igd、igm型(以及其亚型)的免疫球蛋白,其中免疫球蛋白的轻链可以是κ或λ型。由两个相同的重(h)链和两个相同的轻(l)链多肽组装的免疫球蛋白-γ(igg)抗体的整体结构被完全建立并且在哺乳动物中高度保守(padlan1994molimmunol31:169-217)。在骆驼科的血清中发现常规抗体结构的例外。除常规抗体外,这些血清还具有特异的igg抗体。称为重链抗体(hcab)的这些igg抗体缺乏l链多肽,并且缺少第一恒定结构域(ch1)。在其n末端区域,同二聚体蛋白的h链含有专门的免疫球蛋白链可变结构域,称为vhh,其用于与其同源抗原缔合(muyldermans2013annurevbiochem82:775-797,hamers-casterman等人1993nature363(6428):446-448,muyldermans等人1994proteineng7(9):1129-1135,其通过整体引用并入本文)。vhh或vh中的氨基酸残基的总数可以在105-140的范围内,适当地是108-130,并且最适当地是110-125。本文所用的抗原结合片段(或“抗体片段”、“免疫球蛋白片段”或“抗原结合多肽”)是指特异性结合靶标的抗体的一部分(例如其中一个或多个免疫球蛋白链不是全长,但其特异性结合靶标的分子)。抗原结合片段包含免疫球蛋白可变结构域。术语抗原结合片段中包含的结合片段的例子包括:(i)fab片段(由vlc、vhc、cl和ch1结构域组成的单价片段);(ii)f(ab')2片段(包含通过铰链区上的二硫桥连接的两个fab片段的二价片段);(iii)fd片段(由vhc和ch1结构域组成);(iv)fv片段(由抗体的单臂的vlc和vhc结构域组成);(v)scfv片段(由使用重组方法通过合成接头连接的vlc和vhc结构域组成,使得它们能够制成其中的vlc和vhc区域配对以形成单价分子的单个蛋白质链);(vi)vh(由vhc结构域组成的免疫球蛋白链可变结构域(ward等人nature1989341:544-546);(vii)vl(由vlc结构域组成的免疫球蛋白链可变结构域);(viii)v-nar(由来自软骨鱼纲(chondrichthyes)ignar的vhc结构域组成的免疫球蛋白链可变结构域(roux等人,1998procnatlacadsciusa95:11804-11809和griffiths等人,2013antibodies2:66-81,通过整体引用引入本文)(ix)vhh。适当地,本发明的多肽由免疫球蛋白链可变结构域组成。适当地,本发明的多肽是抗体,含有其它抗体结合区的修饰抗体或抗体片段如vhh、vh、vl、v-nar、scfv、fab片段或f(ab')片段。本发明的多肽可以例如通过使用核酸合成技术制备编码多肽的核酸,随后表达由此获得的核酸来获得(如本文进一步详述的)。本文提供的实例涉及免疫球蛋白链可变结构域本身。然而,本文公开的本发明的原理同样至少适用于任何包含免疫球蛋白链可变结构域的多肽,例如抗体和抗体片段。例如,本文公开的免疫球蛋白链可变结构域可以掺入到诸如全长抗体的多肽中。mccoy等人retrovirology201411:83中阐述了这种方法,其提供了一种抗hivvhh,其被改造为具有人fc区(包括铰链、ch2和ch3结构域)的融合物,作为二聚体构建体表达。多肽和多核苷酸序列如本文所用,多肽序列的编号和cdr和fr的定义根据kabat系统(kabat等人,同上)来定义。第一和第二多肽序列之间的“相应”氨基酸残基是第一序列中的、根据kabat系统与第二序列中的氨基酸残基共有相同位置的氨基酸残基,尽管第二序列与第一序列的氨基酸残基的同一性可能不同。如果根据kabat定义框架和cdr具有相同的长度,则适当地相应的残基将共有相同的数字(和字母)。对准可以手动实现,或者通过使用例如使用标准设置的已知用于序列比对的计算机算法(例如ncbiblastv2.0(blastp或blastn))来实现。如果两个或更多个多肽共有相同的序列,但是有任何说明的改变,则它们是“相应的”。适当地,本发明多肽的cdr中的至少一个,例如两个,例如三个精氨酸和/或赖氨酸残基被组氨酸残基取代。适当地,一个精氨酸和/或赖氨酸残基被取代。适当地,取代在至少一个,例如至少两个,例如三个cdr中进行。适当地,在本发明的多肽的所有三个、两个或一个cdr中进行1至3,例如1至2,例如1个取代。适当地,不超过3个,例如不超过2个赖氨酸和/或精氨酸残基被取代。适当地,本发明多肽的cdr1、cdr2和/或cdr3中的每个赖氨酸和/或精氨酸残基已被至少一个,更适当地一个组氨酸残基取代。适当地,包括取代的本发明多肽的每个cdr不短于3,更适当地不短于4,更适当地不短于5,更适当地不短于6,更适当地不短于7,更适当地不小于8,更适当地不短于9,更适当地不短于10,更适当地不短于11,更适当地不短于12,更适当地不短于13个氨基酸。适当地,包括取代的本发明多肽的每个cdr不超过35,更适当地不超过30,更适当地不超过25,更适当地不超过23,更适当地不超过21,更适当地不超过20,更适当地不超过19,更适当地不超过18,更适当地不超过17个氨基酸。适当地,本发明的多肽不超过2000,更适当地不超过1500,更适当地不超过1200,更适当地不超过900,更适当地不超过700,更适当地不超过600,更适当地不超过500,更适当地不超过400,更适当地不超过300,更适当地不超过250,更适当地不超过200,更适当地不超过150个氨基酸。cdr内定义的窗口cdr内的残基可以被认为属于该cdr的特定部分。例如,由15个氨基酸组成的cdr(arnecdqghilkmfp,seqidno:26)可以被认为是由三个三分之一组成:第一个三分之一(由arnec组成的窗口,seqidno:27),第二个三分之一(由dqghi组成的窗口,seqidno:28)和第三个三分之一(由lkmfp组成的窗口,seqidno:29)。类似地,该cdr可被认为由五个五分之一组成:第一个五分之一(由arn组成的窗口),第二个五分之一(由ecd组成的窗口),第三个五分之一(由qgh组成的窗口),第四个五分之一(由ilk组成的窗口)和第五个五分之一(由mfp组成的窗口)。cdr的部分的编号是从n-至c-末端。如果cdr由许多残基组成,从而分成多个部分将导致非整数的残基驻留在每个部分中(例如由arnecdqghilkmfp,seqidno:26,组成的cdr的七分之一),则(a)如果cdr由奇数个残基组成,则中心部分中的残基数(例如第二个三分之一或第三个五分之一等等)被向上舍入至最近的奇数,或者(b)如果cdr由偶数个残基组成,则中心部分中的残基数被向上舍入并至最接近的偶数。例如,由arnecdqghilkmfp组成的cdr的第四个七分之一是由qgh组成的窗口,而由arnecdqg(seqidno:30)组成的cdr的第二个三分之一是由necd(seqidno:31)组成的窗口。适当地,至少一个赖氨酸和/或精氨酸残基存在于窗口中,该窗口定义为cdr1的第二个三分之一和/或cdr2的第二个三分之一和/或cdr3的第二个三分之一和/或cdr1的第三个五分之一和/或cdr2的第三个五分之一和/或cdr3的第三个五分之一和/或cdr1的第四个七分之一和/或cdr2的第四个七分之一和/或cdr3的第四个七分之一。根据具体实施方案,根据本发明的多肽不具有与天然存在的多肽的氨基酸序列完全相同(即,共有100%序列同一性)的氨基酸序列。在一个实施方案中,提供了包含免疫球蛋白链可变结构域的多肽,所述结构域包含三个互补决定区(cdr1-cdr3)和四个框架区,其具有:(a)至少一个组氨酸残基代替cdr1、cdr2和/或cdr3中的至少一个赖氨酸残基,和/或(b)至少一个组氨酸残基代替cdr1、cdr2和/或cdr3中的至少一个精氨酸残基;其中所述多肽相对于不具有所述组氨酸取代的相应祖细胞多肽具有增加的肠稳定性。祖细胞多肽适当地是未经历本文公开的本发明组氨酸取代的多肽。适当地,相应的祖细胞多肽是由动物直接产生的“野生型”多肽(例如抗体),例如通过v(d)j重组和体细胞突变(如美洲驼,如免疫接种后),并且其可以在进行本文公开的本发明的组氨酸取代之前任选地经历其它的合成修饰。特异性、亲和力(affinity)和亲合力(avidity)特异性是指特定抗原结合多肽可以结合的不同类型的抗原或抗原决定簇的数目。抗原结合多肽的特异性是抗原结合多肽将特定抗原识别为独特的分子实体并将其与另一种区分开来的能力。由抗原与抗原结合多肽解离的平衡常数(kd)表示的亲和力是抗原决定簇与抗原结合多肽上的抗原结合位点之间的结合强度的量度:kd值越小,抗原决定簇与抗原结合多肽之间的结合强度越强(或者亲和力也可以表示为亲和常数(ka),它是1/kd)。取决于具体的目的抗原,可以通过已知方法测定亲和力。亲合力是抗原结合多肽与相关抗原之间结合强度的量度。亲合力与抗原决定簇与抗原结合多肽上的抗原结合位点之间的亲和力以及存在于抗原结合多肽上的相关结合位点的数量二者有关。适当地,本发明的多肽以10-6至10-12m的解离常数(kd)与它们的靶标结合,更适当地为10-7至10-12m,更适当地为10-8至10-12m,以及更适当地为10-9至10-12m。任何小于10-6的kd值被认为表示特异性结合。抗原结合多肽与抗原或抗原决定簇的特异性结合可以以任何合适的已知方式测定,包括例如scatchard分析和/或竞争性结合测定法,例如放射免疫测定法(ria),酶免疫测定法(eia)和夹心竞争测定法,以及本领域已知的其不同变型。效力、抑制及中和效力是以产生给定强度的效果所需的量来表示的治疗剂的活性的量度。与在低浓度下引起较小反应的较低效力的活性剂相比,高效活性剂在低浓度下引起更大的反应。效力是亲和力和功效的函数。功效是指治疗剂在与靶配体结合时产生生物反应的能力以及该反应的定量幅度。术语半最大有效浓度(ec50)是指在指定的暴露时间后引起反应到基线和最大值之间的一半时的治疗剂的浓度。治疗剂可引起抑制或刺激。它常常被用作,并且在本文中被用作效力的量度。用于本发明目的的中和多肽是与活性剂(例如tnf-α)结合的多肽,所述多肽抑制抑制该活性剂与一种或多种其同源受体(例如tnfr1和tnfr2)结合,如通过elisa测量的。备选地或另外,用于本发明目的的中和多肽是通过例如抑制活性剂的生物学作用来保护细胞免受活性剂(例如tnf-α)的作用的多肽。例如,为了本发明的目的,中和多肽是通过例如抑制毒素的生物效应来保护细胞免受毒素(例如艰难梭菌毒素a或b-“tcda/tcdb”)的作用的多肽。备选地或另外,用于本发明目的的中和多肽是与il-6r(因此il-6r/il-6复合物)结合、抑制il-6r/il-6复合物结合至gp130的多肽,通过elisa测量。治疗剂的有效性(例如中和能力)可以使用效力测定来确定。特别适当的效力测定是使用alamarblue测量vero细胞活力(fieldsandlancasteramericanbiotechnologylaboratory1993,11(4):48-50)。使用一系列已知浓度的毒素,可以进行该测定以确定治疗性多肽通过产生剂量-反应曲线和/或通过确定该治疗性多肽的半最大有效浓度(ec50)来中和毒素的作用的能力。本文使用此vero细胞细胞毒性标准测定,并在下面的实施例部分进一步详细说明。另一种特别适当的效力测定是标准tnfr2/tnf干扰elisa测定(在下面的实施例部分中进一步详述),其产生剂量-反应曲线和/或通过确定治疗性多肽的半最大有效浓度(ec50),测试关于一定范围的已知浓度的活性剂,治疗剂阻断tnf-α与tnfr2结合中的有效性。另一个特别适当的效力测定是标准gp130elisa测定(在下面的实施例部分中进一步详述),其产生剂量-反应曲线和/或通过确定治疗性多肽的半最大有效浓度(ec50),测试关于一定范围的已知浓度的活性剂,治疗剂阻断与gp130结合的sil-6/il-6r复合物的有效性。适当地,本发明多肽的效力基本上与不具有本发明组氨酸取代的相应多肽的效力相同。适当地,本发明的多肽或本发明方法的多肽在标准tnf/tnfr2干扰elisa测定中抑制结合剂与结合配偶体如tnf-α对tnfr2的结合,ec50为300nm或更小,更适当地为200nm或更小,更适当地为100nm或更小,更适当地为80nm或更小,更适当地为60nm或更小,更适当地为40nm或更小,更适当地为20nm或更小,更适当为10nm或更少,更适当地为5nm或更小。适当地,在标准tnf/tnfr2干扰elisa测定中,例如对于抑制tnf-α与tnfr2的结合,相对于不具有本发明的组氨酸取代的相应多肽,本发明多肽或本发明方法的多肽的ec50增加不超过300pm,更适当地不超过200pm,更适当地不超过100pm,更适当地不超过50pm,更适当地不超过25pm,更适当地不超过10pm,更适当地不超过5pm。适当地,在标准tnf/tnfr2干扰elisa测定中,例如对于抑制tnf-α与tnfr2的结合,相对于不具有本发明的组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽的ec50增加不超过500%,更适当地400%,更适当地300%,更适当地200%,更适当地100%,更适当地70%,更适当地60%,更适当地50%,更适当地40%,更适当地30%,更适当地25%,更适当地20%,更适当地15%,更适当地10%,更适当地5%,更适当地2%,更适当地1%。适当地,在vero细胞细胞毒性标准测定中,本发明的多肽或本发明方法的多肽中和毒素如tcda或tcdb的细胞毒性,ec50为100nm或更小,更适当地为80nm或更小,更适当地为60nm或更小,更适当地为40nm或更小,更适当地为30nm或更小,更适当地为20nm或更小,更适当地为10nm或更小,更适当地为9nm或更小,更适当地为8nm或更小,更适当地为7nm或更小,更适当地为6nm或更小,更适当地为5nm或更小,更适当地为4nm或更小,更适当地为3nm或更小,更适当地为2nm或更小,更适当地为1nm或更小。适当地,在vero细胞毒性标准测定中中和毒素如tcda或tcdb的细胞毒性时,相对于不具有本发明的组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽的ec50增加不多于200nm,更适当地150nm,更适当100nm,更适当地80nm,更适当地60nm,更适当地40nm,更适当地20nm,更适当地10nm,更适当地5nm。适当地,在vero细胞毒性标准测定中,中和毒素(例如tcda或tcdb)的细胞毒性时,相对于不具有本发明的组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽的ec50增加不超过500%,更适当地400%,更适当地300%,更适当地200%,更适当地100%,更适当地70%,更适当地60%,更适当地50%,更适当地40%,更适当地30%,更适当地25%,更适当地20%,更适当地15%,更适当地10%,更适当地5%,更适当地2%,更适当地1%。适当地,在标准gp130elisa测定中,本发明的多肽或本发明方法的多肽抑制结合剂与结合配偶体的结合,例如sil-6/il-6r复合物与gp130结合,ec50为300nm或更小,更适当地200nm或更小,更适当地100nm或更小,更适当地80nm或更小,更适当地60nm或更小,更适当地40nm或更小,更适当地20nm或更小,更适当地10nm或更小,更适当地5nm或更小,更适当地1nm或更小,更适当地0.5nm或更小,更适当地0.3nm或更小,更适当地0.2nm或更小,更适当地0.15nm或更小。适当地,在标准gp130elisa测定中,例如抑制结合剂与结合配偶体的结合,例如sil-6/il-6r复合物与gp130结合,相对于不具有本发明组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽ec50增加不超过300pm,更适当地不超过200pm,更适当地不超过100pm,更适当地不超过80pm,更适当地不超过70pm,更适当地不超过60pm,更适当地不超过50pm,更适当地不超过25pm,更适当地不超过20pm,更适当地不超过15pm,更适当地不超过10pm,更适当地不超过5pm。适当地,在标准gp130elisa测定中,例如sil-6/il-6r复合物与gp130结合,相对于不具有本发明组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽的ec50增加不超过600%,更适当地不超过500%,更适当地400%,更适当地300%,更适当地200%,更适当地100%,更适当地70%,更适当地60%,更适当地50%,更适当地40%,更适当地30%,更适当地25%,更适当地20%,更适当地15%,更适当地10%,更适当地5%,更适当地2%,更适当地1%。可以对多肽进行取代,目的是引入ph敏感性,例如当抗体进入酸性胞内体时,显著降低抗体对抗原的亲和力。然而,本发明的取代适当地不引起实质的ph敏感性。适当地,本发明多肽的取代或本发明方法的多肽的取代不是用于改造靶标结合的ph依赖性。适当地,本发明的多肽或本发明方法的多肽的亲和力在3至9的任何ph,更适当地4至8的任何ph下保持基本相同。胃肠道(git)和消化酶git是负责消耗和消化食物、吸收营养并排出废物的器官系统。在人类和其他哺乳动物中,git由口、食管、胃、小肠(十二指肠、空肠和回肠)和大肠(盲肠、结肠、直肠和肛管)组成。肠道(与胃肠道相反)仅由小肠和大肠组成。各种病原体可能定居,并且各种疾病可能在胃肠道的不同区域显示。胃肠道的不同部位各自含有消化酶的复杂混合物。这些消化酶包括蛋白酶、脂肪酶、淀粉酶和核酸酶。蛋白酶包括丝氨酸蛋白酶、苏氨酸蛋白酶、半胱氨酸蛋白酶、天冬氨酸蛋白酶、谷氨酸蛋白酶和金属蛋白酶。通过分离连接氨基酸残基的肽键(蛋白水解),蛋白酶参与将多肽链消化成较短的片段。一些从蛋白质链分开末端氨基酸(外肽酶);另一些攻击蛋白质的内部肽键(内肽酶)。肠道包括大量不同的蛋白酶。肠道中的蛋白水解可以是高度混杂的,使得广泛的蛋白质底物被存在的广泛的水解酶水解。蛋白酶的情况是这样的:将肠道中大量摄取的多肽阵列切割成较小的多肽片段。适当地,相对于不具有本发明的组氨酸取代的相应多肽,对于本发明的多肽或本发明方法的多肽进行的取代使多肽对存在于小肠或大肠中的一种或多种蛋白酶的稳定性增加。适当地,蛋白酶包括源于肠微生物群或病原菌的蛋白酶,例如其中蛋白酶是细胞膜附着的蛋白酶、分泌的蛋白酶和/或在细胞裂解中释放的蛋白酶。适当地,一种或多种蛋白酶选自胰蛋白酶、糜蛋白酶、宿主炎性蛋白酶、源自微生物群的蛋白酶和源自病原菌的蛋白酶(如艰难梭菌特异性蛋白酶)。适当地,肠道是哺乳动物的肠道,如人、猿、鼠、牛、羊、犬、猫、马或猪肠道。适当地,相对于不具有本发明组氨酸取代的相应多肽,对本发明的多肽进行的取代或对本发明方法的多肽进行的取代使多肽在肠道或在肠道模型中的稳定性增加,例如在小肠和/或大肠中,例如在十二指肠、空肠、回肠盲肠、结肠、直肠和/或肛管中。适当地,肠道模型是标准人粪便上清液肠道模型、标准小鼠小肠上清液肠道模型或标准胰蛋白酶肠道模型。适当地,在标准小鼠小肠上清液肠道模型中孵育6或16小时后,例如当icvd为抗tnf-αicvd时通过标准tnfr2/tnf干扰elisa测定法测定,或者当icvd为抗毒素icvd时通过标准毒素elisa测定,本发明的多肽或本发明方法的多肽中至少20%,更适当地至少25%,更适当地至少30%,更适当地至少35%,更适当地至少40%,更适当地至少50%,更适当地至少60%保持存活的。适当地,例如当icvd为抗tnf-αicvd时通过标准tnfr2/tnf干扰elisa测定法测定,或者当icvd为抗毒素icvd时通过标准毒素elisa测定,在标准小鼠中小肠上清液肠道模型中孵育6或16小时后,相对于不具有本发明组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽的稳定性,增加至少1%,更适当地2%,更适当地3%,更适当地5%,更适当地7%,更适当地10%,更适当地15%,更适当地20%,更适当地30%,更适当地40%,更适当地50%,更适当地60%,更适当地70%。适当地,例如当icvd为抗tnf-αicvd时通过标准tnfr2/tnf干扰elisa测定法、当icvd为抗毒素icvd时通过标准毒素elisa或通过标准western印迹稳定性测定来测定的,在标准人粪便上清液肠道模型中孵育30分钟、1小时、4小时或16小时后,本发明的多肽或本发明方法的多肽中至少20%,更适当地至少25%,更适当地至少30%,更适当地至少35%,更适当地至少40%,更适当地至少50%,更适当地至少60%,更适当地至少70%,更适当地至少80%,更适当地至少90%保持存活的。适当地,例如当icvd为抗tnf-αicvd时通过标准tnfr2/tnf干扰elisa测定法、当icvd为抗毒素icvd时通过标准毒素elisa或通过标准western印迹稳定性测定来测定的,在标准人粪便上清液肠道模型中孵育30分钟、1小时、4小时或16小时后,相对于不具有本发明组氨酸取代的相应多肽,本发明的多肽或本发明方法的多肽的稳定性增加至少1%,更适当地2%,更适当地3%,更适当地5%,更适当地7%,更适当地10%,更适当地15%,更适当地20%,更适当地25%,更适当地30%,更适当地40%,更适当地50%,更适当地60%,更适当地70%。适当地,在标准小鼠小肠上清液肠道模型中孵育4小时后,例如当icvd为抗il-6ricvd时通过标准gp130elisa测定法测定,本发明的多肽或本发明方法的多肽中至少5%,更适当地至少10%,更适当地至少20%,更适当地至少25%,更适当地至少30%,更适当地至少35%,更适当地至少40%,更适当地至少50%,更适当地至少60%保持存活的。适当地,在标准小鼠小肠上清液肠道模型中孵育4小时后,例如当icvd是抗il-6ricvd时,通过标准gp130elisa测定法测定的,相对于不具有本发明组氨酸取代的相应多肽,本发明多肽或本发明方法的多肽的稳定性增加至少1%,更适当地2%,更适当地3%,更适当地5%,更适当地7%,更适当地10%,更适当地15%,更适当地20%,更适当地30%,更适当地40%,更适当地50%,更适当地60%,更适当地70%。适当地,在标准人粪便上清液肠道模型中孵育16小时后,例如当icvd为抗il-6ricvd时,通过标准gp130elisa测定法测定,本发明多肽或本发明方法的多肽中至少20%,更适当地至少25%,更适当地至少30%,更适当地至少35%,更适当地至少40%,更适当地至少50%,更适当地至少60%,更适当地至少70%,更适当地至少80%,更适当地至少90%保持存活的。适当地,在标准人粪便上清液肠道模型中孵育16小时后,当icvd是抗il-6ricvd时,例如通过标准gp130elisa测定法测定的,相对于不具有本发明组氨酸取代的相应多肽,本发明多肽或本发明方法的多肽的稳定性增加至少1%,更多适当地2%,更适当地3%,更适当地5%,更适当地7%,更适当地10%,更适当地15%,更适当地20%,更适当地25%,更适当地30%,更适当地40%,更适当地50%,更适当地为60%,更适当地70%。孵育后剩余的“存活的”icvd的百分比是指完整icvd的比例(例如在标准western印迹稳定性测定法中)或功能性icvd的比例(例如当icvd是抗tnf-αicvd时在标准tnfr2/tnf干扰elisa测定法中或当icvd是抗毒素icvd时在标准毒素elisa测定法中)。或者或另外,孵育后剩余的“存活的”icvd的百分比是指完整icvd的比例(例如在标准western印迹稳定性测定法中)或功能性icvd的比例(例如当icvd是抗il-6ricvd时在标准gp130elisa测定法中)。胃肠道疾病胃肠道的疾病是指涉及胃肠道,即食道、胃、小肠(十二指肠、空肠和回肠)和大肠(盲肠、结肠、直肠和肛管)的疾病。本发明的多肽或本发明方法的多肽可用于治疗或预防这些疾病。适当地,本发明的多肽或本发明方法的多肽用于局部和/或局部治疗或预防这些疾病。下面描述胃肠道的示例性疾病。胃肠道的自身免疫疾病和/或炎性疾病当免疫系统对正常身体组织的反应不利时,自身免疫疾病就会发展。自身免疫疾病可能导致身体组织受损,器官生长异常和/或器官功能变化。该病症可能仅影响一种器官或组织类型,或可影响多种器官和组织。通常受自身免疫病症影响的器官和组织包括血液成分如红血细胞,血管,结缔组织,内分泌腺如甲状腺或胰腺,肌肉,关节和皮肤。炎性疾病是以炎症为特征的疾病。许多炎性疾病是自身免疫疾病,反之亦然。慢性炎症性肠病(ibd)克罗恩氏病和溃疡性结肠炎,其影响儿童和成人,是胃肠道的自身免疫性和炎性疾病的例子(hendrickson等人2002clinmicrobiolrev15(1):79-94,通过整体引用引入本文)。溃疡性结肠炎被定义为其中炎症反应和形态变化保持局限于结肠的病症。95%的患者涉及直肠。炎症在很大程度上局限于粘膜,并且包括沿着结肠长度的溃疡、水肿和出血的可变严重程度的连续涉及(hendrickson等人2002clin.microbiolrev15(1):79-94,通过整体引用并入本文)。溃疡性结肠炎通常表现为与粪便混合的血液和粘液的存在,以及在肠运动过程期间最严重的下腹部痉挛。临床上,血液和粘液腹泻的存在使溃疡性结肠炎与不存在血液的肠易激综合征区分开来。与溃疡性结肠炎不同,克罗恩氏病的呈现通常是微弱的,这导致了延后诊断。诸如涉及的位置、程度和严重性等因素决定了症状的程度。具有回肠结肠涉及的患者通常患有餐后腹痛、右下部压痛和偶发性炎性包块。适当地,本发明的药物组合物或构建体用于治疗胃肠道的自身免疫性和/或炎性疾病,适当地其选自克罗恩氏病、溃疡性结肠炎、肠易激综合征、ii型糖尿病、肾小球性肾炎、自身免疫性肝炎、干燥综合征、乳糜泻和药物或辐射诱导的粘膜炎(最适当地是克罗恩氏病)。胃肠道的感染病毒、细菌、寄生虫和其他致病性感染可发生在胃肠道中。这些可能限于胃肠道或起始于胃肠道然后传播到身体其他部位。本发明的多肽可以用于治疗或预防细菌感染,包括常见的细菌性胃肠道病原体感染,该病原体包括大肠杆菌,沙门氏菌属(salmonella),弯曲杆菌属(campylobacter),霍乱弧菌(vibriocholerae),志贺氏菌属(shigella),产气荚膜梭菌(clostridiumperfringens),艰难梭菌(clostridiumdifficile),蜡状芽孢杆菌(bacilluscereus),副溶血弧菌(vibrioparahaemolyticus)和小肠结肠炎耶尔森氏菌(yersiniaenerocolitica)。本发明的多肽可用于治疗或预防病毒感染,包括常见病毒胃肠道病原体,包括轮状病毒,诺如病毒和小圆形病毒(smallroundviruses)。适当地,本发明的多肽用于治疗或预防医院内感染。适当地,本发明的多肽用于治疗或预防艰难梭菌感染。适当地,本发明的多肽与经由肠道可及的靶标结合(例如肠道内的靶标)。适当地,靶标是源自肠道定居病原微生物的有害剂。适当地,靶标是可诱导发病机制的源自宿主微生物群的靶标,宿主细胞,宿主衍生的炎性介质或参与疾病发病机制的蛋白质。适当地,靶标选自:tnf-α、艰难梭菌毒素a、或艰难梭菌毒素b。备选地,靶标选自il-6r、tnf-α、艰难梭菌毒素a或艰难梭菌毒素b。接头和多聚体根据本发明的构建体包含多个多肽,并因此可以适当地是多价的。这样的构建体可以包含至少两个根据本发明的相同多肽。由根据本发明的两个相同多肽组成的构建体是“同型双头(homobihead)”。在本发明的一个方面,提供了包含本发明多肽的构建体。在又一个方面,提供了包含两个或多个(可能相同的)本发明多肽的构建体。或者,构建体可以包含至少两个不同的但仍都是根据本发明多肽的多肽(“异型双头”)。或者,这样的构建体可以包含(a)根据本发明的至少一个多肽和(b)不是本发明多肽的至少一个多肽,例如抗体或其抗原结合片段,(也是“异型双头”)。(b)的至少一个多肽可以结合tnf-α、tcda或tcdb(例如通过与(a)不同的表位),或者可以一起结合另一靶标。适当地,不同的多肽(b)结合例如:另一种促炎细胞因子或趋化因子或其各自的受体,参与人类病理过程的其它炎症介质或免疫相关的配体。构建体可以是多价和/或多特异性的。多价构建体(例如二价构建体)包含两个或更多个结合多肽,因此呈现两个或更多个可以发生与一种或多种抗原附着的位点。多价构建体的例子可以是同型双头或异型双头。多特异性构建体(例如双特异性构建体)包含两个或更多个不同的结合多肽,其呈现两个或更多个位点,在该位点(a)可以发生向两种或更多种不同抗原的附着或(b)可以发生在相同抗原上的两个或更多个不同的表位的附着。多特异性构建体的例子可以是异型双头。多特异性构建体是多价的。适当地,包含在构建体内的多肽是抗体片段。更适当地,构建体中包含的多肽选自由vhh、vh、vl、v-nar、scfv、fab片段或f(ab')2片段组成的列表。更适当地,构建体内包含的多肽是vhh。本发明的多肽可以直接(即不使用接头)或通过接头彼此连接。接头适当地是多肽,并且将被选择以允许多肽与其表位的结合。如果用于治疗目的,在施用多肽的受试者中,接头适当地是非免疫原性的。适当地,多肽均通过接头连接。适当地,接头为(g4s)x形式。最适当地,x是6。治疗用途和递送适当地,本发明的多肽通用作过口服施用的药物,适当地,用于治疗或预防胃肠道疾病(参见上文)。本发明的多肽或本发明方法的多肽也可以用于通过口服施用来治疗或预防其它医学病症例如代谢紊乱如肥胖。在一个实施方案中,本发明的多肽旨在在肠道中具有局部作用。在一个实施方案中,本发明的多肽或本发明方法的多肽不用于通过以治疗有效量递送至循环来治疗或预防疾病。在本发明的一个方面,提供了一种治疗胃肠道疾病的方法,包括向有需要的人施用治疗有效量的本发明的多肽。本发明的多肽的治疗有效量是在对受试者单剂量或多剂量施用时,在受试者中中和所选靶标的生物学作用至显著程度的有效的量。治疗有效量可以根据诸如个体的疾病状态、年龄、性别和体重等因素以及多肽在个体中引起所需反应的能力而变化。治疗有效量还是其中多肽的治疗有益效果优于任何毒性或不利影响的量。本发明的多肽可掺入到适合口服施用于受试者的药物组合物中。本发明的多肽可以是药学上可接受的盐的形式。在本发明的一个方面,提供了包含本发明的多肽和一种或多种药学上可接受的稀释剂或运载体的药物组合物。本发明的药物组合物可以配制用于口服递送。本发明的药物组合物可以是各种形式。这些包括例如液体、半固体和固体剂型,例如液体溶液,分散体或悬浮液,片剂,丸剂和散剂。固体剂型是优选的。药物组合物可以包含药学上可接受的赋形剂,以及适当地以可摄入片剂,口腔片剂,锭剂,胶囊剂,酏剂,悬浮液,糖浆剂,膜片(wafer)等的形式使用。通常,本发明的组合物或本发明的药物组合物包括本发明的多肽以及药学上可接受的赋形剂例如运载体。药学上可接受的运载体的例子包括水、盐水、磷酸缓冲盐溶液、右旋糖、甘油、乙醇等中的一种或多种,以及它们的组合。药学上可接受的运载体还可以包含少量辅助物质,例如润湿剂或乳化剂,防腐剂或缓冲剂,其增强本发明多肽的保质期或有效性。药物组合物可以包括抗粘附剂,粘合剂,包衣,崩解剂,香料,颜料,润滑剂,吸附剂,防腐剂,甜味剂,冷冻干燥赋形剂(包括冻干保护剂)或压缩助剂。适当地,本发明的多肽在掺入到药物组合物之前被冻干。本发明的多肽还可以用肠包衣提供。肠包衣是应用于口服药物的聚合物屏障,其保护多肽免受胃的低ph。用于肠包衣的材料包括脂肪酸、蜡、紫胶、塑料和植物纤维。合适的肠包衣组分包括丙烯酸甲酯-甲基丙烯酸共聚物,乙酸琥珀酸纤维素,邻苯二甲酸羟丙基甲基纤维素,醋酸琥珀酸羟丙基甲基纤维素(醋酸琥珀酸羟丙甲纤维素),聚乙烯醋酸邻苯二甲酸酯(pvap),甲基丙烯酸甲酯-甲基丙烯酸共聚物,藻酸钠和硬脂酸。合适的肠包衣包括ph依赖性释放聚合物。这些是在胃中发现的高酸性ph下不溶,但是其在较小酸性ph下快速溶解的聚合物。因此,适当地,肠包衣将不会在酸性胃液(ph~3)中溶解,但是将在小肠中(ph高于6)或结肠中(ph高于7.0)存在的较高ph环境下溶解。选择ph依赖性释放聚合物,使得本发明的多肽大概在剂量达到肠道的靶标区域时被释放。本发明的组合物可以在缓冲液中配制,以便将组合物的ph稳定在5-50或更适当地15-40或更适当地25-30g/升的浓度。适当的缓冲组分的例子包括生理盐,例如柠檬酸钠和/或柠檬酸。适当的缓冲液含有100-200,更适当地125-175mm的生理盐如氯化钠。适当地选择缓冲液使pka接近组合物的ph或患者的生理ph。药物组合物中的示例性多肽浓度可以为约10ng/ml至约200mg/ml的范围,例如约50ng/ml至约100mg/ml,例如约1μg/ml至约80mg/ml,例如约10μg/ml至约50mg/ml,例如约50μg/ml至约30mg/ml,例如约100μg/ml至约20mg/ml,或约1mg/ml至约200mg/ml,或约50mg/ml至约200mg/ml,或约150mg/ml至约200mg/ml。本发明的多肽的水性制剂可以在ph缓冲溶液中制备,例如ph为约4.0至约7.0,或约5.0至约6.0,或可选地约5.5。适当的缓冲液的例子包括磷酸盐,组氨酸,柠檬酸盐,琥珀酸盐,乙酸盐缓冲液和其它有机酸缓冲液。缓冲液浓度可以为约1mm至约100mm,或约5mm至约50mm,这取决于例如缓冲液和制剂所需的张力。药物组合物的张力可以通过包括张力调节剂来改变。这种张力调节剂可以是带电或不带电的化学物质。典型的不带电张力调节剂包括糖或糖醇或其它多元醇,优选海藻糖,蔗糖,甘露醇,甘油,1,2-丙二醇,棉子糖,山梨糖醇或乳糖醇(特别是海藻糖,甘露醇,甘油或1,2-丙二醇)。典型的带电张力调节剂包括盐,诸如钠、钾或钙离子与氯化物,硫酸盐,碳酸盐,亚硫酸盐,硝酸盐,乳酸盐,琥珀酸盐,乙酸盐或马来酸盐离子的组合(特别是氯化钠或硫酸钠);或氨基酸如精氨酸或组氨酸。适当地,水性制剂是等渗的,尽管高渗或低渗溶液可能是适当的。术语“等渗”表示与其比较的其它溶液(例如生理盐溶液或血清)具有相同张力的溶液。张力剂可以以约5mm至约350mm的量使用,例如以1mm至500nm的量使用。适当地,组合物中包含至少一种等渗剂。还可以向药物组合物中加入表面活性剂以减少配制的多肽的聚集和/或使制剂中的微粒的形成最小化和/或减少吸附。示例性的表面活性剂包括聚氧乙烯山梨坦-脂肪酸酯(tween),聚氧乙烯烷基醚(brij),烷基苯基聚氧乙烯醚(triton-x),聚氧乙烯-聚氧丙烯共聚物(泊洛沙姆,pluronic)和十二烷基硫酸钠(sds)。合适的聚氧乙烯山梨坦-脂肪酸酯的例子是聚山梨醇酯20和聚山梨醇酯80。表面活性剂的示例性浓度可以在约0.001%至约10%w/v的范围内。为了保护本发明的多肽免受冻干过程中的不稳定条件的影响,还可加入冻干保护剂。例如,已知的冻干保护剂包括糖(包括葡萄糖、蔗糖、甘露糖和海藻糖);多元醇(包括甘露醇、山梨醇和甘油);和氨基酸(包括丙氨酸、甘氨酸和谷氨酸)。冻干保护剂可以以约10mm至500mm的量被包括。本发明的药物组合物的施用剂量范围,是产生所需治疗效果的剂量范围。所需的剂量范围取决于药物组合物的准确性质,肠道的靶标区域,制剂的性质,患者的年龄,患者状况的性质、程度或严重程度,禁忌症,如果有的话,以及主治医师的判断。可以使用标准经验程序调整这些剂量水平的变化以进行优化。本发明的多肽的肠稳定性的增加意味着可以口服递送更低剂量,相比在不具有本发明的组氨酸取代的相应多肽的情况下,本来是需要口服递送的那些。适当地,本发明的多肽或本发明的药物组合物的日剂量在50ng-50mg/kg,例如50μg-40mg/kg,例如5-30mg/kg(例如人)体重的范围内,例如小于25,例如小于20,例如小于15,例如小于10mg,如小于50μg,例如小于50ng/kg体重。单位剂量通常将在每剂量250-2000mg的范围内,例如小于1000mg,例如小于700mg,例如小于400mg,例如小于100mg,例如小于100μg,例如小于50μg,例如小于10μg,例如小于100ng,例如小于50ng。剂量可以每天或更频繁地施用,例如每天2或3次或4次或更少频率,例如每隔一天、每周一次、每两周一次或每月一次。疾病的治疗还包括治疗其恶化,以及还包括治疗患者的疾病症状缓解,以预防疾病症状的复发。联合治疗本发明的药物组合物还可以包含一种或多种活性剂(例如适用于治疗疾病如本文提及的疾病的活性剂)。将本发明的药物组合物用于治疗细菌感染、自身免疫和/或炎性疾病的治疗方法在本发明的范围内,作为通常用于治疗细菌感染、自身免疫和/或炎性疾病的其它已有疗法的辅助物或与其结合使用。对于炎性肠病(例如克罗恩氏病或溃疡性结肠炎)的治疗,可能的组合包括与例如选自以下列表的一种或多种活性剂的组合:包括:5-氨基水杨酸或其前药(如柳氮磺吡啶、奥沙拉嗪或bisalazide);皮质类固醇(例如泼尼松龙、甲泼尼龙或布地奈德);免疫抑制剂(例如环孢菌素,他克莫司,甲氨蝶呤,硫唑嘌呤或6-巯基嘌呤);抗tnf-α抗体(例如,英夫利昔单抗,阿达木单抗,certolizumabpegol或戈利木单抗);抗il12/il23抗体(例如优特克诺单抗);抗il-6r抗体或小分子il12/il23抑制剂(例如阿匹莫德);抗α-4-β-7抗体(例如,vedolizumab);madcam-1阻断剂(例如pf-00547659);针对细胞粘附分子α-4-整联蛋白的抗体(例如那他珠单抗);针对il2受体α亚基的抗体(例如,达克珠单抗或巴利昔单抗);jak3抑制剂(例如托法替尼或r348);syk抑制剂及其前药物(例如,fostamatinib和r-406);磷酸二酯酶-4抑制剂(如替托司特);hmpl-004;益生菌;dersalazine;塞马莫德/cpsi-2364;和蛋白激酶c抑制剂(例如aeb-071)。最适当的组合剂是英夫利昔单抗、阿达木单抗、certolizumabpegol或戈利木单抗。对于诸如艰难梭菌感染的细菌感染的治疗,可能的组合包括与例如选自以下列表的一种或多种活性剂的组合,所述列表包括艰难梭菌类毒素疫苗,氨苄青霉素,阿莫西林,万古霉素,甲硝唑,fidaxomicin,利奈唑胺,硝唑沙奈,利福昔明,雷莫拉宁,二亚胺嘧啶(difimicin),克林霉素,头孢菌素(如第二代和第三代头孢菌素),氟喹诺酮类(如加替沙星或莫西沙星),大环内酯类(如红霉素,克拉霉素,阿奇霉素),青霉素,氨基糖苷类,甲氧苄啶-磺胺甲唑,氯霉素,四环素,亚胺培南,美罗培南,抗菌剂,杀菌剂或抑菌剂。可能的组合还包括与一种或多种活性剂(是益生菌)的组合,例如布鲁氏酵母(saccharomycesboulardii)或鼠李糖乳杆菌gg。因此,本发明的另一方面提供了本发明的药物组合物与一种或多种其它活性剂,例如上述一种或多种活性剂的组合。在本发明的另一方面,药物组合物或多肽与选自上述列表中的至少一种活性剂顺序、同时或分开地施用。类似地,本发明的另一方面提供一种组合产品,包括:(a)本发明的药物组合物;和(b)一种或多种其它活性剂,其中组分(a)和(b)中的每一种与药学上可接受的佐剂,稀释剂或运载体混合配制。在本发明的这个方面,组合产品可以是单一(组合)制剂或套盒。因此,本发明的该方面包括与药学上可接受的佐剂、稀释剂或运载体混合的、包括了本发明的药物组合物和另一种治疗剂的组合制剂。本发明还包括套盒,包括组分:(i)与药学上可接受的佐剂、稀释剂或运载体混合的本发明的药物组合物;和(ii)与药学上可接受的佐剂、稀释剂或运载体混合的、包含一种或多种其它活性剂的制剂,其中组分(i)和(ii)各自以适于与另一种结合的施用的形式提供。因此,套盒的组分(i)是与药学上可接受的佐剂、稀释剂或运载体混合的上述组分(a)。类似地,组分(ii)是与药学上可接受的佐剂、稀释剂或运载体的混合的上述组分(b)。一种或多种其它活性剂(即上述组分(b))可以是例如上述关于治疗细菌感染例如艰难梭菌感染、自身免疫和/或炎性疾病如ibd(例如克罗恩氏病和/或溃疡性结肠炎)的任何活性剂。如果组分(b)是多于一种其它活性剂,则这些其它活性剂可以彼此配制或与组分(a)配制,或者它们可以分开配制。在一个实施方案中,组分(b)是另一种治疗剂。在另一个实施方案中,组分(b)是另外两种治疗剂。本发明该方面的组合产品(组合制品或套盒)可用于治疗或预防自身免疫疾病(例如本文提及的自身免疫疾病)。载体和宿主本文所用的术语“载体”是指能够转运与其连接的另一种核酸的核酸分子。一种类型的载体是“质粒”,其是指可以连接额外的dna区段的环状双链dna环。另一种类型的载体是病毒载体,其中额外的dna区段可以连接到病毒基因组中。某些载体能够在其被引入的宿主细胞中自主复制(例如,具有细菌复制起点的细菌载体和附加型(episomal)哺乳动物和酵母载体)。其它载体(例如非附加型哺乳动物载体)可以在引入宿主细胞后整合到宿主细胞的基因组中,从而与宿主基因组一起复制。此外,某些载体能够引导与它们有效连接的基因的表达。这些载体在本文中称为“重组表达载体”(或简称为“表达载体”)。通常,用于重组dna技术的表达载体通常是质粒的形式。在本说明书中,“质粒”和“载体”可以互换使用,因为质粒是最常用的载体形式。然而,本发明旨在包括这样的其它形式的表达载体,例如用于相同功能的病毒载体(例如复制缺陷型逆转录病毒,腺病毒和腺伴随病毒),以及噬菌体和噬菌粒系统。本发明还涉及编码本发明多肽的核苷酸序列。本文所用的术语“重组宿主细胞”(或简称“宿主细胞”)旨在表示引入了重组表达载体的细胞。这些术语旨在不仅指特定的受试细胞,而且指这种细胞的后代。在本发明的一个方面,提供了编码本发明多肽的多核苷酸。在本发明的又一方面,提供了包含所述多核苷酸或含有所述多核苷酸的cdna的载体。在本发明的另一方面,提供了用所述载体转化的宿主细胞,其能够表达本发明的多肽。适当地,宿主细胞是哺乳动物细胞、植物细胞、酵母细胞,如属于曲霉属、酵母属、克鲁维酵母属、汉逊酵母属或毕赤酵母属的酵母细胞,如酿酒酵母或巴斯德毕赤酵母;或细菌细胞如大肠杆菌。制备方法可以使用例如green和sambrook2012molecμlarcloning:alaboratorymanual第四版coldspringharborlaboratorypress中公开的技术获得并操作本发明的多肽。适当地,对本发明的多肽进行的取代或在本发明的方法中进行的取代是通过合成引入。适当地,不是通过v(d)j重组或体细胞突变引入取代。特别地,可以使用人造基因合成(nambiar等人1984science223:1299-1301,sakamar和khorana1988nucl.acidsres14:6361-6372,wells等人1985gene34:315-323和grundstrom等人1985nucl.acidsres13:3305-3316,通过整体引用并入本文)。编码本发明多肽的基因可以通过例如固相dna合成来合成产生。整个基因可以从头合成,而不需要前体模板dna。为了获得所需的寡核苷酸,构建单元按照产物序列所要求的顺序依次偶联到生长的寡核苷酸链上。在链装配完成后,将产物从固相中释放到溶液中,去保护并收集。可以通过高效液相色谱(hplc)分离产物以获得高纯度的所需寡核苷酸(verma和eckstein1998annurevbiochem67:99-134)。本发明的构建体可以在dna水平上遗传融合,即编码包含一个或多个多肽的完整构建体的多核苷酸构建体。通过遗传途径连接多个多肽的一种方法是通过经不稳定肽接头编码序列来连接多肽编码序列。例如,第一多肽的羧基末端可以通过不稳定肽接头编码序列连接到下一个多肽的氨基末端。可以扩展这种连接模式以连接多肽用于构建三、四、等等、功能构建体。在wo96/34103中公开了多价(如二价)vhh多肽构建体的制备方法(通过整体引用并入本文)。可以对编码多肽的dna或cdna进行突变,所述突变对于多肽的氨基酸序列是沉默的,但是为在特定宿主中翻译提供优选的密码子。在例如大肠杆菌和酿酒酵母中翻译核酸的优选密码子是已知的。可以例如通过对编码多肽的核酸的取代、添加或缺失来实现多肽的突变。取代是在相同、相应位置用不同残基对残基的替换。可以通过许多合成方法引入对编码多肽的核酸的取代、添加或缺失,包括例如易错pcr,改组,寡核苷酸定向诱变,装配pcr,pcr诱变,体内诱变,盒诱变,递归整体诱变,指数整体诱变,位点特异性诱变(ling等人1997analbiochem254(2):157-178,通过整体引用并入本文),基因重装配,基因位点饱和诱变(gssm),合成连接重装配slr)或这些方法的组合。还可以通过包括重组,递归序列重组,磷酸硫酸酯修饰的dna诱变,含有尿嘧啶的模板诱变,间隙双链突变,点错配修复诱变,修复缺陷型宿主菌株诱变,化学诱变,辐射诱变,缺失诱变,限制性选择诱变,限制性纯化诱变,整体诱变,嵌合核酸多聚体产生或其组合的方法引入对核酸的修饰、添加或缺失。包含免疫球蛋白链可变结构域如vh和vhh的多肽的表达可以使用适当的表达载体例如原核细胞如细菌例如大肠杆菌(例如根据wo94/04678和wo96/34103中公开的方案,其通过整体引用并入本文)来实现。免疫球蛋白链可变结构域如vh和vhh的表达也可以使用真核细胞,例如昆虫细胞,cho细胞,vero细胞或适当的酵母细胞如属于曲霉属、酵母属、克鲁维酵母属、汉逊酵母属或毕赤酵母属的酵母来实现。适当地使用酿酒酵母(例如根据wo94/025591中公开的方案,其通过引用并入本文)。适当地,本发明的多肽可以在真菌例如酵母(例如酿酒酵母)中生产,包括根据wo02/48382中公开的方法,在包含碳源的培养基上生长真菌,其中50-100wt%的所述碳源是乙醇。条款定义本发明及其优选方面的一组条款如下:1.一种包含免疫球蛋白链可变结构域的多肽,所述可变结构域包含三个互补决定区(cdr1-cdr3)和四个框架区,其中:(a)cdr1、cdr2和/或cdr3中的至少一个赖氨酸残基已被至少一个组氨酸残基取代,和/或(b)cdr1、cdr2和/或cdr3中的至少一个精氨酸残基已被至少一个组氨酸残基取代;其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。2.一种增加包含免疫球蛋白链可变结构域的多肽的肠稳定性的方法,其中所述免疫球蛋白链可变结构域包含三个互补决定区(cdr1-cdr3)和四个框架区,其中所述方法包括以下步骤:(a)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个赖氨酸残基,和/或(b)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个的精氨酸残基。3.一种制备包含免疫球蛋白链可变结构域的多肽的方法,其中所述免疫球蛋白链可变结构域包含三个互补决定区(cdr1-cdr3)和四个框架区,其中所述方法包括以下步骤:(a)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个赖氨酸残基,和/或(b)用至少一个组氨酸残基取代cdr1、cdr2和/或cdr3中的至少一个精氨酸残基其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。4.根据条款1至3中任一项的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中相对于不具有所述组氨酸取代的相应多肽,所述取代增加了所述多肽在肠道中的稳定性,例如在小肠和/或大肠中,例如在十二指肠、空肠、回肠盲肠、结肠、直肠和/或肛管中。5.根据条款1至4中任一项的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中相对于不具有所述组氨酸取代的相应多肽,所述取代增加了多肽在肠道模型中的稳定性,例如在小肠和/或大肠中,例如在十二指肠、空肠、回肠盲肠、结肠、直肠和/或肛管中。6.根据条款5的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中肠道模型是标准人粪便上清液肠道模型。7.根据条款6的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中在标准人粪便上清液肠道模型中孵育16小时后,当免疫球蛋白链可变结构域为抗tnf-α免疫球蛋白链可变结构域时,通过标准tnfr2/tnf干扰elisa测定法,或者当免疫球蛋白链可变结构域是抗il-6r免疫球蛋白链可变结构域时,通过标准gp130elisa测定法测定的多肽的稳定性相对于不具有所述组氨酸取代的相应多肽,增加至少1%,更适当5%,更适当地10%。8.根据条款1至7中任一项的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中相对于不具有所述组氨酸取代的相应多肽,所述取代增加多肽对在小肠或大肠中产生的一种或多种蛋白酶的稳定性。9.根据条款1至8中任一项的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中多肽的效力与不具有所述组氨酸取代的相应多肽的效力基本相同。10.根据条款1至9中任一项的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中所述至少一个赖氨酸和/或精氨酸残基存在于定义为cdr1的第二个三分之一和/或cdr2的第二个三分之一和/或cdr3的第二个三分之一。11.根据条款11的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中cdr1、cdr2和/或cdr3中的每个赖氨酸和/或精氨酸残基各自被一个组氨酸残基取代。12.根据条款1至12中任一项的多肽、增加多肽的肠稳定性的方法或制备多肽的方法,其中所述多肽是抗体,含有额外的抗体结合区的修饰抗体或抗体片段如scfv、fab片段、f(ab')2片段或免疫球蛋白链可变结构域如vhh、vh、vl、v-nar。13.根据条款1至12条中任一项的多肽,增加多肽的肠稳定性的方法或制备多肽的方法,其中所述多肽与经由肠道可及的靶标结合。14.一种包含根据条款1至13项中任一项的多肽或构建体的药物组合物,用作口服施用的药物。15.根据条款14的药物组合物,其中所述组合物以肠包衣形式存在。其他条款定义本发明及其优选方面的一组其他条款如下。如下所述的根据权利要求4至61所述的特征任选地作必要修改的适用于这些其他条款1至3。1.一种多肽,包含能够以高亲和力结合靶标的区域,其中:(a)该区域中至少一个赖氨酸残基已被至少一个组氨酸残基取代,和/或(b)该区域中至少一个精氨酸残基已被至少一个组氨酸残基取代;其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。2.一种增加包含能够以高亲和力结合靶标的区域的多肽的肠稳定性的方法,其中所述方法包括以下步骤:(a)用至少一个组氨酸残基取代该区域中至少一个赖氨酸残基,和/或(b)用至少一个组氨酸残基取代该区域中至少一个精氨酸残基。3.一种制备包含能够以高亲和力结合靶标的区域的多肽的方法,其中所述方法包括以下步骤:(a)用至少一个组氨酸残基取代该区域中至少一个赖氨酸残基,和/或(b)用至少一个组氨酸残基取代该区域中至少一个精氨酸残基。其中相对于不具有所述组氨酸取代的相应多肽,所述多肽具有增加的肠稳定性。现在将通过以下非限制性实施例进一步描述本发明。实施例实施例1:标准肠道模型、标准肠稳定性测定法和标准效力测定法可以使用以下方法测定包含免疫球蛋白链可变结构域的多肽的肠稳定性和效力。以下方法是指icvd,但同样适用于包含icvd的任何多肽,例如抗体。1.1标准肠道模型来自人粪便和小鼠小肠样品的离体样品是评估人肠道稳定性的高度相关的基质。这些样品含有天然宿主产生的和相关微生物产生的蛋白酶以及可能在蛋白酶存在下影响icvd稳定性的任何离液剂或表面活性剂。存在于小鼠和人类源的小肠中的至少一些蛋白酶的酶切位点被充分表征并且在两种物种之间是保守的。通过比较来自猪的小肠样品与临床来源的人类小肠的灌洗样品,发现小鼠小肠上清液就总蛋白酶活性而言是特别严格的挑战。下文详述了肠道模型,其利用来自人类粪便和小鼠小肠的离体样品,因此允许测定在高度代表肠道状况的环境中包含icvd的多肽的稳定性。在肠道模型中使用适当的测定法(例如标准western印迹稳定性测定法(用于测定完整icvd的比例))或标准tnfr2/tnf干扰elisa测定法或标准毒素elisa测定法(两者都用于测定功能icvd的比例)来评估孵育后剩余的存活icvd的百分比。需指出,从小鼠或人类采样点到在icvd稳定性测定中使用点,所有粪便/肠样品,浆液和上清液应保持在冰上冷冻或操作,例如在4℃下进行离心。一旦产生,上清液样品可以在-80℃下冷冻并在使用前解冻一次(或两次)。重复冻融可能导致蛋白酶稳定性的丧失。在-80℃下长时间存储(>1年)似乎不会降低总蛋白酶活性。然而,随着时间的推移,应根据具体情况监测浆液和上清液。1.1.1标准人粪便上清液肠道模型粪便上清液池产生为了产生用于稳定性测试的上清液,将1xpbs以每克粪便1或2ml的1xpbs的比例加入到粪便样品中。然后将样品涡旋至均匀。所得到的材料被称为粪便浆液(在以下实施例中使用的非常有限数量的特别牢固的样品的情况下,必须向每克粪便中加入3ml的1xpbs以产生均匀的粪便浆液)。为了产生用于测试的上清液,将浆液以4.5krpm或13.5krpm(4℃)离心1-5分钟以除去大部分固体材料和所有细胞材料。然后将第一次旋转的上清液以13.5krpm(4℃)重新离心5分钟,仅留下可溶性级分,包括蛋白酶。将来自多个个体的上清液集合在一起,使得每个池代表来自多个个体的粪便的组合的蛋白酶输出。为了下面的实施例的目的,获得了医院来源的人类粪便样品(并且确定了样品中存在艰难梭菌),然后如上所述产生上清液池。池的特征如表1所示。表1进行测定通过向5ml蛋白酶终止缓冲液(1×pbs,2%bsa,5mmedta)中加入1片sigmafast蛋白酶抑制剂混合物(sigmas8830,含有aebsf(4-(2-氨基乙基)苯磺酰氟,bestatin,e-64,pepstatina,phosphoramidon,leupeptin,aprotinin)制备20×蛋白酶抑制剂溶液。该溶液可以在2-8℃下存储2周。在测定当天,短暂地涡旋上清液基质以确保均匀性。在冰上进行反应并保持冷冻,直到首次孵育测定。通过在蛋白酶终止缓冲液中稀释20×蛋白酶抑制剂溶液并在2x终止缓冲液(0.1m溶液sigma93482的1/100稀释液)中加入pmsf至浓度为1mm来制备2x蛋白酶终止液。使用前始终将此溶液在冰上冷却。在0.1%bsa中制备250μg/ml的icvd(或抗体)溶液。在冰上,在薄壁pcr管或板中,将250μg/ml的icvd稀释到上清液基质中,得到20μg/ml的最终icvd浓度(在时间零时)。通过移液将所得溶液在冰上混合,确保溶液不升温。一旦均匀,立即取出一体积的样品基质加20μg/ml的icvd,并与等体积的2x蛋白酶终止溶液混合。将终止的基质溶液在冰上混合,立即在-80℃冷冻。这是时间零样品。在pcr仪器或类似设备中,在37℃下孵育剩余的测试基质样品加20μg/ml的icvd。在所需的时间点,重复上述步骤以产生终止的上清液样品,用于与时间零样品进行比较。此外,通过加入来自时间零的一体积的基质样品(不含icvd)以及等体积的2x蛋白酶终止溶液,生成不含icvd的蛋白酶终止的基质对照。这将用作评估基质对例如elisa或western印迹谱图的影响的下游分析中的对照。使用标准western印迹稳定性测定法,标准tnfr2/tnf干扰elisa测定法或标准毒素elisa测定法进行测量后,将在给定时间点在基质样品中孵育后剩余的存活icvd量除以在零时间点存在的量。然后将所得到的数乘以100以给出稳定性%。在标准western印迹稳定性测定法的情况下,这提供了完整icvd的比例。在标准tnfr2/tnf干扰elisa测定法或标准毒素elisa测定法的情况下,这提供了功能icvd的比例。1.1.2标准小鼠小肠上清液肠道模型粪便上清液池产生处死c57bl/6('黑6')小鼠。从小鼠的体腔小心地排出包括十二指肠、空肠和回肠在内的小肠,以最小化不必要的组织损伤。收集小肠的固体内容物,用1ml0.9%盐水冲洗小肠内表面(以保持肠内容物的天然ph值)。然后将1ml肠清洗溶液和肠内容物样品混合在一起,通过涡旋充分匀浆以产生小肠浆液。为了产生用于测试的上清液,将浆液以13.5krpm(4℃)离心2分钟以除去大部分固体材料和所有细胞材料。然后将第一次旋转的上清液以13.5krpm(4℃)重新离心5分钟,仅留下可溶性级分,包括蛋白酶。将来自多只小鼠(平均每池5只)的上清液混合在一起,使得每个池代表来自多只小鼠的小肠的组合的蛋白酶输出。在下面的实施例中,发现使用的小鼠小肠上清液不同的池随着时间的推移表现出类似的蛋白水解活性。进行测定以与上述标准人粪便上清液肠道模型的“进行测定”下相同的方式使用上清液。1.2标准western印迹稳定性测定法用于评估在肠道模型中孵育后剩余的存活icvd的百分比用于sds-page的样品制备(还原条件下):1)制备用于还原sds-page的样品缓冲液:将还原剂0.5m二硫苏糖醇(dtt)(novexnp0004)以1:9的比例加入到novex4xlds样品缓冲液(np0007)中。例如,将10μl的0.5mdtt加入到90μl的4x样品缓冲液中。所得到的溶液将从这一点向前称为“4x上样染料”。2)可以将4x上样染料储备液以1:3用无菌h2o稀释来制备1x上样染料。3)从时间0或30分钟,将15μl的每个在消化基质中含有icvd的实验样品,加入到5μl的4x上样染料中。目的是从终止的零时间点加载100-200ngicvd的最终量。将来自30分钟时间点的样品体积与为了零时间点加入的体积进行匹配,以便使icvd随着时间的任何损失/降解都将在最终印迹上通过眼睛显见(同样适用于其他时间点,如15分钟,1小时,2小时等,也可以使用)。如果可能,包括测试icvd的未经处理的标准(100和10ng),以确认转移和检测系统正确执行。4)将含有icvd的所有样品加热至95℃持续5-10分钟(均匀处理所有样品),使蛋白质变性,并用上样染料中存在的lds包被它们。使样品冷却,在离心机中短暂旋转,收集所有的液体。5)准备可以在印迹后显现的合适的参照梯度(supersignalmw蛋白梯度(pierce))。加入6.5μl蛋白梯度+13μl的1x上样染料。需指出,在凝胶加载之前,不需要加热参照梯度(参见供应商说明)。电泳使用novex10%bis-tris凝胶(np0302box)与1xsds-mes运行缓冲液(novexnp0002-02)组合,通过sds-page可视化icvd。1)制备1xsds-mes溶液(来自novexnp0002-02,20x储备液),并在适当的电泳槽中安装novex10%bis-tris凝胶。2)使用凝胶加样移液管吸头,在每个凝胶泳道上加载15μl上面制备的样品。3)在200v下运行凝胶直到染料前端到达凝胶边缘,而不再前进。印迹1)电泳后,使用iblot半干转移装置(invitrogen,7分钟半干转移程序3)将蛋白质转移到硝酸纤维素膜(ib3010,invitrogen)上。2)在室温下通过用25ml封闭溶液(1%bsa,2%marvel,0.05%tween20,1xpbsph7.4)轻轻振荡孵育2小时来封闭膜。3)对于主要检测抗体,在封闭溶液(1%bsa,2%marvel,0.05%tween20,1xpbsph7.4)中制备1/1000稀释的pab1952兔α-vhh(使用vhh免疫原在eurogentech产生-另一种pab兔α-icvd,例如pab兔α-vh,也可以使用)。在4℃用25ml该溶液将印迹孵育,轻轻振荡过夜。4)第二天,将印迹置于25mlpbst(1xpbs,0.1%tween20)中,并在室温下在摇床上孵育5分钟。重复此过程5次,每次使用新鲜体积的pbst洗涤任何非特异性结合的一级抗体。共完成6次洗涤。5)对于二级检测抗体,在封闭溶液中以1/1000稀释制备hrp-缀合的pab猪α-兔(dako,p0217)。将正常山羊血清(dako)加入到该溶液中至1%的终浓度(例如在50ml二级抗体溶液中为500μl山羊血清)。在室温下将该印迹用25ml该溶液孵育2小时,轻轻振荡。6)将印迹放置到25mlpbst(1×pbs,0.1%tween20)中,并在摇床上孵育5分钟。重复此过程5次,每次使用新鲜体积的pbst以洗去任何非特异性结合的二级抗体。共完成6次洗涤。7)为了使印迹显影,用2mlsupersignalwestpicochemiluminescent(ecl,pierce34087)孵育1-2分钟,确保印迹的全部表面被覆盖在底物中。8)使用imagequantlas4000仪器或等同物,5-10分钟曝光,可视化印迹上存在的icvd。改变用于获得最佳icvd信号的曝光时间。使用imagequanttl软件或等同物确定带密度。在给定时间点的基质样本中的存活icvd的量除以在零时间点存在的量。然后将所得到的数乘以100以给出稳定性%。1.3标准毒素elisa测定用于评估抗-tcda或抗-tcdbicvd的效力,以及用于评估在肠道模型中孵育后剩余的存活抗-tcda或抗-tcdbicvd百分比。材料:·96孔,平底,nuncmaxisorpimmunoplates·在1×pbs中的来自菌株r20291(核型027)的重组n-末端his10标记的艰难梭状芽孢杆菌tcdb细胞结合结构域(cbd-b)。克隆该蛋白质,从大肠杆菌中表达,并用fplc纯化his标签。·来自菌株vpi10463(核型087)的纯化的全长艰难梭菌毒素a。细菌在静置的厌氧培养物中生长并通过fplc离子交换层析纯化分泌的tcda。·抗vhh多克隆兔抗体:6cp(也可使用等效的抗icvd,如抗vh多克隆兔抗体)。·猪抗兔多克隆免疫球蛋白-hrp缀合的(dako,p0217)·对elisa超敏的tmb:sigma(t4444)·0.5m硫酸·封闭缓冲液:1pbs中1%bsa(ph7.2-7.5)。·封闭缓冲液加2×蛋白酶抑制剂(在1×pbs中1%bsa,ph7.3-7.5,2×蛋白酶抑制剂混合液,2.5mmedta,0.5mmpmsf)。·pbst:1xpbs加0.05%tween20。当icvd样品存在于诸如小鼠小肠上清液或人粪便上清液之类的、本来会干扰elisa性能的消化基质时,使用封闭缓冲液加2×蛋白酶抑制剂作为测定稀释剂以制备icvd溶液,随后加入elisa板。可以使用1/200稀释的0.1mpmsf溶液sigma93482来实现0.5mmpmsf。还必须加入edta至终浓度为2.5mm。在该缓冲液中使用sigmafast蛋白酶抑制剂混合物(sigmas8830,含有aebsf(4-(2-氨基乙基)苯磺酰氟,bestatin,e-64,pepstatina,phosphoramidon,leupeptin,aprotinin)。可以通过向5ml蛋白酶终止缓冲液(1×pbs,2%bsa,5mmedta)中加入1片sigmafast蛋白酶抑制剂混合物(sigmas8830)来制备20×蛋白酶抑制剂溶液的储备液。该溶液可以在2-8℃下存储2周,在elisa当天被稀释成封闭缓冲液。通过elisa检测抗-tcdaicvd该测定被设计用于测试抗-tcda特异性icvds的结合到与elisa板结合的艰难梭菌毒素a的能力。用于该测定的包被板的毒素是全长tcdavpi10463(087)。方法:1.在1xpbs中稀释艰难梭菌tcda以制备2μg/ml包被溶液。将其以每孔50μl加入nuncmaxisorp板,密封板并在2-8℃过夜孵育。不用2μg/ml溶液tcda的相同储备液制备大量的板(超过3个)。2.用洗板器用380μlpbst将板清洗×4。敲板以确保留下最少的残留物。3.加入每孔200μl的封闭缓冲液,密封并放置在室温下振荡孵育至少1小时。如果需要,还可以在2-8℃将板过夜封闭。4.如果主要测定样品来自消化基质,则使用封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂来制备icvd参考标准的连续稀释系列作为稀释剂。稀释范围应根据测试的每个icvd的结合进行调整,使其覆盖从背景信号到饱和的完整测定信号范围,其线性范围明确定义。制备足够体积的每种稀释液,铺板50μl,一式三份5.如果样品来自消化基质,在封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂中制备适当稀释的含有待测试的icvd的样品作为稀释剂。制备稀释液,使其预计的浓度将落在测定法检测的线性范围内。稀释范围应根据测试的每个icvd的结合进行调整。这些稀释也应在微板中连续进行,使得在最终的elisa板上有足够体积用于一式三份的50μl重复。包括检测空白(无icvd)。对于消化分析,elisas包括蛋白酶抑制剂-终止时间零基质对照(不含icvd)以在测定中检测背景信号。这应该在封闭缓冲液加2x蛋白酶抑制剂中稀释,并且应该匹配包含在板上测试的icvd样品的基质的顶部浓度。如果从消化基质制备样品,则保持样品在制备过程中冷却。制备足够的每个样品以50μl/孔向板中加入一式三份。6.将elisa板上的封闭缓冲液移到废弃物中,将任何残留物敲到纸巾上,并向每个孔中加入50μl稀释样品。包括1)无基质,无icvd(空白孔)和2)仅基质(无icvd)孔。密封板并在室温下振荡孵育2小时。7.按照步骤2清洗x4。8.在封闭缓冲液中加入50μl/孔的稀释至1/2000的兔抗vhhpab,密封板并在室温下振荡孵育1小时。9.按照步骤2清洗x4。10.使用封闭缓冲液加入每孔50μl的稀释至1/2000的猪抗兔hrp,密封板并在室温下振荡孵育1小时。11.按照步骤2清洗x4。12.每孔加入100μl的tmb,密封板,并在室温孵育不超过30分钟,振荡。tmb是光敏感的,应该用银箔覆盖板。13.向每孔中加入50μl的0.5m硫酸,并在450nm处读板。14.使用graphpadprism软件(或等同物),使用icvd标准校准曲线,插值未知样品浓度。通过elisa检测抗-tcdbicvd该测定法被设计用于测试抗-tcdb特异性icvds与结合到elisa板的艰难梭菌tcdb细胞结合结构域(cbd-b)结合的能力。在运行该测定法之前,检查正测试的icvd并未结合在tcdb上的其他位置是非常重要的,否则将不会观察到任何信号。方法:1.在pbs中稀释艰难梭菌cbd-b(027),制备0.5-1μg/ml的包被溶液。将其以每孔50μl加入nuncmaxisorp板,用膜密封并在2-8℃过夜孵育。不用0.5-1μg/ml的cbd-b溶液的相同储备液制备大量的板(超过3个)。2.加入每孔200μl的封闭缓冲液,密封并放置在室温下振荡孵育至少1小时。如果需要,还可以在2-8℃将板过夜封闭。3.如果主要测定样品来自消化基质,则使用封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂来制备icvd参考标准的连续稀释系列作为稀释剂。稀释范围应根据测试的每个icvd的结合进行调整,使其覆盖从背景信号到饱和的完整测定信号范围,其线性范围明确定义。制备足够体积的每种稀释液,铺板50μl,一式三份4.如果样品来自消化基质,在封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂中制备适量稀释的待测试的含有icvd的样品作为稀释剂。制备稀释液,使其预计的浓度将落在测定法检测的线性范围内。稀释范围应根据测试的每个icvd的结合进行调整。这些稀释也应在微量培养板中连续进行,使得在最终的elisa板上有足够体积用于一式三份的50μl重复。包括测定空白(无icvd)。对于消化分析,elisas包括蛋白酶抑制剂-终止时间零基质对照(不含icvd)以在测定中检测背景信号。这应该在封闭缓冲液加2x蛋白酶抑制剂中稀释,并且应该匹配包含在板上测试的icvd样品的基质的顶部浓度。如果从消化基质制备样品,则保持样品在制备过程中冷却。制备足够的每个样品以50μl/孔向板中加入一式三份。5.将elisa板上的封闭缓冲液移到废弃物中,将任何残留物敲到纸巾上,并向每个孔中加入50μl样品稀释液。包括1)无基质,无icvd(空白孔)和2)仅基质(无icvd)孔。密封板并在室温下振荡孵育2小时。6.按照步骤2清洗x4。7.在封闭缓冲液中加入50μl/孔的稀释至1/2000的兔抗vhhpab,密封板并在室温下振荡孵育1小时。8.按照步骤2清洗x4。9.使用封闭缓冲液加入每孔50μl的稀释至1/2000的猪抗兔hrp,密封板并在室温下振荡孵育1小时。10.按照步骤2清洗x4。11.每孔加入100μl的tmb,密封板,并在室温孵育不超过30分钟,振荡。tmb是光敏感的,应该用银箔覆盖。12.向每孔中加入50μl的0.5m硫酸,并在450nm处读板。13.使用graphpadprism软件(或等同物),使用icvd标准校准曲线插值未知样品浓度。1.4标准tnfr2/tnf干扰elisa测定用于评估抗-tnficvd的效力和用于评估在肠道模型中孵育后剩余的存活的抗-tnficvd百分比1.原理该测定检测重组人tnf与融合蛋白enbrel(依那西普)的结合。该蛋白质由与人igg的fc区结合的可溶性tnrf2组成,并且可以用于捕获tnfα。这种相互作用可以通过抗-tnficvd进行竞争,导致tnfα与enbrel的结合降低。然后用抗-htnfα抗体检测结合的tnf。因此,该elisa中的高信号代表低浓度的抗-tnficvd,反之亦然。由于用主要检测抗体的过夜孵育步骤,该测定通常需要约一天半的时间来完成。2.材料需要的溶液:·0.5m硫酸(h2so4)·1xpbs·pbst(1×pbs,0.05%tween20)·封闭缓冲液(1pbs中1%bsa,ph7.3-7.5)·封闭缓冲液加2×蛋白酶抑制剂(1×pbs中1%bsa,ph7.3-7.5,2×蛋白酶抑制剂混合物,2.5mmedta,0.5mmpmsf)。当icvd样品存在于诸如小鼠小肠上清液或人粪便上清液之类的、本来会干扰elisa表现的消化基质时,使用封闭缓冲液加2×蛋白酶抑制剂作为测定稀释剂以制备icvd和tnf溶液,随后混合并加入elisa板。可以使用1/200稀释的0.1mpmsf溶液sigma93482来实现0.5mmpmsf。还必须加入edta至终浓度为2.5mm。在该缓冲液中使用sigmafast蛋白酶抑制剂混合物(sigmas8830,含有aebsf(4-(2-氨基乙基)苯磺酰氟,bestatin,e-64,pepstatina,phosphoramidon,leupeptin,aprotinin)。20×蛋白酶抑制剂溶液的储备液可以通过向5ml蛋白酶终止缓冲液(1×pbs,2%bsa,5mmedta)中加入1片sigmafast蛋白酶抑制剂混合物(sigmas8830)来制备。该溶液可以在2-8℃下存储2周,在elisa当天被稀释到封闭缓冲液中。需要的试剂:·已知浓度的enbrel储备液(例如pbs中2mg/ml)·已知浓度的重组人tnf储备液(lifetechnologies,目录nophc3015)在pbs中的1%bsa中以10μg/ml组成,并以小等分试样保持在-80℃(≤20μl)·已知浓度的抗-tnfαicvd标准·兔抗人tnfα抗体(peprotech,500-p31abt,300μg/ml)·extravidinhrp(sigma,e2886)·tmb底物(microwell过氧化物酶底物系统2-c,kpl,50-70-00)3.程序制备:确定测定所需的板数。用1xpbs中的50μl/孔1μg/mlenbrel包被maxisorb96孔elisa板(nunc)。短暂振荡板,密封并在4℃孵育过夜。测定:1.使用洗板机(4x~380μlpbst)洗涤elisa板。将板绑在毛巾上以除去残留液体。2.应用200μl/孔封闭缓冲液。密封并在旋转板振荡器上孵育>1小时。3.如果主要测定样品来自消化基质,则使用封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂制备在100μl的最小最终体积中为0.04nm至10nm之间的连续稀释系列的icvd参考标准作为稀释剂。稀释范围应根据测试的每个icvd的效力进行调整。实例如表2所示。表24.如果样品来自消化基质,在封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂中制备适当稀释的待测试的含有icvd的样品作为稀释剂。制备连续稀释系列。稀释范围应根据测试的每个icvd的效力进行调整,使其覆盖从背景信号到饱和的完整测定信号范围,其线性范围明确定义。这些稀释也应在微量培养板中连续进行,使得在最终的elisa板上有足够体积用于一式三份的50μl重复。对于消化分析elisas,包括蛋白酶抑制剂-终止时间零基质对照(不含icvd)。这应该在封闭缓冲液加2x蛋白酶抑制剂中稀释,并且应该匹配包含在板上测试的icvd样品的基质的顶部浓度。如果从消化基质制备样品,则保持样品在制备过程中冷却。5.如果测定样品来自消化基质,则在封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂中制备5ng/ml的hrtnfα溶液。6.在一个单独的96孔板中,用封闭缓冲液或封闭缓冲液加2x蛋白酶抑制剂填充空白孔(例如,孔h1)。用85μl的tnf溶液填充剩余的相关孔。7.将来自制备板的85μl每种icvd稀释液与第二板中的85μl的hrtnfα溶液混合。包括一个含有封闭缓冲液或仅有封闭缓冲液加2x蛋白酶抑制剂(空白)的孔。包括另一个孔,其中hrtnfα用封闭缓冲液,或仅封闭缓冲液加2x蛋白酶抑制剂(仅tnf的对照孔)稀释。包括一个孔,其中hrtnfα被“终止的”消化基质稀释,如上所述。密封,并在旋转板振荡器上孵育1小时。8.如步骤1所示洗涤被封闭的elisa板。9.将50μl的icvd-tnf混合物(加适当对照;1)无tnf,无icvd,2)tnf,但无icvd3)tnf加“终止的”消化基质,无icvd)转移至洗涤的elisa板,一式三份。密封并在旋转板振荡器上孵育2小时。10.按照步骤1洗涤被封闭的elisa板。11.制备以封闭缓冲液制成的5ml/板1/1000稀释的抗人tnfα抗体(peprotech,p31a)。加入50μl/孔,密封,短暂地在旋转板振荡器上振荡,然后在冷室/冰箱(4℃)中孵育过夜。注意:这个步骤可以在rt下在振荡器上减少到2h,但信号会降低,结果是灵敏度降低。12.如步骤1所示洗涤被封闭的elisa板。13.制备5ml/板1/1000稀释的extravidin连接的hrp(sigma,e2886)。加入50μl/孔,密封并在旋转板振荡器上孵育>30分钟。14.如步骤1所示洗涤封闭的elisa板。15.制备10ml/板tmb底物(底物a和b的比例为1:1)。加入100μl/孔,密封并在旋转板振荡器上孵育>30分钟。避光。16.用50μl/孔0.5mh2so4终止反应。16.在450nm读板。18.使用graphpadprism软件(或等同物),使用icvd标准校准曲线,插值未知样品浓度。在步骤6中,将等体积的稀释的icvd和tnfα在加入elisa板之前混合。该步骤将icvd和tnfα浓度有效地稀释了两倍。因此,板上tnfα的最终浓度为2.5ng/ml,并且icvd标准曲线的最终浓度为0.02nm至5nm。在评估适当的样品稀释因子时应考虑此稀释。tmb底物反应可能快速进展。应定期检查板的颜色,如果在30min之前出现非常亮的蓝色,则应终止反应,因为非常高的吸光度会导致高背景。合适的对照应包括以下三重孔:仅bsa,无icvd(即,仅2.5ng/ml的tnfα),以及如果需要,不含tnfα(即,仅5nm的icvd)。对于消化分析elisas,应将通过加入2x蛋白酶终止溶液而终止的无icvd基质样品加入tnf中。对照中背景基质的最低稀释(或最高浓度)应与在与应用于板的tnf/混合的最高icvd浓度中的消化基质的最低稀释(或最高浓度)相匹配。1.5vero细胞细胞毒性标准测定用于评估抗-毒素icvd的效力vero细胞在使用前的培养和维持vero细胞的常规继代培养可以如下实现:1.一旦一瓶细胞生长到完全汇合,吸出所有的细胞培养基,并施加2ml1x胰蛋白酶(溶于0.02%edta,sigmae8008)。一旦胰蛋白酶被施用就迅速作用以防止洗涤期间的细胞损失。2.洗涤细胞表面上的首先的胰蛋白酶施用,然后完全吸出以除去所有痕量的细胞培养基(培养基中任何痕量的血清将抑制胰蛋白酶活性)。3.施用2ml胰蛋白酶并洗涤细胞表面。4.从烧瓶中取出大约1.5-1.7ml的胰蛋白酶。5.倾斜烧瓶,以便剩余的300-500μl覆盖板表面上的vero细胞。6.将细胞在37℃,5%co2中孵育10-12分钟。7.终止胰蛋白酶活性,加入10mlvero细胞培养基。8.用移液管轻轻地将悬浮液对烧瓶底部喷射,直到培养基变浑浊(表示细胞团块消散),重悬细胞。3-4次应足够了。避免过度的吸液,因为这可能会伤害细胞。9.在75cm2的细胞培养瓶(corning)中,将0.2至0.5ml的细胞悬浮液加入到25-30ml新鲜的vero细胞培养基中。将烧瓶在37℃5%co2下孵育,以允许细胞生长至完全汇合。这应该在3-5天内进行,取决于接种量和细胞计数。为了获得对该过程的更好控制,可以使用血细胞计数器计数细胞,如下所述,并以固定的接种物加入培养基。一旦处于汇合状态,细胞单层应保持健康,持续1-2天,无需培养基替换。为了延长汇合单层的使用寿命,通常更新1/3-1/2的培养基是有帮助的(不要将所有的培养基替换,因为它将已经用来自生长的veros的细胞因子进行了调理)。细胞在变圆前应该分传,并且开始发生脱离。制备测定用板(第-1天)理想地,应在使用前一天在细胞毒性测定中制备板。然而,如果需要,也可以在使用当天制备板。如果是后者的情况,在早上制备板(供下午使用),并确保使用前有至少3小时使细胞附着于微量培养板。应使用完全汇合的vero细胞瓶,使细胞悬浮液用于铺板。1.将150μl无菌h2o加入到孔间空间中,并将300μl加入到96孔平底微量培养板的顶部和底部。这确保培养的细胞在微量培养板中在生长期间水合。2.如上所述,将汇合的vero细胞瓶用胰蛋白酶消化并重悬(在10mlvero细胞培养基中)。3.使用血细胞计数器和光学显微镜(取四个独立计数并使用平均值,例如使用单个血球计数器载玻片的四个栅格角)计算细胞。如果在胰蛋白酶消化后存在细胞存活力的问题,在列举(1:1v/v)之前将台盼蓝染料加入到细胞中,并将活细胞计数乘以x2。4.将细胞稀释至在所需体积(允许每个测定板8ml)的vero细胞培养基中的5×104个细胞/ml。5.使用多通道移液管,每孔加入100μl细胞悬浮液。这相当于5000个细胞/孔。如果正在制备多个板,则在连续铺板之间保持旋转和/或吹打(pipetting)细胞悬浮液以确保细胞均匀分布。6.在室温下以1000rpm离心微量培养板2分钟,将细胞均匀地固定在板的底部。在离心机的每个臂上最多旋转2个板,以避免臂向内倾斜并溢出孔间的水。7.用光学显微镜目视确认细胞分布和数量是按预期的。8.在37℃,5%co2孵育板。设置测定(第0天)注意:本节中描述的所有溶液均在vero细胞培养基中制备。应该计算毒素和icvd的所需的最终体积以覆盖测定开始前的板/组合数。在稀释步骤之间充分混合所有溶液(通过涡旋和/或多次颠倒)。1.以最终测定浓度两倍(2x)制备所需体积的毒素。所需的测定浓度应事先确定(参见下面的初步工作)。2.以在测试中要测试的最高浓度的两倍(2x)制备测试icvd。目标是icvd的最高浓度将在测定中显示明确的剂量反应毒素中和关系(参见下面的实施例图)。3.在稀释槽中制备2×icvd储备液的10个连续稀释液(包括未稀释的顶部浓度)。通常,1/3稀释产生有用的数据范围。4.使用96孔圆底微量培养板制备混合溶液,然后加入到含有vero细胞的板中。5.一式三份,制备仅培养基、仅毒素(1x稀释)和triton-x100(0.01%)对照的溶液,并将其各加入空板孔中。6.将10μl移液器吸头连接到8通道抽吸器的中央6排。小心地从在第0天制备的vero细胞微量培养板中取出所有培养基(每孔约100μl)。7.使用多通道移液器,将来自制备板的一排的100μl加入到测定板上的细胞。重复两次以填充测定板上的两个相邻排(总共3个重复排):8.一旦板进料完成,在37℃孵育3天。处理测定(第3天)1.在光学显微镜下观察板。检查在仅对照孔的培养基中的汇合生长以及在仅毒素对照孔中良好的毒素反应。2.使用多通道移液器,在黑暗中,向每个孔中加入10μl的alamar蓝试剂(光敏感)。3.振荡板30秒,确保将alamar蓝混合到培养基中。4.在37℃,5%co2下孵育板1小时30分钟5.孵育后,在黑暗中加入50μl的3%sds。6.使用读板器(例如fluostaromega),激发滤光器544,发射滤光器590,底部光学器件读板。将空白(针对它来校正数据)设置为用tritonx100处理的三个板孔。7.计算板上每次处理的三次重复的平均值。使用以下公式计算毒素中和值%:中和%=(icvd处理-毒素对照)*100/(培养基对照-毒素对照)。预备工作:确定主要中和试验中使用的毒素的最佳量为了在主要测定中易于解释,应通过在vero细胞上进行毒素剂量反应实验来预先确定使用毒素的适当浓度。在12孔稀释槽中制备10次连续稀释的毒素。使用剩余的两个孔用于0.01%的triton和仅培养基对照。在稀释槽中制备最少为330μl的每种溶液(这允许三次重复,每次100μl)。如果事先没有提示毒素制剂如何有效,那么选择广泛的稀释范围进行初步实验。如果需要,这可以在更精细的浓度范围内重复。将这些溶液施用于平底微量培养板中的vero细胞,孵育并处理板,如上所述。为了测定icvd或全抗体对于给定浓度的毒素的中和活性,选择能诱导细胞活力最大降低的每种毒素制剂的最小浓度。vero细胞上的示例性毒素剂量-反应曲线在图1中提供。水平条表示适合用于主要中和测定的毒素浓度。1.6标准gp130elisa测定用于评估抗il-6ricvd的效力该测定的目的是通过测量对sil-6/il-6r复合物的gp130结合的干扰来测量抗il-6ricvd的效力。该测定检测hil-6r/hil-6复合物与重组人gp130的结合。这种相互作用可以被抗il-6ricvd竞争性地抑制,导致hil-6r-hil-6复合物与gp130的结合减少。因此,该elisa中的高信号代表低浓度的抗il-6ricvd,反之亦然。材料所需要的解决方案:1xpbspbst(1×pbs,0.05%tween20)封闭缓冲液(1×pbs中1%bsa,ph7.3-7.5)0.5m硫酸(h2so4)所需要的试剂:已知浓度的重组可溶性人gp130已知浓度的icvd原料已知浓度的重组可溶性人il-6已知浓度的重组可溶性人il-6r生物素化山羊抗il-6r多克隆抗体(r&d系统baf227);以250ug/ml重悬于无菌pbs中。extravidin-过氧化物酶(sigmae2886)tmb底物(microwell过氧化物酶底物系统2-c,kpl,50-70-00)程序制备:1.确定测定所需的板数。2.制备相关体积(一次高达3个板)的在pbs中的0.2μg/ml重组可溶性人gp130以及在1xpbs中的0.5μg/mlbsa3.迅速作用,将50μl/孔分配到maxisorp96孔elisa板(nunc)中,一批加载最多3个板。4.短暂振荡板,密封并在4℃孵育过夜。分析:1.使用洗板机(4x~380μl的pbst)洗涤elisa板。将板绑在毛巾上以除去残留液体。2.施用200μl/孔封闭缓冲液。密封并在旋转板振荡器上孵育>1小时。3.使用封闭缓冲液作为稀释剂,制备在70μl的最小最终体积中为0.004nm至80nm的稀释系列的icvd标准。4.在封闭缓冲液中制备待测试样品的适当稀释液,使其在板上的估计终浓度将落在0.001nm至250nmicvd的范围内。5.在封闭缓冲液中制备40ng/mlil-6r溶液。6.在单独的96孔板中,将50μl的各icvd稀释液与50μl的il-6r溶液混合。每个稀释系列包括一个没有icvd的孔。在旋转板振荡器上孵育1小时。7.在封闭缓冲液中制备100ng/mlil-6溶液。8.在又另外的96孔板中,将来自步骤6的85μl的icvd-il-6r混合物与步骤7中制备的85μl的il-6溶液混合。包括仅含有封闭缓冲液的孔,使得将以下对照施用到每个板:只有il-6,和没有icvd(仅il-6+il-6r)。在旋转板振荡器上孵育10分钟。9.如步骤1洗涤被封闭的elisa板。10.将50μl在步骤8中制备的混合物转移到洗涤的elisa板上,一式三份。密封并在旋转板振荡器上孵育2小时。11.如步骤1所示洗涤封闭的elisa板。12.制备5.2ml/板125μg/ml的以封闭缓冲液制成的baf227抗hil-6r抗体。加入50μl/孔,密封,短暂摇匀,室温孵育1小时或4℃过夜。13.如步骤1所示洗涤封闭的elisa板。14.在封闭缓冲液中制备5.2ml/板的1/1,000-1/3000稀释的extravidin。加入50μl/孔,密封并在旋转板振荡器上孵育30min。15.如步骤1所示洗涤封闭的elisa板。16.制备10ml/板tmb底物(底物a和b的比例为1:1)。加入100μl/孔,密封并在旋转板振荡器上孵育直到在最低稀释孔中演变出中度蓝色或孵育高达30分钟。避光。17.用50μl/孔0.5mh2so4终止反应。18.在450nm读板。19.使用标准曲线来插值活性icvd的浓度。原始od450值用从空白对照孔读取的数据进行调整。使用适当的软件(例如使用log(抑制剂)对反应-可变斜率(四个参数)的graphpadprism)绘制标准曲线。使用标准曲线在软件中计算测试样品中的icvd浓度。用于评估在肠道模型中孵育后剩余的存活抗il-6ricvd百分比该测定的目的是测量先前在蛋白水解物质例如小鼠小肠上清液或人粪便提取物存在下孵育的活性抗il-6ricvd的剩余浓度,从而阐明对在孵育期间可能发生的任何蛋白水解的icvd的影响,以及因此抗il-6ricvd的蛋白水解稳定性。该测定检测hil-6r/hil-6复合物与重组人gp130的结合。这种相互作用可以被抗il-6ricvd竞争性地抑制,导致hil-6r-hil-6复合物与gp130的结合减少。因此,该elisa中的高信号代表保留活性的抗il-6ricvd的低浓度或低亲和力,反之亦然。存活率%是活性icvd的百分比浓度,使用标准曲线进行插值,维持在消化前后的样品之间。材料所需要的溶液1xpbs在pbs中的1%bsapbst(1×pbs,0.05%tween20)封闭缓冲液(1×pbs中1%bsa,ph7.3-7.5)测定缓冲液(1%bsa,1xpbs中的2x蛋白酶抑制剂*)0.5m硫酸(h2so4)*2x蛋白酶抑制剂=每50ml缓冲液1片所需要的试剂:已知浓度的重组可溶性人gp130sigmafast蛋白酶抑制剂片剂(s8820)已知浓度的icvd储存液已知浓度可溶性人il-6已知浓度的可溶性人il-6r生物素化山羊抗il-6r多克隆抗体(r&d系统baf227);以250ug/ml重悬于无菌pbs中。extravidin-过氧化物酶(sigmae2886)tmb底物(microwell过氧化物酶底物系统2-c,kpl,50-70-00)程序制备:1.确定测定所需的板数。2.在pbs+5μg/mlbsa中制备相关体积(一次高达3个板)的0.2μg/ml重组可溶性人gp1303.迅速作用,将50μl/孔分配到maxisorp96孔elisa板(nunc)中,一批加载最多4个板。4.短暂振荡板,密封并在4℃孵育过夜。分析:1.使用洗板机(4x~380μl的pbst)洗涤elisa板。将板绑在毛巾上以除去残留液体。2.施用200μl/孔封闭缓冲液。密封并在旋转板振荡器上孵育>1小时。3.使用测定缓冲液作为稀释剂,制备在70μl的最小最终体积中为0.004nm至1000nm的稀释系列的icvd标准。4.在封闭缓冲液中制备待测试样品的适当稀释液,使其在板上的估计终浓度将落在0.001nm至250nmicvd的范围内。确保含有gi/粪便的样品尽可能地保持在冰上。5.在测定缓冲液中制备400ng/mlil-6r溶液。6.在测定缓冲液中制备40ng/mlil-6r溶液。7.在单独的96孔板中,将50μl的各icvd稀释液与50μl的il-6r溶液混合。每个稀释系列包括一个没有icvd的孔。8.在又另外的96孔板中,将来自步骤7的85μl的icvd-il-6混合物与步骤6中制备的85μl的il-6r溶液混合。包括仅含有测定缓冲液的孔,使得将以下对照施用到每个板:只有il-6,和没有icvd(仅il-6+il-6r)。在旋转板振荡器上孵育5分钟。9.如步骤1洗涤被封闭的elisa板。10.将50μl在步骤8中制备的混合物转移到洗涤的elisa板上,一式三份。密封并在旋转板振荡器上孵育2小时。11.如步骤1所示洗涤封闭的elisa板。12.制备5ml/板125ng/ml的以封闭缓冲液制成的baf227抗hil-6r抗体。加入50μl/孔,密封,短暂摇匀,室温孵育1小时或4℃过夜。13.如步骤1所示洗涤封闭的elisa板。14.在封闭缓冲液中制备5ml/板1/1000-1/3000稀释的extravidin。加入50μl/孔,密封并在旋转板振荡器上孵育<30min。15.如步骤1所示洗涤封闭的elisa板。16.制备10ml/板tmb底物(底物a和b的比例为1:1)。加入100μl/孔,密封并在旋转板振荡器上孵育直到在最低稀释孔中演变出中度蓝色或孵育高达30分钟。避光。17.用50μl/孔0.5mh2so4终止反应。18.在450nm读板。19.使用标准曲线来插值活性icvd的浓度。原始od450值用从空白对照孔读取的数据进行调整。使用适当的软件(例如使用log(抑制剂)对反应-可变斜率(四个参数)的graphpadprism)绘制标准曲线。使用标准曲线在软件中计算测试样品中的icvd浓度。测试样品中的活性icvd浓度表示为0h样品中的活性icvd浓度%以给出存活率%。实施例2:在抗tnf-αicvd的cdr2中用丙氨酸、组氨酸或谷氨酰胺取代赖氨酸残基q65b1是从用可溶性人重组tnf-α免疫的美洲驼分离、克隆并纯化的抗tnf-αicvd。用丙氨酸、组氨酸或谷氨酰胺取代q65b1多肽序列的残基k59,并测试每个取代对肠道稳定性和效力的影响。将编码每个icvd的dna克隆到载体pmek222中,表达并从大肠杆菌的周质(通过talon或镍nta柱)纯化。所有在此测试的icvd都携带相同的c-末端flag-his6标签。残基k59位于q65b1的cdr2中。具有k59a取代的q65b1被标记为“id43f”,具有k59h取代的q65b1被标记为“id8f-ev”,并且具有k59q取代的q65b1被标记为“id44f”。2.1.1效力-标准tnfr2/tnf干扰elisa测定-实验1使用标准tnfr2/tnf干扰elisa测定法产生每个icvd的剂量-反应曲线,其用于产生ec50值(图2a和表3)。表3构建体取代ec50(pm)q65b1无(k59)98.4id8f-evk59h139.3id43fk59a602.6id44fk59q245.472.1.2效力-标准tnfr2/tnf干扰elisa测定-实验2在重复实验中,使用标准tnfr2/tnf干扰elisa测定法再次产生q65b1和id8f-ev的剂量-反应曲线(图2b)。2.2.1肠道稳定性-标准小鼠小肠上清液肠道模型-实验1根据标准小鼠小肠上清液肠道模型,将icvds在小鼠小肠材料中消化6小时。使用标准tnfr2/tnf干扰elisa测定法计算icvd的稳定性百分比。结果如图3a所示。2.2.2肠道稳定性-标准小鼠小肠上清液肠道模型-实验2根据标准小鼠小肠上清液肠道模型,将q65b1和id8f-ev在小鼠小肠材料中消化16小时。使用标准tnfr2/tnf干扰elisa测定法计算icvd的稳定性百分比。结果示于图3b的右侧。2.2.3肠道稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,将q65b1和id8f-ev在人粪便上清液中消化16小时。使用标准tnfr2/tnf干扰elisa测定法计算icvd的稳定性百分比。结果示于图3b的左侧。2.3结论与k59和k59h相比,k59a和k59q的效力降低(分别参见图2a,id43f和id44f对q65b1和id8f-ev)。从图2a和2b可以看出,id8f-ev(k59h)相对于q65b1(k59)的效力的任何观察到的变化可能都下降到实验变化,并且这些icvd具有基本上相同的效力。与k59相比(参见图3a,分别是id43f和id44f对q65b1),并与k59h相比(参见图3a,id8f-ev),k59a和k59q在6小时孵育后降低在小鼠肠道材料中的稳定性。与k59(参见图3a和图3b,id8f-ev与q65b1)相比,k59h在6小时孵育后和16小时孵育后增加在小鼠小肠材料中的稳定性。在人粪便上清液测定中16小时孵育后id8f-ev和q65b1的稳定性未分化(图3b)。实现k59h的稳定性增加而不显著影响效力。实施例3:在抗tnf-αicvd的cdr2和cdr3中用组氨酸残基取代赖氨酸残基q65b1的残基k59和k101都用组氨酸取代(产生“id34f”)。残基k59位于q65b1的cdr2中,而残基k101位于q65b1的cdr3中。克隆编码id34f的dna并在酵母中表达。再次生产被k59h残基取代的q65b1(如实施例2中),具有与上述id8f-ev相同的序列。然而,在这种情况下,克隆编码该icvd的dna并在酵母中表达(因此缺少c-末端flag-his6标签),因此在本实施例中标记为“id32f”。3.1效力-标准tnfr2/tnf干扰elisa测定使用标准tnfr2/tnf干扰elisa测定法产生每个icvd的剂量-反应曲线。使用0-3nm的浓度范围(图4)。3.2.1肠稳定性-标准小鼠小肠上清液肠道模型根据标准小鼠小肠上清液肠道模型,将icvd在小鼠小肠材料中消化16小时。使用标准tnfr2/tnf干扰elisa测定法计算icvd的稳定性百分比。结果示于图5a中。3.2.2肠道稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,将icvds在人粪便上清液中消化16小时。使用标准tnfr2/tnf干扰elisa测定法计算icvd的稳定性百分比。结果如图5b所示。3.3结论根据标准小鼠小肠上清液肠道模型(图5a)和标准人粪便上清液肠道模型(图5b),id34f的cdr3中的额外的k101h取代进一步增加icvd的肠稳定性,而不显著影响效力(图4)。实施例4:在抗tcdbicvd的cdr3中用丙氨酸、组氨酸、谷氨酰胺、苯丙氨酸或色氨酸残基取代精氨酸残基id45b是源自祖细胞icvd(q31b1)的经修饰的抗tcdbicvd。从通过纯化的tcdb的福尔马林灭活制备的tcdb类毒素免疫的美洲驼中分离、克隆并纯化q31b1。id45b多肽序列的残基r107用丙氨酸、组氨酸、谷氨酰胺、苯丙氨酸或色氨酸取代,并测试每种取代对肠稳定性和效力的影响。将编码每个icvd的dna克隆到载体pmek222中,表达并从大肠杆菌的周质(通过talon或镍nta柱)纯化。所有在此测试的icvd都携带相同的c-末端flag-his6标签。残基r107位于id45b的cdr3中。根据表4对取代的icvd进行标记。表4icvd取代id45b无(r107)id46br107hid47br107aid48br107qid49br107fid50br107w4.1效力-vero细胞细胞毒性标准测定使用vero细胞细胞毒性标准测定法产生每个icvd的剂量-反应曲线(图6a)。4.2肠道稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,将icvd在人粪便上清液池4中消化30分钟。使用标准western印迹稳定性测定法计算icvd的存活百分比。结果如图6b所示。4.3结论所有取代相对于'未取代的'id45b的效力降低。然而,r107h和r107f取代(id46b和id49b)仅导致效力降低微小,而r107a、r107q和r107w取代(id47b、id48b和id50b)导致显著的效力降低(图6a)。虽然r107h和r107f取代都导致类似的微小功能降低,但是r107h导致测试的所有取代的肠稳定性增加最高(参见图6b,id46b-与id45br107,0分钟相比,恢复的增加约35%)。相比之下,r107f取代导致相比r107约10%的降低(图6b,id49b)。r107h提供了最大的稳定性增长,而对效力影响不大。实施例5:在抗tcdbicvdid2b的cdr2中用组氨酸残基取代多个精氨酸残基,以及id2b的cdr3内的取代位置的影响id2b是来自祖细胞icvd(q31b1)的经修饰的抗tcdbicvd。id2b多肽序列的cdr2中的残基r53和r56都用组氨酸残基取代(产生“id20b”)。独立地,id2b多肽序列的cdr3中的残基r107和r109各自用组氨酸残基取代(唯一的r107h取代产生“id21b”和唯一的r109h取代产生“id22b”)。这些icvd总结在表5中。测试了这些取代对胰蛋白酶稳定性、肠稳定性和效力的影响。表5将编码id2b的dna克隆到载体pmek222中,表达并从大肠杆菌的周质中纯化。id2b带有c-末端flag-his6标签。克隆编码id20b、id21b和id22b的dna并在酵母中表达。5.1效力-vero细胞细胞毒性标准测定在vero细胞毒性标准测定法中使用027艰难梭菌核糖型的tcdb产生每个icvd的剂量-反应曲线(图7)。5.2.1标准胰蛋白酶肠道模型测定icvds的胰蛋白酶稳定性。将tpck处理的胰蛋白酶-琼脂糖珠(来自牛胰腺的胰蛋白酶;t4019;sigmaaldrich)的缓冲的(10mm乙酸,ph3.2,含0.01%硫柳汞)水悬浮液用于测定。将珠用水(250μl珠+1.25ml水)洗涤3次,然后用胰蛋白酶缓冲液(tryp缓冲液;1mmtris-hcl,20mmcacl2[ph8.0])洗涤5次。最后,将树脂重悬于tryp缓冲液中作为50%(v/v)悬浮液。将100μl的2mg/ml构建体溶液与225μl的50%(v/v)固定化的tpck处理的胰蛋白酶在tryp缓冲液中混合。在振荡器中在37℃下孵育0、10、15、30、45和60分钟的时间间隔后,取样如下:将树脂通过500×g离心1分钟步骤沉淀,并从上清液中取40μl样品并与2x上样缓冲液(例如laemmli缓冲液)混合。将剩余的悬浮液再次混合,并在37℃下放回振荡器中。为了分析,将15μl每个样品与5μl的4x上样染料混合,煮沸10分钟,并在聚丙烯酰胺凝胶上每个泳道上加载15μl(如nupage10%丙烯酰胺bis-tris凝胶)。凝胶在sds-mes缓冲液中在200v下运行35分钟。将凝胶固定在40%甲醇,7%乙酸中30分钟,并在胶体考马斯亮蓝染色中过夜染色。凝胶在成像之前在水中脱色(例如使用imagequantlas4000,曝光7秒)(图8a-c)。相对于切割的组成型多肽的完整构建体的量可以通过比较每个时间点泳道中相应的条带来评估。电泳凝胶图中的星号和#表示含有切割片段的条带。5.2.2肠道稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,将id2b和id21b在粪便池3和4中消化1小时(图9)。使用标准毒素elisa测定法计算icvd的稳定性百分比。5.3结论单个cdr3取代导致效力降低微小(图7,id21b和id22b),而双cdr2取代导致效力更显著降低(图7,id20b)。由于id2b中his标签的存在,图8a中电泳凝胶的结果不清楚。更中心的r107h取代(图8b,id21b)提供比更外周的r109h取代更大的胰蛋白酶稳定性增加(图8c,id22b)。这表明当在cdr的中心“窗口”中产生取代时,这种取代可更稳定。与未取代的id2b相比,池3(艰难梭菌阳性患者粪便)和池4(艰难梭菌阴性患者粪便)中id21b(r107h)的粪便上清液稳定性都显著增加(图9)。实施例6:在抗tcdbicvdid1b的cdr2中用组氨酸残基取代精氨酸残基,以及id1b的cdr3内的取代位置的影响id1b是源自祖细胞icvd(b10f1)的经修饰的抗tcdbicvd。从用100ug通过纯化的tcdb的福尔马林灭活制备的tcdb类毒素免疫的美洲驼中分离,克隆和纯化b10f1。id1b多肽序列的cdr2中的残基r58用组氨酸残基取代(产生“id24b”)。独立地,id1b多肽序列的cdr3中的残基r105和r108各自用组氨酸残基取代(r105h取代产生“id27b”,并且r108h取代产生“id25b”)。这些icvd总结在表6中。测试了这些取代对肠稳定性和效力的影响。表6克隆编码id1b、id24b、id25b和id27b的dna并在酵母中表达。6.1效力-vero细胞细胞毒性标准测定在vero细胞毒性标准测定中,使用来自027艰难梭菌核糖型的tcdb产生每个icvd的剂量-反应曲线(图10a)。6.2.1肠稳定性-标准人粪便上清液肠道模型根据标准人类粪便上清液肠道模型,将id1b、id24b、id25b和id27b在粪便池2中消化1小时(图10b)。使用标准毒素elisa测定计算icvd的存活百分比。6.2.2肠道稳定性-标准胰蛋白酶肠道模型以上述实施例5所述的方式测定icvds的胰蛋白酶稳定性(图11a-c)。6.3结论单个cdr3取代导致效力的轻微降低(图10a)。在测试的时间段内,id1b凝胶中的主带的密度(图11a)似乎比被取代的icvds的密度(图11b-11c)降低程度更大,因此,在此胰蛋白酶测定中被取代的icvd似乎比未被取代的id1b更稳定。所有取代的icvd的粪便上清液稳定性增加(图10b)。较中心的r105hcdr3取代(图10b,id27b)提供了比更外周的r108hcdr3取代更大的粪便上清稳定性增加(图10b,id25b)。这表明当在cdr的中心“窗口”中进行取代时,这种取代可能更稳定。实施例7:在抗tcdb二价构建体的一个臂的cdr3中用组氨酸残基取代精氨酸残基id41b是由修饰形式的野生型icvdsq31b1和b10f1组成的抗tcdb二价构建体。在id41b的b10f1臂中进行r108h(cdr3)取代(产生“id43b”)。测试了该取代对效力和肠稳定性的影响。克隆编码id41b和id43b的dna并在酵母中表达。7.1效力-vero细胞细胞毒性标准测定在vero细胞毒性标准测定法中,使用来自017艰难梭菌核糖型的tcdb产生每个构建体的剂量-反应曲线(图12a)。7.2肠稳定性-标准毒素elisa测定根据标准人粪便上清液肠道模型,将构建体在粪便池2、3和4中消化4小时。对每个粪便池运行三次重复elisa。使用标准毒素elisa测定计算存活百分比(图12b-12d)。7.3结论r108h取代(id43b)对效力影响非常小(图12a)。在大多数粪便上清液测定(所有粪便池中9个里有6个)中,id43b中的r108h取代导致稳定性增加(图12b-12d)。实施例8:在抗tcda二价icvd的cdr3中用组氨酸残基取代精氨酸残基id17a是由修饰形式的野生型icvdb4f10和q34a3组成的抗tcda二价构建体(b4f10和q34a3是从用通过纯化的tcda的福尔马林灭活制备的tcda类毒素免疫的美洲驼中分离、克隆和纯化的)。在id17a的b4f10臂中进行r109h(cdr3)取代(产生“id29a”)。测试该取代对效力和肠稳定性的影响。克隆编码id17a和id29a的dna并在酵母中表达。8.1效力-vero细胞细胞毒性标准测定在vero细胞毒性标准测定法中使用tcda产生每种构建体的剂量-反应曲线(图13a)。8.2肠稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,将粪便在粪便池2、3和4中消化1小时。使用标准毒素elisa测定法计算存活百分比(图13b)。8.3结论该抗tcda双头的一个臂中的r109h(cdr3)取代对效力的影响较小(图13a)。在测试的所有粪便池中,这种取代导致稳定性的高度增加(图13b)。实施例9:在抗il-6ricvd7f6的cdr3中用组氨酸残基取代精氨酸残基7f6是一种抗il-6ricvd。从可溶性人重组il-6r免疫的美洲驼分离、克隆并纯化7f6。7f6多肽序列的cdr3中的残基r102用组氨酸残基取代(产生“id-3v”),并测试该取代对效力和肠稳定性的影响。编码7f6和id-3v的dna在大肠杆菌中克隆并表达。9.1效力-标准gp130elisa测定使用标准gp130elisa测定法产生剂量-反应曲线,并用它们产生ec50值(表7,曲线图未示出)。构建体取代ec50(nm)7f6无(r102)0.15id-3vr102h(在cdr3中)0.169.2肠稳定性-标准小鼠小肠上清液肠道模型根据标准小鼠小肠上清液肠道模型,icvd在小鼠小肠材料中消化4小时。使用标准gp130elisa测定法计算icvd的稳定性百分比。结果示于表8。表8构建体取代稳定性%7f6无(r102)1%id-3vr102h(在cdr3中)12%9.3肠稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,icvds在人粪便上清液中消化16小时。使用标准gp130elisa测定法计算icvd的稳定性百分比。结果示于表9。表9构建体取代稳定性%7f6无(r102)28%id-3vr102h(在cdr3中)41%9.4结论根据标准小鼠小肠上清液肠道模型,7f6的cdr3中的该r102h取代进一步增加了icvd的肠稳定性(参见表8和9),没有显著影响效力(表7)。实施例10:在抗il-6ricvd5g9的cdr3中用组氨酸残基取代精氨酸残基5g9是一种抗il-6ricvd。从可溶性人重组il-6r免疫的美洲驼分离、克隆并纯化5g9。5g9多肽序列的cdr3中的残基r105用组氨酸残基取代(产生“id-54v”),并测试该取代对效力和肠稳定性的影响。编码5g9和id-54v的dna在大肠杆菌中克隆并表达。10.1效力-标准gp130elisa测定使用标准gp130elisa测定法产生剂量-反应曲线,并用它们产生ec50值(表10,曲线图未示出)。构建体取代ec50(nm)5g9无(r105)0.09id-54vr105h(在cdr3中)0.1510.2肠稳定性-标准小鼠小肠上清液肠道模型根据标准小鼠小肠上清液肠道模型,icvd在小鼠小肠材料中消化4小时。使用标准gp130elisa测定法计算icvd的稳定性百分比。结果示于表11。表11构建体取代稳定性%5g9无(r105)5%id-54vr105h(在cdr3中)36%10.3肠稳定性-标准人粪便上清液肠道模型根据标准人粪便上清液肠道模型,icvds在人粪便上清液中消化16小时。使用标准gp130elisa测定法计算icvd的稳定性百分比。结果示于表12。表12构建体取代稳定性%5g9无(r105)40%id-54vr105h(在cdr3中)48%10.4结论根据标准小鼠小肠上清液肠道模型,5g9的cdr3中的该r105h取代进一步增加了icvd的肠稳定性(参见表11和12),对效力仅有轻微影响(表10)。在整个说明书和所附权利要求中,除非上下文另有要求,措词“包括”和诸如“包含”和“含有”之类的变型将被理解为暗示包括所指定的整数、步骤、整数组或步骤组,但不排除任何其他整数、步骤、整数组或步骤组。在本说明书中提及的所有专利和专利申请的全部内容通过引用并入本文。本发明包括优选的和更优选的组的所有组合以及适当的和更适当的上述组的实施方案。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1