结晶型HCV抑制剂及其制备方法和应用与流程
本发明属于药物开发
技术领域:
,具体涉及一种结晶型hcv抑制剂及其制备方法和应用。
背景技术:
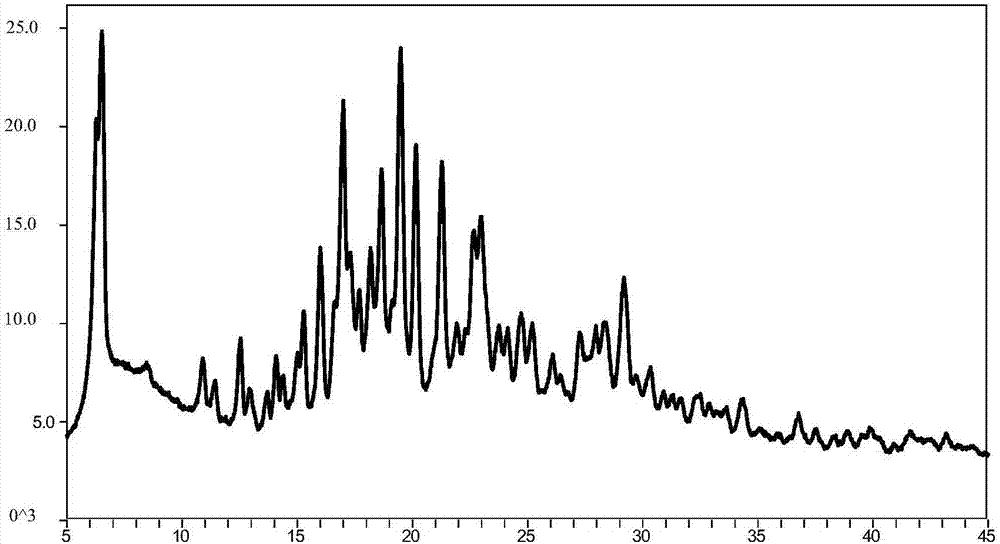
:黄病毒科家族的病毒包括至少三种不同的属:瘟病毒属(pestiviruses),其在牛和猪中引起疾病;黄病毒属(flavivruses),其为诸如登革热和黄热病等疾病的主要原因;以及丙型肝炎病毒属(hepaciviruses),其唯一成员为hcv。黄病毒属包括的成员超过68个,基于血清学亲缘关系进行分组。临床症状各异并且包括发热、脑炎和出血热。全球所关注的与人类疾病有关的黄病毒属包括登革出血热病毒(dhf)、黄热病病毒、休克综合征病毒和日本脑炎病毒。由于hcv基因组在结构和表型特征上与人黄病毒和瘟病毒相类似,将其归为黄病毒科hcv。丙型肝炎病毒是正链rna病毒,在核衣壳外包绕含脂质的囊膜,囊膜上有刺突。hcv仅有huh7,huh7.5,huh7.5.1三种体外细胞培养系统。丙型肝炎病毒于1974年被首次发现,1989年美国科学家迈克尔·侯顿(michaelhoughton)和他的同事们利用分子生物学方法找到了该病毒的基因序列,并克隆出了丙肝病毒,命名本病及其病毒为丙型肝炎(hepatitisc)和丙型肝炎病毒(hcv)。丙型肝炎病毒(hcv)已经严重危及人类健康,其在大量的受感染个体(据估计为全世界人口的2-15%)中导致慢性肝脏疾病如肝硬化和肝细胞癌。根据美国疾病控制中心估计,仅在美国就有四百五十万人受感染。根据世界卫生组织,全世界有超过2亿的受感染个体,每年至少有3至4百万人被感染。一旦被感染后,大约20%的人能清除该病毒,但是剩余的人可能在他们的余生中携带hcv。10%至20%的慢性感染个体最终发展成肝脏破坏性的硬化或癌症。该病毒性疾病在胃肠外通过被污染的血液和血液制品、被污染的针传播;或者通过性传播;以及从被感染的母亲或携带者母亲垂直传播给她们的后代。当前用于hcv感染的治疗限于重组干扰素α单独或与核苷类似物利巴韦林相结合免疫疗法,其具有有限的临床益处。现今已批准的治疗选择是重组干扰素α单独或与核苷类似物利巴韦林相结合的免疫疗法受其临床效果的限制,并且仅有50%的受治疗患者对该疗法有响应。因此,需要发展更为有效和新型的疗法,以解决由hcv感染造成的未被满足的医疗需求。目前已经能够确认的可以作为抗hcv治疗剂的药物开发的一些潜在的分子靶点,包括但不限于ns2-ns3自体蛋白酶(autoprotease)、n3蛋白酶、n3解旋酶和ns5b聚合酶。rna依赖性rna聚合酶对单链rna基因组复制绝对重要,该聚合酶已引起了药物化学家的显著兴趣。2015年,江苏豪森公司在专利wo2015101183a1中公开了一类尿嘧啶核苷酸类似物,其中代表性化合物(式i化合物,化学名称为:异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸)结构如下:该化合物具有非常好的药代动力学特征,可作为rna依赖性rna病毒复制的抑制剂,并且可用作hcvns5b聚合酶的抑制剂、hcv复制的抑制剂,用于治疗哺乳动物的丙型肝炎感染。但是,由于专利wo2015101183a1实施例二十四公开的式化合物在ch2cl2中重结晶纯化得到的还是无定型固体,而无定型api难以制备成适宜临床应用的药物制剂,因此,迫切需要开发一种稳定的、溶解性好的结晶体来满足药物临床开发的需要。技术实现要素:为了解决现有技术存在的问题,发明人通过深入研究式ⅰ化合物的不同的聚集状态,最终得到了一种结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸,大大改善了专利wo2015101183a1公开的无定型固体的理化性质。本发明提供了一种结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸。优选的,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图包括位于19.98±0.2°,7.38±0.2°,25.06±0.2°和17.52±0.2°的衍射角(2θ)处的峰或者包括位于6.52±0.2°,19.50±0.2°,6.28±0.2°和17.00±0.2°的衍射角(2θ)处的峰。作为进一步优选的方案,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图包括位于19.98±0.2°,7.38±0.2°,25.06±0.2°,17.52±0.2°,23.02±0.2°,14.92±0.2°,18.76±0.2°,18.40±0.2°和25.44±0.2°的衍射角(2θ)处的峰,所述结晶型态指定为式i化合物晶型i。作为更进一步优选的方案,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图还包括位于22.80±0.2°,26.80±0.2°,15.44±0.2°,5.52±0.2°,21.58±0.2°和20.84±0.2°的衍射角(2θ)处的峰。作为最优选的方案,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图还还包括位于16.24±0.2°,15.86±0.2°,11.08±0.2°,22.42±0.2°,24.74±0.2°的衍射角(2θ)处的峰,其化学位移(2θ值)和峰强度如表1所示:表12θ(°)强度%2θ(°)强度%5.5223.619.981007.3888.320.8420.611.0817.721.5823.114.9230.222.4212.115.4423.822.802515.8618.223.0246.416.2419.224.7411.317.525625.066818.4029.625.4428.518.7630.226.8024.1作为进一步优选的方案,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图包括位于6.52±0.2°,19.50±0.2°,6.28±0.2°,17.00±0.2°,20.16±0.2°,21.28±0.2°,18.66±0.2°,16.00±0.2°,22.96±0.2°和22.66±0.2°的衍射角(2θ)处的峰,所述结晶型态指定为式i化合物晶型ii。作为更进一步优选的方案,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图还包括位于17.30±0.2°,29.18±0.2°,15.28±0.2°,18.18±0.2°,12.54±0.2°和23.20±0.2°的衍射角(2θ)处的峰。作为最优选的方案,所述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸粉末x射线衍射图还还包括位于27.96±0.2°,24.78±0.2°,25.2±0.2°,16.62±0.2°的衍射角(2θ)处的峰,其化学位移(2θ值)和峰强度如表2所示:表22θ(°)强度%2θ(°)强度%6.2875.619.5082.86.5210020.1661.112.542421.2857.915.2826.422.6637.3164222.9639.816.6216.523.20181775.524.7817.617.3030.825.217.518.1825.527.9617.718.664529.1830.5正如本领域技术人员理解,峰位置(2θ)将由于xrpd仪器不同,而造成测量值有所变化,有时这种变化达有时多达0.2°因此,这种变化或偏移也可以使用的“基本上相同的”术语来表达x射线衍射峰位置和强度可变性。此外,本领域技术人员也能理解,由于xrpd样品制样方法,xrpd仪器,样品结晶度,样品用量以及晶体择优取向等因素也将导致样品xrpd衍射图中相对峰强度的改变。相比于比无定形物,本发明所得结晶体无论化学上,还是物理性上都更加稳定,有利于药物生产、运输和贮存;也更加适合药物开发需要。对于药物提纯、脱色、过滤以及其他工艺操作也更具优势。因此,本发明结晶型化合物具有重大改进意义,符合临床开发要求。本发明另一方面提供一种前述的结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸的制备方法,包括如下步骤:1)将异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸溶解在含水溶剂或在适合的有机溶剂中;2)搅拌下加入适量反溶剂或加入少量晶种至溶液出现浑浊,继续搅拌析晶;3)固液分离得到结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸。作为进一步优选的方案,所述制备方法中步骤1)所述有机溶剂包括但不限定于以下溶剂:甲醇、乙醇、正丙醇、异丙醇、正丁醇、乙腈、丙酮、甲乙酮、四氢呋喃、二氧六环、n,n-二甲基甲酰胺、二甲基亚砜、乙酸乙酯、乙酸异丙酯、二氯甲烷、三氯乙烷、四氯化碳、甲基叔丁基醚、异丙醚、苯、甲苯、二甲苯或其组合物。所述反溶剂包括但不限定于以下溶剂:正庚烷、正己烷、异辛烷、戊烷、环己烷、环戊烷、乙醚或其组合物。所述的溶解是指本领域的普通技术人员一般的操作,通常可以适当的加热使原料溶解或溶清,或者加大溶剂的用量来使原料溶解或溶清,或者其技术方案进行修改或者等同替换,其均应涵盖在本发明的
发明内容之内。作为更进一步优选的方案,所述制备方法中步骤3)分离得到的结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸的粉末x射线衍射图包括位于19.98±0.2°,7.38±0.2°,25.06±0.2°和17.52±0.2°的衍射角(2θ)处的峰或者包括位于6.52±0.2°,19.50±0.2°,6.28±0.2°和17.00±0.2°的衍射角(2θ)处的峰。作为更进一步优选的方案,所述制备方法中步骤3)分离得到的结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸的粉末x射线衍射图包括位于19.98±0.2°,7.38±0.2°,25.06±0.2°,17.52±0.2°,23.02±0.2°,14.92±0.2°,18.76±0.2°,18.40±0.2°和25.44±0.2°的衍射角(2θ)处的峰或者包括位于6.52±0.2°,19.50±0.2°,6.28±0.2°,17.00±0.2°,20.16±0.2°,21.28±0.2°,18.66±0.2°,16.00±0.2°,22.96±0.2°和22.66±0.2°的衍射角(2θ)处的峰。作为更进一步优选的方案,所述制备方法中步骤3)分离得到基本上纯的结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸;所述基本上纯的结晶体是指晶型纯度大于90%的结晶体。优选的,所述制备方法中步骤3)分离得到结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸晶型纯度大于95%。最优选的,所述制备方法中步骤3)分离得到结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸晶型纯度大于98%。本发明再一方面提供一种药物组合物,其包括治疗有效剂量的前述的结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸及可药用的载体。本发明再一方面提供了前述结晶型异丙基((s)-(4-环丙苯氧基)(((2r,3r,4r,5r)-5-(2,4-二氧-3,4-二氢嘧啶-1(2h)-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)磷酰基)-l-丙氨酸或前述药物组合物在制备用于治疗丙型肝炎病毒、甲型肝炎病毒、西尼罗病毒、黄热病病毒、登革病毒、鼻病毒、脊髓灰质炎病毒、牛病毒性腹泻病毒或日本脑炎病毒感染所引起的疾病的药物中的应用。附图说明图1为式i化合物晶型i的粉末x射线衍射图;x轴为衍射峰角度2θ(°),y轴为峰的强度。图2为式i化合物晶型ii的粉末x射线衍射图;x轴为衍射峰角度2θ(°),y轴为峰的强度。图3为式i化合物晶型i的dsc图;x轴为温度(℃),y轴为热流(w/g)。图4为式i化合物晶型i的tga图;x轴为温度(℃),y轴为失重百分比(%)。图5为式i化合物晶型ii的dsc图;x轴为温度(℃),y轴为热流(w/g)。图6为式i化合物晶型ii的tga图。x轴为温度(℃),y轴为失重百分比(%)。具体实施方式1、术语本文所用的术语"可药用"是指在合理医疗判断的范围内适合与人类和动物的组织接触而没有过度的毒性、刺激、过敏反应或其它问题并发症的与合理的受益/风险比相称的那些化合物、材料、组合物和/或剂型。本文所用的术语"基本纯净"是指在本发明的某些优选实施方案中式i化合物的结晶结构为基本纯净形式,hplc纯度或晶型纯度基本在90%以上(包含本数),优选95%以上,更优选98%以上,最优选99.5%以上。本文所用的“结晶型”或“结晶型物”是指具有相同化学组成但构成该晶体的分子、原子和/或离子的不同空间排列的晶型。尽管结晶型物具有相同的化学组成,但它们的堆积和几何排列不同,并可能表现出不同的物理性质,如熔点、形状、颜色、密度、硬度、可形变性、稳定性、溶解度、溶出速率和类似性质。根据他们的温度-稳定性关系,两种多晶型物可以是单变性或互变性的。对于单变性体系,在温度变化时,两种固相之间的相对稳定性保持不变。相反,在互变性体系中,存在一个过渡温度,在此两种相的稳定性调换((theoryandoriginofpolymorphismin"polymorphisminpharmaceuticalsolids"(1999)isbn:)-8247-0237)。本发明的结晶样品可以以基本纯净的相均一性提供,意味着存在主导量的单一结晶结构和任选次要量的一种或多种其它结晶结构。样品中多于一种的本发明的结晶结构的存在可以通过如粉末x-射线衍射(xrpd)或因态核磁共振能谱法(ssnmr)之类的技术测定。例如,在实验测得的xrpd图(观察的)与模拟的xrpd图(计算的)的比较中,额外峰的存在可以表明样品中多于一种的结晶结构。模拟xrpd可以由单晶x-射线数据计算(参看smith,d.k.,"afortranprogramforcalculatingx-raypowderdiffractionpatterns,"lawrenceradiationlaboratory,livermore,california,ucrl-7196,1963年4月;也参看yin.s.,scaringe,r.p.,dimarco,j.,galella,m和gougoutas,j.z.,americanpharmceuticalreview.2003.6.2.80)。优选地,该结晶结构具有如在实验测得的xrpd图中由模拟xrpd图中不存在的额外峰产生的为总峰面积的小于10%,优选小于5%,更优选小于2%所示的基本纯净的相均一性。最优选的是在实验测得的xrpd图中由模拟xrpd图中不存在的额外峰产生的小于总峰面积的1%的具有基本纯净的相均一性的本发明的结晶结构。本文所述的本发明的各种结晶结构可以使用本领域普通技术人员已知的各种分析技术彼此区分。这类技术包括但不限于,固态核磁共振(ssnmr)光谱法、x-射线粉末衍射(xrpd)、差示扫描量热法(dsc)和/或热重分析法(tga)。本发明的结晶结构可以通过各种方法制备,包括,例如,从合适的溶剂中结晶或重结结晶、升华、从熔融体中生长、从另一相固态转化、从超临界流体中结晶、和射流喷雾。结晶结构从溶剂混合物中结晶或重结晶的技术包括,例如,溶剂蒸发、降低溶剂混合物的温度、该分子和/或盐的过饱和溶剂混合物的引晶、冻干溶剂混合物、向溶剂混合物中加入抗溶剂(反萃溶剂)。可以使用高生产量的结晶技术制备结晶结构,包括多晶型物。药物晶体,包括多晶型物,药物晶体的制备方法和表征公开在solid-statechemistryofdrugs,s.r.byrn,r.r.pfeiffer,和j.g.stowell,第2版,ssci,westlafayette,indiana,1999中。可以在任何结晶混合物中加入晶种以促进结晶。如技术人员清楚的那样,使用晶种作为控制特定结晶结构生长的方式或作为控制结晶产物的粒度分布的方式。相应地,如"programmedcoolingofbatchcrystallizers,"j.w.mullin和j.nyvlt,chemicalengineeringscience,1971,26,369-377中所述,所需晶种量的计算取决于可得晶种的尺寸和平均产物粒子的所需尺寸。一般而言,需要小尺寸的品种以有效控制该批中晶体的生长。小尺寸晶种可以通过较大晶体的筛分、研磨或微粉化或通过溶液的微结晶产生应该注意,晶体的研磨或微粉化不能造成所需晶体结构的结晶度的任何改变(即变成无定形或变成另一多晶型)。本发明公开或要求保护的晶体结构对等的晶体结构可能根据试验条件、纯度、设备和本领域技术人员已知的其它常几变量在合理误差范围内表现出类似但不完全相同的分析特性。相应地,本领域技术人员显而易几的是,可以在不背离本发明的范围和精神的情况下在本发明内作出各种修改和变动。在考虑本文公开的本发明的说明书和实践的基础上,本发明的其它实施方案是本领域技术人员显而易见的。申请人期望该说明书和实施例被视为示例性的,而非限制其范围。本文所用的术语"室温"或"rt"是指20至25℃(68-77°f)的环境温度。2、实验材料本发明实施例中所用试剂为市售工业级或分析级试剂,所选式i化合物原料为根据豪森专利wo2015101183a1实施例二十四制备得到的无定型固体。3、分析方法3.1、x-射线粉末衍射本领域普通技术人员会认识到,粉末x-射线衍射图可以在测量误差下获得,该测量误差取决于所用测量条件。特别地,通常己知的是,x-射线粉末衍射图中的强度可能随所用材料条件波动。应该进一步理解,相对强度也可能随实验条件而变,并相应地,确切强度不应该被计入考虑。另外,传统的粉末x-射线粉末衍射角的测量误差通常为大约5%或更低,且这种测量误差度应该被视为属于上述衍射角。因此,要理解的是,本发明的晶体结构不限于提供与本文公开的附图中所绘的x-射线粉末衍射图完全相同的x-射线衍射图的晶体结构。提供与附图中公开的那些基本相同的粉末x-射线衍射图的任何晶体结构都落在本发明的范围内。确定x-射线粉末衍射图基本相同的能力在本领域普通技术人员的能力范围内。本领域技术人员已知的其它合适的标准校准。但是,相对强度可能随晶体大小和形状而变。式i化合物多晶型由它们的x射线粉末衍射图表征。因此,在具有使用cukα辐射以反射方式操作的gadds(一般面积衍射检测器系统)cs的brukerd8discoverx射线粉末衍射仪上采集所述盐的x射线粉末衍射图。管电压和电流量分别设置为40kv和40ma采集扫描。在3.0°至40°的2θ范围内扫描样品60秒的时期。针对2θ表示的峰位置,使用刚玉标准品校准衍射仪。在通常是20℃-30℃的室温下实施所有分析。使用用于4.1.14t版wnt软件的gadds,采集和积分数据。使用2003年发行的具有9.0.0.2版eva的diffracplus软件,分析衍射图。xrpd样品的制备,通过是将样品至于单晶硅片上,用玻璃片或等效物压样品粉末以确保样品的表面平坦并有适当的高度。然后将样品支架放入brukerxrpd仪器,并使用上文描述的仪器参数采集粉末x射线衍射图。由包括以下的多种因素产生与这类x射线粉末衍射分析结果相关的测量差异:(a)样品制备物(例如样品高度)中的误差,(b)仪器误差,(c)校准差异,(d)操作人员误差(包括在测定峰位置时出现的那些误差),和(e)物质的性质(例如优选的定向误差)。校准误差和样品高度误差经常导致所有峰在相同方向中的位移。一般地说,这个校准因子将使测量的峰位置与预期的峰位置一致并且可以在预期的2θ值±0.2°的范围中。本发明实施例所得各多晶型的2θ(°)值和强度值(作为最高峰值的%)已列入表1-表2中。3.2、热重分析热重分析(tga)实验在tainstrumentstm型号q5000中进行。将样品(大约10-30毫克)装在预先称皮重的铂盘中。通过仪器精确测量样品重量并记录至千分之一毫克(athousandofamilligram)。将该炉用氮气以100毫升/分钟吹扫。在室温至300℃之间以10℃/分钟加热速率收集数据。3.3、差示扫描量热法差示扫描量热法(dsc)实验在tainstrumentstm模型q2000中进行。将样品(大约1-6毫克)在铝盘中称重并精确记录至百分之一毫克,并转移到dsc中。将该仪器用氮气以50毫升/分钟吹扫。在室温至300℃之间以10℃/分钟加热速率收集数据。在吸热峰朝下的情况下绘图。但是,本领域技术人员会注意到,在dsc测量中,根据加热速率、晶体形状和纯度和其它测量参数,实测的开始温度和最高温度具有一定程度的可变性。以下提供的具体实施例以及制备方法例将进一步举例说明本发明实施方案的特定方面。下列实施例的范围将不以任何方式限制本发明的范围。实施例1称取50mg式i化合物(无定形)置于10.0ml玻璃瓶中,然后加入3.0ml正溶剂甲基叔丁基醚,搅拌溶清,缓慢加入5.0ml反溶剂正庚烷,室温下(20-25℃)磁力搅拌48小时,固液分离得到结晶型式i化合物(晶型i),其粉末x射线衍射图如图1所示。所得到的式i化合物晶型i分别用差示扫描量热仪(dsc),热重分析仪(tga)进行热分析,所用dsc仪器型号为taq2000,tga仪器型号为taq5000。dsc,tga分析方法参数如下:温度范围为室温至300摄氏度,扫描速率为10摄氏度每分钟,保护气体为氮气(流速25毫升/分钟)。dsc分析图见图3,tga分析图见图4。实施例2称取30mg式i化合物(无定形)置于10.0ml玻璃瓶中,然后加入2.0ml正溶剂乙酸乙酯,搅拌溶清,缓慢加入5.0ml反溶剂正庚烷,室温下(20-25℃)磁力搅拌48小时,固液分离得到结晶型式i化合物(晶型ii),其粉末x射线衍射图如图2所示。所得到的式i化合物晶型ii分别用差示扫描量热仪(dsc),热重分析仪(tga)进行热分析,所用dsc仪器型号为taq2000,tga仪器型号为taq5000。dsc,tga分析方法参数如下:温度范围为室温至300摄氏度,扫描速率为10摄氏度每分钟,保护气体为氮气(流速25毫升/分钟)。dsc分析图见图5,tga分析图见图6。实施例3称取200mg式i化合物(无定形)置于20.0ml玻璃瓶中,然后加入5.0ml正溶剂异丙醚,搅拌溶清,缓慢加入10.0ml反溶剂正己烷,室温下(20-25℃)磁力搅拌48小时,固液分离得到结晶型式i化合物(晶型i),其粉末x射线衍射图与如图1基本一致。实施例4称取3.0g式i化合物(无定形)置于300.0ml圆底烧瓶中,然后加入80.0ml正溶剂乙酸异丙酯,搅拌溶清,缓慢加入200.0ml反溶剂乙醚,室温下(20-25℃)磁力搅拌48小时,固液分离得到结晶型式i化合物(晶型ii),其粉末x射线衍射图与如图2基本一致。实施例5称取50mg式i化合物(无定形)置于10.0ml玻璃瓶中,然后加入2.0ml甲基叔丁基醚,搅拌溶清,加入少量实施例1制备得到的式i化合物晶型i作为晶种,在4℃下继续搅拌48小时,固液分离得到结晶型式i化合物(晶型i),其粉末x射线衍射图与如图1基本一致。实施例6称取50mg式i化合物(无定形)置于10.0ml玻璃瓶中,然后加入1.0m乙酸乙酯,加热搅拌溶清,加入少量实施例2制备得到的式i化合物晶型ii作为晶种,在4℃下继续搅拌48小时,固液分离得到结晶型式i化合物(晶型ii),其粉末x射线衍射图与如图2基本一致。实施例7:晶型i样品稳定性试验前后的晶型比较研究取本发明的式i化合物晶型i制备样品,在长期稳定性(温度30℃,湿度65%)条件下放置六个月后作x-射线粉末衍射,分析x-射线粉末衍射图并与起始数据进行比较。数据比较分析见表3:表3:晶型i样品长期稳定性的x-射线粉末衍射数据对比表序号起始的2θ(°)六个月后的2θ(°)15.525.5427.387.4311.0811.1414.9214.94515.4415.46615.8615.88716.2416.26817.5217.54918.4018.421018.7618.791119.98201220.8420.861321.5821.61422.4222.441522.8022.821623.0223.041724.7424.761825.0625.081925.4425.462026.8026.82结论:对比上述x-射线粉末衍射数据,衍射峰的2θ角度未发生明显改变。本发明的式i化合物晶型i样品在长期稳定性(温度30℃,湿度65%)条件下放置六个月后,晶型未发生改变。实施例8:晶型i样品研磨前后的晶型比较研究取本发明的式i化合物晶型i制备样品,并进行如下试验:(1)直接研磨5分钟;(2)原料直接微粉。将上述两种晶型i样品分别进行x-射线粉末衍射,分析x-射线粉末衍射图并与起始数据进行比较。数据比较分析见表4:表4:晶型i样品研磨、微粉前后的x-射线粉末衍射数据对比表序号起始的2θ(°)研磨后的2θ(°)微粉后的2θ(°)15.525.515.5427.387.377.4311.0811.0711.1414.9214.9114.94515.4415.4315.46615.8615.8515.88716.2416.2316.27817.5217.5117.54918.4018.3918.421018.7618.7518.781119.9819.97201220.8420.8320.861321.5821.5721.61422.4222.4122.441522.8022.7922.821623.0223.0123.041724.7424.7324.761825.0625.0525.081925.4425.4325.462026.8026.7926.82结论:对比上述x-射线粉末衍射数据,衍射峰的2θ角度未发生明显改变。本发明的式i化合物晶型i样品在直接研磨和微粉条件下,晶型未发生改变。实施例9:晶型ii样品稳定性试验前后的晶型比较研究取本发明的式i化合物晶型ii制备样品,在长期稳定性(温度30℃,湿度65%)条件下放置六个月后作x-射线粉末衍射,分析x-射线粉末衍射图并与起始数据进行比较。数据比较分析见表5:表5:晶型ii样品长期稳定性的x-射线粉末衍射数据对比表结论:对比上述x-射线粉末衍射数据,衍射峰的2θ角度未发生明显改变。本发明的式i化合物晶型ii样品在长期稳定性(温度30℃,湿度65%)条件下放置六个月后,晶型未发生改变。实施例10:晶型ii样品研磨前后的晶型比较研究取本发明的式i化合物晶型ii制备样品,并进行如下试验:(1)直接研磨5分钟;(2)原料直接微粉。将上述两种晶型ii样品分别进行x-射线粉末衍射,分析x-射线粉末衍射图并与起始数据进行比较。数据比较分析见表6:表6:晶型ii样品研磨、微粉前后的x-射线粉末衍射数据对比表结论:对比上述x-射线粉末衍射数据,衍射峰的2θ角度未发生明显改变。本发明的式i化合物晶型ii样品在直接研磨和微粉条件下,晶型未发生改变。实施例11:样品引湿性研究取本发明的式i化合物无定型、晶型i、晶型ii制备样品,分别进行如下试验:(1)将样品放置于温度25℃,湿度75%条件下;(2)将样品放置于温度25℃,湿度92.5%条件下。将式i化合物无定型、晶型i、晶型ii的上述两种样品在不同时间点取样检测,考察样品吸湿增重情况,数据对比见表7:表7:样品吸湿增重数据对比表结论:式i化合物的晶型i、晶型ii样品没有引湿性,无定型有引湿性。实验例12:样品制备片剂的溶出结果研究取本发明的式i化合物无定型、晶型i、晶型ii制备的样品,制备成api片剂(常规压片),分别在0.1mol/lhcl溶液、ph4.5醋酸缓冲液、ph6.8磷酸缓冲液和纯化水中考察样品溶出度,数据对比见表6:表8:20mg规格产品的溶出数据表结论:本发明的式i化合物晶型i、晶型ii样品制备成片剂的溶出结果表明,i化合物晶型片剂产品在0.1mol/lhcl溶液、ph4.5醋酸缓冲液、ph6.8磷酸缓冲液和纯化水中,15min的溶出度均大于90%,显著优于无定型片剂产品。从样品稳定性考察数据可以看出,本发明晶型稳定性表现很好,适合医药质量标准。实验例13:生物利用度研究将体重为200-250g的sd大鼠18只,雌雄各半,按体重随机分成3组,每组6只,雌雄各半。三组分别单次经口给予等摩尔的式i化合物无定型、晶型i、晶型ii3mg/kg。给药前和给药后的0.5、1.0、2.0、3.0、4.0、5.0、7.0、10、24和48h从眼眶采取0.5ml的血液进行血浆中式i化合物的含量检测,研究结果见表9。表9:大鼠给予式i化合物无定型、晶型i、晶型ii后的药代参数*中位数实验结果表明,与给予等摩尔的式i化合物无定型相比,给予式i化合物晶型i后血浆中的式i化合物cmax和auc分别提高了81%和82%,给予式i化合物晶型ii后血浆中的式i化合物cmax和auc分别提高了80%和81%,而且这种差异存在统计上的显著性。这说明了式i化合物的晶型i和晶型ii与无定型相比,生物利用度更高。最后应当说明的是,以上实施例仅用以说明本发明的技术方案而非限制本发明,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1