一种蛋白基表面活性剂的制备方法与流程
本发明涉及一种表面活性剂的制备方法,具体涉及一种蛋白基表面活性剂的制备方法。
技术背景
皮革产业的迅速发展在给人们带来舒适与便利的同时,也产生了越来越多的制革废弃物,铬革屑便在其中占有很大的比例。铬革屑主要是由胶原蛋白和三价铬组成,对其进行资源化利用具有巨大的环境和经济价值。
传统化学表面活性剂会受到原料、价格、产品性能等因素的影响,同时在使用过程中常常会造成严重的环境污染以及对人体的危害。目前对表面活性剂的研究主要集中在从天然产物中制备出安全、温和、易生物降解的表面活性剂,如蛋白基表面活性剂。该表面活性剂以蛋白质水解产物胶原多肽或氨基酸为原料,使其中反应活性较高的侧链氨基接枝疏水性基团构成双亲分子,具有对人体温和、生物降解快、性能优异、与别的表面活性剂具有协同效应的优点,能够在诸多领域取代传统表面活性剂,应用前景十分广阔。
化学法是合成蛋白基表面活性剂最常用的方法,此法原料易得,对操作设备要求低、工艺流程简单,但国内合成的蛋白基表面活性剂普遍存在转化率较低、产品纯度不高的问题。因此,在制备蛋白基表面活性剂的过程中常常加入缚酸剂和亲核催化剂来提高缩合的转化率,提高产品的纯度。
缚酸剂的加入可以减少副反应的发生、减缓对设备的腐蚀,而传统缚酸剂如三乙胺、吡啶等吸收气体后形成固态盐,溶剂用量大,产品分离困难,不利于工业化生产。传统亲核催化剂的催化效果不够明显且原料成本较高、合成过程中存在着毒物污染,同样不利于工业化生产。
技术实现要素:
本发明所要解决的技术问题是,提供一种蛋白基表面活性剂的制备方法,利用废弃铬革屑制备出低刺激性、高表面活性、优良的配伍性及环境相容性的蛋白基表面活性剂,提高脱铬速度,并提高蛋白基表面活性剂的产率和特性粘数。
本发明采用了以下技术方案:
一种蛋白基表面活性剂的制备方法,其特征在于按照以下步骤进行:
(a)、制备革屑一次碱水解液:将烘干铬革屑、烘干铬革屑质量7%~9%的氧化钙、烘干铬革屑质量9~11倍的水以及烘干铬革屑质量3%~5%的壳聚糖4-(二乙氨基)水杨醛希夫碱加入到装有冷凝管、温度计和搅拌器的容器中,在30~40℃条件下用40khz的超声波处理5~8次,每次处理50~90min,相邻两次处理时间间隔为5~10min,将水解液离心、抽滤后得到革屑一次碱水解液;
(b)、制备蛋白多肽水解液:向步骤(a)所得革屑一次碱水解液中加入革屑质量15%~20%的碳酸钠进行脱钙,离心抽滤后再加入革屑一次碱水解液固含量5~18%的氢氧化钠并于70~90℃继续水解2~4h,减压蒸馏浓缩至波美度15~20,得到蛋白多肽水解液;
(c)、制备蛋白基表面活性剂:将步骤(b)所得蛋白多肽水解液、蛋白多肽水解液质量1‰~10‰的亲核催化剂置于容器中,将与蛋白多肽水解液游离氨基摩尔比为1:1的油酰氯溶于体积为油酰氯0.5~2倍的乙酸乙酯溶剂中,通过恒压滴液漏斗滴加到所述容器中,半小时内滴加完毕,滴加过程中用缚酸剂调节反应体系的ph值维持在8~10,然后升高温度至40~80℃,继续搅拌反应4~8h,反应结束后减压蒸出溶剂,得到蛋白基表面活性剂。
优选地,壳聚糖4-(二乙氨基)水杨醛希夫碱的合成方法如下:
将壳聚糖以及壳聚糖质量60~65倍的1%~3%的醋酸溶液加入到反应容器中,于50~70℃条件下溶胀20~40min;将与壳聚糖质量比为178:4~6溶有10%~15%4-(二乙氨基)水杨醛的乙醇溶液滴加到该反应容器中,于70~90℃超声波清洗器中回流反应4~6h;抽滤后产物使用乙醇、乙醚溶液(体积比为1:1)洗涤5~8次,恒温干燥后得到粗产品,用乙醇在索氏提取器上回流萃取5~6h,70~80℃下真空干燥得到壳聚糖4-(二乙氨基)水杨醛希夫碱。
优选地,亲核催化剂的合成方法如下:
取4-吡啶甲酸及4-吡啶甲酸质量5‰~10‰的聚苯乙烯-二乙烯苯接枝季铵盐于反应容器中,加入与4-吡啶甲酸摩尔比为1:1~1.5的二氯亚砜,40~70℃加热回流4h,反应结束后,于减压蒸馏装置中升温至70~80℃将二氯亚砜蒸干,得到黄色无定形固体;将该固体加入到溶有1~5‰吗啉环己烯胺的氨水溶液中,于冰水浴中反应1~2h,过滤后将所得滤液浓缩结晶,用体积比10:1的石油醚和乙酸乙酯进行层析纯化得到异烟酰胺;另取反应容器,加入200~300ml30%~40%的次溴酸钠溶液,将制备好的异烟酰胺加入到该次溴酸钠溶液中,直至溶液变澄清,将该溶液加热20~30min,蒸出来的粗品用盐酸溶解,再加聚醚链叔胺调ph为12~14,将析出的絮状物抽滤干燥后用体积比为10:1的石油醚和乙酸乙酯进行层析纯化,得到微红色晶体;在温度-10~5℃条件下,将该晶体加入到浓hcl中,并通入hcl气体至饱和,然后滴加亚硝酸钠溶液,滴加结束静止5~6h,然后将溶液水分蒸干得到固体;将该固体加入到-5~0℃的50~60%二甲胺水溶液中,升温回流1~3h,再用naoh、甲苯反复萃取,用甲苯重结晶后得亲核催化剂。
优选地,缚酸剂的合成方法如下:
在反应容器中用氮气置换空气5~8次,加入三乙胺、三乙胺质量0.5~1倍的四甘醇单甲醚、三乙胺质量2~3倍的甲苯,于冰水浴中滴加与三乙胺质量比为40:75~80的65~70%乙基磺酰氯的甲苯溶液,滴加结束后反应9~11h,抽滤除去固体后将滤液减压蒸出甲苯及三乙胺,得到四甘醇单甲醚乙基磺酸酯;量取二甲胺加入到另一反应容器中,开启搅拌并加热升温至60~80℃,将体系抽真空后连续通入与二甲胺质量比为58:40~50的环氧丙烷气体,继续维持体系温度60~80℃反应2~4h,反应结束后进行减压蒸馏,收集95~102℃的馏分即为n,n-二甲基丙醇胺;
另取反应容器抽真空后氮气置换5~8次,加入金属钠、金属钠质量20~25倍的甲苯后搅拌升温至80~100℃,待金属钠熔融形成钠砂后停止加热,冷却5~10min后转移至冰水浴中,向该反应容器中滴加与金属钠质量比为50:10~15合成的n,n-二甲基丙醇胺,滴加结束后于室温下继续反应直至金属钠完全溶解;将该反应容器转移至冰水浴中,向内滴加与金属钠质量比为100:7~8的四甘醇单甲醚乙基磺酸酯,滴加结束后转移至室温继续反应17~19h,加水后分液,用饱和氢氧化钠溶液洗涤有机相,水相用二氯甲烷萃取,合并有机相用无水氯化钙干燥,过滤后将所得滤液于50~70℃减压蒸馏除去甲苯,再升温收集160~180℃馏分即为缚酸剂。
本发明的积极效果在于:
(1)、利用氧化钙、超声波、自制壳聚糖4-(二乙氨基)水杨醛希夫碱联合脱铬的方法将铬革屑进行水解脱铬,得到铬含量极低的蛋白多肽水解液。采用的壳聚糖4-(二乙氨基)水杨醛希夫碱改善了壳聚糖分子中氨基的质子化现象,提高了其絮凝、吸附重金属离子作用;在超声波作用下,会产生瞬时的高温高压,空化泡崩溃所形成的特殊条件足以使水中o-h键断裂,形成自由基,并进而形成氧与过氧化氢,提高脱铬的速度。
(2)、在蛋白多肽水解液与油酰氯缩合的过程中通过以下方法大大提高了缩合的产率及产品的纯度:①、加入乙酸乙酯作为反应溶剂提高了缩合产率;②、加入自制高效亲核催化剂提高缩合产率③、加入自制缚酸剂调节反应体系ph,提高了缩合转化率。
(3)、本发明利用价廉且易得的4-吡啶甲酸为原料合成高效亲核催化剂,摈弃了传统的合成方法,操作步骤简单易于工业化生产,并且可以显著提高缩合的转化率。
(4)、采用的缚酸剂在吸收hcl后形成的胺盐是室温离子液体不溶于非极性有机溶剂。用本发明缚酸剂代替传统的缚酸剂,反应过程中不再有固态盐生成而造成操作不便,反应结束后通过液-液相分离即可将胺盐离子液体分出,利于工业化生产。
(5)、合成的蛋白基表面活性剂产品具有天然性、安全性、低刺激性、高表面活性、优良的配伍性及环境相容性,具有十分广阔的应用前景。
附图说明
图1是本发明实例1制备的表面活性剂(a)及其水解液(b)的红外光谱图。
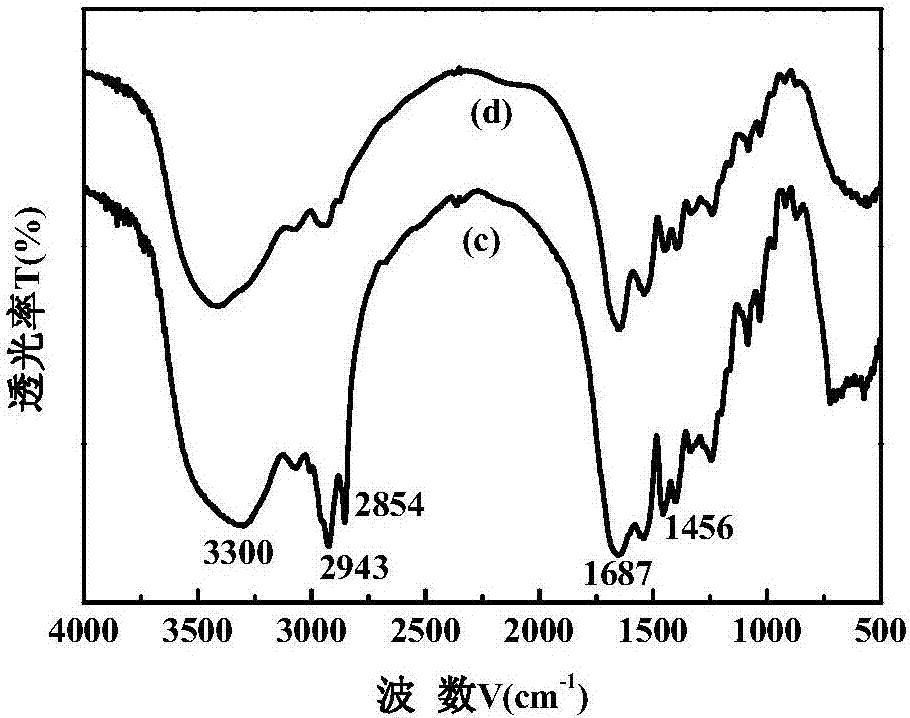
图2是本发明实例2制备的表面活性剂(c)及其水解液(d)的红外光谱图。
图3是本发明实例3制备的表面活性剂(e)及其水解液(f)的红外光谱图。
图4是本发明实例4制备的表面活性剂(g)及其水解液(h)的红外光谱图。
图5是本发明实例5制备的表面活性剂(i)及其水解液(j)的红外光谱图。
图6是本发明实例制备的表面活性剂析水率随时间变化曲线。
图7是本发明实例制备的表面活性剂乳化稳定指数柱状图。
具体实施方式
下面结合附图和实施例详细说明本发明。
实例1
一种蛋白基表面活性剂的制备方法,其特征在于按照以下步骤进行:
(a)、制备革屑一次碱水解液:取60g烘干铬革屑、4.8g氧化钙、600ml去离子水加入到装有冷凝管、温度计和搅拌器的容器中,并加入3g壳聚糖4-(二乙氨基)水杨醛希夫碱进行脱铬吸附,在35℃条件下,每次用40khz的超声波处理铬革屑90min,共8次,相邻两次处理时间间隔为5min,将水解液离心、抽滤后即得铬含量极低的革屑一次碱水解液。
(b)、制备革屑二次碱水解液:向步骤(a)所得水解液中加入10.2g碳酸钠进行脱钙、抽滤后,测得该革屑一次碱水解液固含量为0.07036g/ml,取500ml该革屑一次碱水解液,加入1.8g氢氧化钠于80℃继续水解3h,水解结束后减压蒸馏浓缩至波美度15保存备用。
(c)、制备蛋白基表面活性剂:取步骤(b)所得蛋白多肽水解液40ml置于四口烧瓶中,加入水解液质量5‰的自制亲核催化剂,取3.1ml油酰氯溶于3.1ml乙酸乙酯溶剂中,通过恒压滴液漏斗滴加到所述容器中,半小时内滴加完毕,滴加过程中用缚酸剂调节反应体系的ph值维持在8.5,然后升高温度至60℃,继续搅拌反应6h,反应结束后减压蒸出溶剂乙酸乙酯,即可得到蛋白基表面活性剂1(pbs1),收率为77.61%。
其中壳聚糖4-(二乙氨基)水杨醛希夫碱作为高效脱铬吸附剂,其合成工艺如下:
称取5g壳聚糖于1000ml三口烧瓶中,加入300ml2%的醋酸溶液,60℃溶胀30min;将溶解有20g4-(二乙氨基)水杨醛的200ml乙醇溶液滴加到该三口烧瓶中,于80℃超声波清洗器中回流反应5h。抽滤后产物使用乙醇、乙醚溶液(1:1,体积比)洗涤5~8次,恒温干燥后得到粗产品,用乙醇在索氏提取器上回流萃取6h,80℃下真空干燥即可得到壳聚糖4-(二乙氨基)水杨醛希夫碱,该产品改善了壳聚糖分子中氨基的质子化现象,提高了其絮凝、吸附重金属离子作用。
其中亲核催化剂的合成工艺如下:
取10g4-吡啶甲酸、0.05g聚苯乙烯-二乙烯苯接枝季铵盐(ptc)、9g二氯亚砜于容器中,60℃加热回流4h,反应结束后,于减压蒸馏装置中升温至75℃将二氯亚砜蒸干,得到黄色无定形固体。将该固体加入到200ml溶有0.5g吗啉环己烯胺的氨水溶液中,于冰水浴中反应1h,过滤后将所得滤液浓缩结晶,用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化即可得到异烟酰胺。另取反应容器,加入200ml40%的次溴酸钠溶液,将制备好的异烟酰胺加入到该次溴酸钠溶液中,直至溶液变澄清,将该溶液加热30min,蒸出来的粗品用盐酸溶解,再加聚醚链叔胺调ph为13,将析出的絮状物抽滤干燥后用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化,得到微红色晶体。在温度-10~5℃条件下,将该晶体加入到浓hcl中,并通入hcl气体至饱和,然后滴加亚硝酸钠溶液,滴加结束静止6h,然后将溶液水分蒸干得到固体。将该固体加入到0℃的50%二甲胺水溶液中,升温回流2h,再用naoh、甲苯反复萃取,用甲苯重结晶后即得高效亲核催化剂。
其中缚酸剂的合成工艺如下:
取500ml三口烧瓶抽真空后氮气置换6次,加入35g四甘醇单甲醚、40g三乙胺、100ml甲苯,于冰水浴中滴加溶有50g乙基磺酰氯的30ml甲苯溶液,控制滴速为1滴/3秒,滴加结束后反应约10h,抽滤除去固体后将滤液减压蒸出甲苯及三乙胺,得到四甘醇单甲醚乙基磺酸酯。
称取45g二甲胺加入到三口烧瓶中,开启搅拌并加热升温至70℃,将体系抽真空后连续通入58g环氧丙烷气体,继续维持体系温度70℃反应3h,反应结束后在三口烧瓶上安装蒸馏装置进行减压蒸馏,收集95~102℃的馏分即为n,n-二甲基丙醇胺。另取500ml三口烧瓶抽真空后氮气置换6次,加入200ml甲苯、8g金属钠后搅拌升温至90℃,待金属钠熔融形成钠砂后停止加热,冷却5min后转移至冰水浴中,向该三口烧瓶中滴加37g合成的n,n-二甲基丙醇胺,控制滴速为1滴/3秒,滴加结束后于室温下继续反应直至金属钠完全溶解。将该三口烧瓶转移至冰水浴中,向内滴加105g四甘醇单甲醚乙基磺酸酯,控制滴速为1滴/3秒,滴加结束后转移至室温继续反应18h,加入100ml水,分液,用饱和氢氧化钠溶液洗涤有机相,水相用二氯甲烷萃取,合并有机相用无水氯化钙干燥,过滤后将所得滤液于70℃减压蒸馏除去甲苯,再升温收集160~180℃馏分即为缚酸剂。
实例2
一种蛋白基表面活性剂的制备方法,其特征在于按照以下步骤进行:
(a)、制备革屑一次碱水解液:取70g烘干铬革屑、5.6g氧化钙、700ml去离子水加入到装有冷凝管、温度计和搅拌器的容器中,并加入3.5g壳聚糖4-(二乙氨基)水杨醛希夫碱进行脱铬吸附,在35℃条件下,每次用40khz的超声波处理铬革屑90min,共8次,相邻两次处理时间间隔为5min,将水解液离心、抽滤后即得铬含量极低的一次水解液。
(b)、制备革屑二次碱水解液:向步骤(a)所得水解液中加入11.9g碳酸钠进行脱钙、抽滤后,测得该革屑一次碱水解液固含量为0.07036g/ml,取600ml该革屑一次碱水解液,加入3.38g氢氧化钠于80℃继续水解3h,水解结束后减压蒸馏浓缩至波美度15保存备用。
(c)、制备蛋白基表面活性剂:取步骤(b)所得蛋白多肽水解液80ml置于四口烧瓶中,加入水解液质量5‰的自制亲核催化剂,取7ml油酰氯溶于7ml乙酸乙酯溶剂中,通过恒压滴液漏斗滴加到所述容器中,半小时内滴加完毕,滴加过程中用缚酸剂调节反应体系的ph值维持在9.0,然后升高温度至40℃,继续搅拌反应4h,反应结束后减压蒸出溶剂乙酸乙酯,即可得到蛋白基表面活性剂2(pbs2),收率为81.29%。
其中壳聚糖4-(二乙氨基)水杨醛希夫碱作为高效脱铬吸附剂,其合成工艺如下:
称取5g壳聚糖于1000ml三口烧瓶中,加入300ml2%的醋酸溶液,60℃溶胀30min;将溶解有20g4-(二乙氨基)水杨醛的200ml乙醇溶液滴加到该三口烧瓶中,于80℃超声波清洗器中回流反应5h。抽滤后产物使用乙醇、乙醚溶液(1:1,体积比)洗涤5~8次,恒温干燥后得到粗产品,用乙醇在索氏提取器上回流萃取6h,80℃下真空干燥即可得到壳聚糖4-(二乙氨基)水杨醛希夫碱,该产品改善了壳聚糖分子中氨基的质子化现象,提高了其絮凝、吸附重金属离子作用。
其中亲核催化剂的合成工艺如下:
取10g4-吡啶甲酸、0.05g聚苯乙烯-二乙烯苯接枝季铵盐(ptc)、9g二氯亚砜于容器中,60℃加热回流4h,反应结束后,于减压蒸馏装置中升温至75℃将二氯亚砜蒸干,得到黄色无定形固体。将该固体加入到200ml溶有0.5g吗啉环己烯胺的氨水溶液中,于冰水浴中反应1h,过滤后将所得滤液浓缩结晶,用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化即可得到异烟酰胺。另取反应容器,加入200ml40%的次溴酸钠溶液,将制备好的异烟酰胺加入到该次溴酸钠溶液中,直至溶液变澄清,将该溶液加热30min,蒸出来的粗品用盐酸溶解,再加聚醚链叔胺调ph为13,将析出的絮状物抽滤干燥后用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化,得到微红色晶体。在温度-10~5℃条件下,将该晶体加入到浓hcl中,并通入hcl气体至饱和,然后滴加亚硝酸钠溶液,滴加结束静止6h,然后将溶液水分蒸干得到固体。将该固体加入到0℃的50%二甲胺水溶液中,升温回流2h,再用naoh、甲苯反复萃取,用甲苯重结晶后即得高效亲核催化剂。
其中缚酸剂的合成工艺如下:
取500ml三口烧瓶抽真空后氮气置换6次,加入35g四甘醇单甲醚、40g三乙胺、100ml甲苯,于冰水浴中滴加溶有50g乙基磺酰氯的30ml甲苯溶液,控制滴速为1滴/3秒,滴加结束后反应约10h,抽滤除去固体后将滤液减压蒸出甲苯及三乙胺,得到四甘醇单甲醚乙基磺酸酯。
称取45g二甲胺加入到三口烧瓶中,开启搅拌并加热升温至70℃,将体系抽真空后连续通入58g环氧丙烷气体,继续维持体系温度70℃反应3h,反应结束后在三口烧瓶上安装蒸馏装置进行减压蒸馏,收集95~102℃的馏分即为n,n-二甲基丙醇胺。另取500ml三口烧瓶抽真空后氮气置换6次,加入200ml甲苯、8g金属钠后搅拌升温至90℃,待金属钠熔融形成钠砂后停止加热,冷却5min后转移至冰水浴中,向该三口烧瓶中滴加37g合成的n,n-二甲基丙醇胺,控制滴速为1滴/3秒,滴加结束后于室温下继续反应直至金属钠完全溶解。将该三口烧瓶转移至冰水浴中,向内滴加105g四甘醇单甲醚乙基磺酸酯,控制滴速为1滴/3秒,滴加结束后转移至室温继续反应18h,加入100ml水,分液,用饱和氢氧化钠溶液洗涤有机相,水相用二氯甲烷萃取,合并有机相用无水氯化钙干燥,过滤后将所得滤液于70℃减压蒸馏除去甲苯,再升温收集160~180℃馏分即为缚酸剂。
实例3
一种蛋白基表面活性剂的制备方法,其特征在于按照以下步骤进行:
(a)、制备革屑一次碱水解液:取80g烘干铬革屑、6.4g氧化钙、800ml去离子水加入到装有冷凝管、温度计和搅拌器的容器中,并加入4g壳聚糖4-(二乙氨基)水杨醛希夫碱进行脱铬吸附,在35℃条件下,每次用40khz的超声波处理铬革屑90min,共8次,相邻两次处理时间间隔为5min,将水解液离心、抽滤后即得铬含量极低的一次水解液。
(b)、制备革屑二次碱水解液:向步骤(a)所得水解液中加入13.6g碳酸钠进行脱钙、抽滤后,测得该革屑一次碱水解液固含量为0.07036g/ml,取700ml该革屑一次碱水解液,加入4.93g氢氧化钠于80℃继续水解3h,水解结束后减压蒸馏浓缩至波美度15保存备用。
(c)、制备蛋白基表面活性剂:取40ml步骤(b)所得蛋白多肽水解液置于四口烧瓶中,加入水解液质量5‰的自制催化剂,取3.4ml油酰氯溶于1.7ml乙酸乙酯溶剂中,通过恒压滴液漏斗滴加到所述容器中,半小时内滴加完毕,滴加过程中用缚酸剂调节反应体系的ph值维持在8.0,然后升高温度至60℃,继续搅拌反应4h,反应结束后减压蒸出溶剂乙酸乙酯,即可得到蛋白基表面活性剂3(pbs3),收率为85.16%。
其中壳聚糖4-(二乙氨基)水杨醛希夫碱作为高效脱铬吸附剂,其合成工艺如下:
称取5g壳聚糖于1000ml三口烧瓶中,加入300ml2%的醋酸溶液,60℃溶胀30min;将溶解有20g4-(二乙氨基)水杨醛的200ml乙醇溶液滴加到该三口烧瓶中,于80℃超声波清洗器中回流反应5h。抽滤后产物使用乙醇、乙醚溶液(1:1,体积比)洗涤5~8次,恒温干燥后得到粗产品,用乙醇在索氏提取器上回流萃取6h,80℃下真空干燥即可得到壳聚糖4-(二乙氨基)水杨醛希夫碱,该产品改善了壳聚糖分子中氨基的质子化现象,提高了其絮凝、吸附重金属离子作用。
其中亲核催化剂的合成工艺如下:
取10g4-吡啶甲酸、0.05g聚苯乙烯-二乙烯苯接枝季铵盐(ptc)、9g二氯亚砜于容器中,60℃加热回流4h,反应结束后,于减压蒸馏装置中升温至75℃将二氯亚砜蒸干,得到黄色无定形固体。将该固体加入到200ml溶有0.5g吗啉环己烯胺的氨水溶液中,于冰水浴中反应1h,过滤后将所得滤液浓缩结晶,用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化即可得到异烟酰胺。另取反应容器,加入200ml40%的次溴酸钠溶液,将制备好的异烟酰胺加入到该次溴酸钠溶液中,直至溶液变澄清,将该溶液加热30min,蒸出来的粗品用盐酸溶解,再加聚醚链叔胺调ph为13,将析出的絮状物抽滤干燥后用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化,得到微红色晶体。在温度-10~5℃条件下,将该晶体加入到浓hcl中,并通入hcl气体至饱和,然后滴加亚硝酸钠溶液,滴加结束静止6h,然后将溶液水分蒸干得到固体。将该固体加入到0℃的50%二甲胺水溶液中,升温回流2h,再用naoh、甲苯反复萃取,用甲苯重结晶后即得高效亲核催化剂。
其中缚酸剂的合成工艺如下:
取500ml三口烧瓶抽真空后氮气置换6次,加入35g四甘醇单甲醚、40g三乙胺、100ml甲苯,于冰水浴中滴加溶有50g乙基磺酰氯的30ml甲苯溶液,控制滴速为1滴/3秒,滴加结束后反应约10h,抽滤除去固体后将滤液减压蒸出甲苯及三乙胺,得到四甘醇单甲醚乙基磺酸酯。
称取45g二甲胺加入到三口烧瓶中,开启搅拌并加热升温至70℃,将体系抽真空后连续通入58g环氧丙烷气体,继续维持体系温度70℃反应3h,反应结束后在三口烧瓶上安装蒸馏装置进行减压蒸馏,收集95~102℃的馏分即为n,n-二甲基丙醇胺。另取500ml三口烧瓶抽真空后氮气置换6次,加入200ml甲苯、8g金属钠后搅拌升温至90℃,待金属钠熔融形成钠砂后停止加热,冷却5min后转移至冰水浴中,向该三口烧瓶中滴加37g合成的n,n-二甲基丙醇胺,控制滴速为1滴/3秒,滴加结束后于室温下继续反应直至金属钠完全溶解。将该三口烧瓶转移至冰水浴中,向内滴加105g四甘醇单甲醚乙基磺酸酯,控制滴速为1滴/3秒,滴加结束后转移至室温继续反应18h,加入100ml水,分液,用饱和氢氧化钠溶液洗涤有机相,水相用二氯甲烷萃取,合并有机相用无水氯化钙干燥,过滤后将所得滤液于70℃减压蒸馏除去甲苯,再升温收集160~180℃馏分即为缚酸剂。
实例4
一种蛋白基表面活性剂的制备方法,其特征在于按照以下步骤进行:
(a)、制备革屑一次碱水解液:取90g烘干铬革屑、7.2g氧化钙、900ml去离子水加入到装有冷凝管、温度计和搅拌器的容器中,并加入4.5g壳聚糖4-(二乙氨基)水杨醛希夫碱进行脱铬吸附,在35℃条件下,每次用40khz的超声波处理铬革屑90min,共8次,相邻两次处理时间间隔为5min,将水解液离心、抽滤后即得铬含量极低的一次水解液。
(b)、制备革屑二次碱水解液:向步骤(a)所得水解液中加入15.3g碳酸钠进行脱钙、抽滤后,测得革屑一次碱水解液固含量为0.07036g/ml,取800ml该革屑一次碱水解液,加入8.44g氢氧化钠于80℃继续水解3h,水解结束后减压蒸馏浓缩至波美度15保存备用。
(c)、制备蛋白基表面活性剂:取步骤(b)所得蛋白多肽水解液40ml置于四口烧瓶中,加入水解液质量5‰的自制催化剂,取4.2ml油酰氯溶于2.1ml乙酸乙酯溶剂中,通过恒压滴液漏斗滴加到所述容器中,半小时内滴加完毕,滴加过程中用缚酸剂调节反应体系的ph值维持在8.5,然后升高温度至50℃,继续搅拌反应8h,反应结束后减压蒸出溶剂乙酸乙酯,即可得到蛋白基表面活性剂4(pbs4),收率为88.65%。
其中壳聚糖4-(二乙氨基)水杨醛希夫碱作为高效脱铬吸附剂,其合成工艺如下:
称取5g壳聚糖于1000ml三口烧瓶中,加入300ml2%的醋酸溶液,60℃溶胀30min;将溶解有20g4-(二乙氨基)水杨醛的200ml乙醇溶液滴加到该三口烧瓶中,于80℃超声波清洗器中回流反应5h。抽滤后产物使用乙醇、乙醚溶液(1:1,体积比)洗涤5~8次,恒温干燥后得到粗产品,用乙醇在索氏提取器上回流萃取6h,80℃下真空干燥即可得到壳聚糖4-(二乙氨基)水杨醛希夫碱,该产品改善了壳聚糖分子中氨基的质子化现象,提高了其絮凝、吸附重金属离子作用。
其中亲核催化剂的合成工艺如下:
取10g4-吡啶甲酸、0.05g聚苯乙烯-二乙烯苯接枝季铵盐(ptc)、9g二氯亚砜于容器中,60℃加热回流4h,反应结束后,于减压蒸馏装置中升温至75℃将二氯亚砜蒸干,得到黄色无定形固体。将该固体加入到200ml溶有0.5g吗啉环己烯胺的氨水溶液中,于冰水浴中反应1h,过滤后将所得滤液浓缩结晶,用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化即可得到异烟酰胺。另取反应容器,加入200ml40%的次溴酸钠溶液,将制备好的异烟酰胺加入到该次溴酸钠溶液中,直至溶液变澄清,将该溶液加热30min,蒸出来的粗品用盐酸溶解,再加聚醚链叔胺调ph为13,将析出的絮状物抽滤干燥后用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化,得到微红色晶体。在温度-10~5℃条件下,将该晶体加入到浓hcl中,并通入hcl气体至饱和,然后滴加亚硝酸钠溶液,滴加结束静止6h,然后将溶液水分蒸干得到固体。将该固体加入到0℃的50%二甲胺水溶液中,升温回流2h,再用naoh、甲苯反复萃取,用甲苯重结晶后即得高效亲核催化剂。
其中缚酸剂的合成工艺如下:
取500ml三口烧瓶抽真空后氮气置换6次,加入35g四甘醇单甲醚、40g三乙胺、100ml甲苯,于冰水浴中滴加溶有50g乙基磺酰氯的30ml甲苯溶液,控制滴速为1滴/3秒,滴加结束后反应约10h,抽滤除去固体后将滤液减压蒸出甲苯及三乙胺,得到四甘醇单甲醚乙基磺酸酯。
称取45g二甲胺加入到三口烧瓶中,开启搅拌并加热升温至70℃,将体系抽真空后连续通入58g环氧丙烷气体,继续维持体系温度70℃反应3h,反应结束后在三口烧瓶上安装蒸馏装置进行减压蒸馏,收集95~102℃的馏分即为n,n-二甲基丙醇胺。另取500ml三口烧瓶抽真空后氮气置换6次,加入200ml甲苯、8g金属钠后搅拌升温至90℃,待金属钠熔融形成钠砂后停止加热,冷却5min后转移至冰水浴中,向该三口烧瓶中滴加37g合成的n,n-二甲基丙醇胺,控制滴速为1滴/3秒,滴加结束后于室温下继续反应直至金属钠完全溶解。将该三口烧瓶转移至冰水浴中,向内滴加105g四甘醇单甲醚乙基磺酸酯,控制滴速为1滴/3秒,滴加结束后转移至室温继续反应18h,加入100ml水,分液,用饱和氢氧化钠溶液洗涤有机相,水相用二氯甲烷萃取,合并有机相用无水氯化钙干燥,过滤后将所得滤液于70℃减压蒸馏除去甲苯,再升温收集160~180℃馏分即为缚酸剂。
实例5
一种性能优良的蛋白基表面活性剂的制备工艺,其特征在于按照以下步骤制成:
(a)、制备革屑一次碱水解液:取100g烘干铬革屑、8g氧化钙、1000ml去离子水加入到装有冷凝管、温度计和搅拌器的容器中,并加入5g壳聚糖4-(二乙氨基)水杨醛希夫碱进行脱铬吸附,在35℃条件下,每次用40khz的超声波处理铬革屑90min,共8次,相邻两次处理时间间隔为5min,将水解液离心、抽滤后即得铬含量极低的一次水解液。
(b)、制备革屑二次碱水解液:向步骤(a)所得水解液中加入17g碳酸钠进行脱钙、抽滤后,测得革屑一次碱水解液固含量为0.07036g/ml,取900ml该革屑一次碱水解液,加入11.4g氢氧化钠于80℃继续水解3h,水解结束后减压蒸馏浓缩至波美度15保存备用。
(c)、制备蛋白基表面活性剂:取步骤(b)所得蛋白多肽水解液40ml置于四口烧瓶中,加入水解液质量5‰的自制催化剂,取4.6ml油酰氯溶于2.3ml乙酸乙酯溶剂中,通过恒压滴液漏斗滴加到所述容器中,半小时内滴加完毕,滴加过程中用缚酸剂调节反应体系的ph值维持在9.0,然后升高温度至60℃,继续搅拌反应6h,反应结束后减压蒸出溶剂,即可得到蛋白基表面活性剂5(pbs5),收率为92.49%。
其中壳聚糖4-(二乙氨基)水杨醛希夫碱作为高效脱铬吸附剂,其合成工艺如下:
称取5g壳聚糖于1000ml三口烧瓶中,加入300ml2%的醋酸溶液,60℃溶胀30min;将溶解有20g4-(二乙氨基)水杨醛的200ml乙醇溶液滴加到该三口烧瓶中,于80℃超声波清洗器中回流反应5h。抽滤后产物使用乙醇、乙醚溶液(1:1,体积比)洗涤5~8次,恒温干燥后得到粗产品,用乙醇在索氏提取器上回流萃取6h,80℃下真空干燥即可得到壳聚糖4-(二乙氨基)水杨醛希夫碱,该产品改善了壳聚糖分子中氨基的质子化现象,提高了其絮凝、吸附重金属离子作用。
其中亲核催化剂的合成工艺如下:
取10g4-吡啶甲酸、0.05g聚苯乙烯-二乙烯苯接枝季铵盐(ptc)、9g二氯亚砜于容器中,60℃加热回流4h,反应结束后,于减压蒸馏装置中升温至75℃将二氯亚砜蒸干,得到黄色无定形固体。将该固体加入到200ml溶有0.5g吗啉环己烯胺的氨水溶液中,于冰水浴中反应1h,过滤后将所得滤液浓缩结晶,用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化即可得到异烟酰胺。另取反应容器,加入200ml40%的次溴酸钠溶液,将制备好的异烟酰胺加入到该次溴酸钠溶液中,直至溶液变澄清,将该溶液加热30min,蒸出来的粗品用盐酸溶解,再加聚醚链叔胺调ph为13,将析出的絮状物抽滤干燥后用石油醚和乙酸乙酯(10:1,体积比,下同)进行层析纯化,得到微红色晶体。在温度-10~5℃条件下,将该晶体加入到浓hcl中,并通入hcl气体至饱和,然后滴加亚硝酸钠溶液,滴加结束静止6h,然后将溶液水分蒸干得到固体。将该固体加入到0℃的50%二甲胺水溶液中,升温回流2h,再用naoh、甲苯反复萃取,用甲苯重结晶后即得高效亲核催化剂。
其中缚酸剂的合成工艺如下:
取500ml三口烧瓶抽真空后氮气置换6次,加入35g四甘醇单甲醚、40g三乙胺、100ml甲苯,于冰水浴中滴加溶有50g乙基磺酰氯的30ml甲苯溶液,控制滴速为1滴/3秒,滴加结束后反应约10h,抽滤除去固体后将滤液减压蒸出甲苯及三乙胺,得到四甘醇单甲醚乙基磺酸酯。
称取45g二甲胺加入到三口烧瓶中,开启搅拌并加热升温至70℃,将体系抽真空后连续通入58g环氧丙烷气体,继续维持体系温度70℃反应3h,反应结束后在三口烧瓶上安装蒸馏装置进行减压蒸馏,收集95~102℃的馏分即为n,n-二甲基丙醇胺。另取500ml三口烧瓶抽真空后氮气置换6次,加入200ml甲苯、8g金属钠后搅拌升温至90℃,待金属钠熔融形成钠砂后停止加热,冷却5min后转移至冰水浴中,向该三口烧瓶中滴加37g合成的n,n-二甲基丙醇胺,控制滴速为1滴/3秒,滴加结束后于室温下继续反应直至金属钠完全溶解。将该三口烧瓶转移至冰水浴中,向内滴加105g四甘醇单甲醚乙基磺酸酯,控制滴速为1滴/3秒,滴加结束后转移至室温继续反应18h,加入100ml水,分液,用饱和氢氧化钠溶液洗涤有机相,水相用二氯甲烷萃取,合并有机相用无水氯化钙干燥,过滤后将所得滤液于70℃减压蒸馏除去甲苯,再升温收集160~180℃馏分即为缚酸剂。
壳聚糖4-(二乙氨基)水杨醛希夫碱的加入能明显降低革屑水解液的铬含量,未加该脱铬剂的革屑水解液铬含量为0.042mg/g,加入脱铬剂后革屑水解液铬含量降低至0.003mg/g。
表1亲核催化剂对表面活性剂缩合产率的影响
由表1可以发现,本发明亲核催化剂可以有效提高缩合反应的转化率、提高表面活性剂产品的纯度。
表2亲核催化剂对表面活性剂产率的影响(未加ptc)
由表2可以发现,聚苯乙烯-二乙烯苯接枝季铵盐(ptc)对亲核催化剂的催化效果有明显的促进作用,可以有效提高缩合反应的转化率。
表3本发明亲核催化剂对表面活性剂产率的影响(未加吗啉环己烯胺)
由表3可以发现,吗啉环己烯胺的加入对亲核试剂的催化效果也有一定的提升作用。
表4本发明亲核催化剂对表面活性剂产率的影响(未加聚醚链叔胺)
由表4可以发现,聚醚链叔胺的加入对亲核催化剂的催化效果有明显的促进作用,实例1-5所得表面活性剂的产率均有明显的提升。
表5缚酸剂对表面活性剂产率的影响
如表5所示,本发明缚酸剂的加入可以显著提高蛋白基表面活性剂的产率。未加缚酸剂时,缩合反应产生的hcl会使体系中活性基团游离氨基呈现质子化状态,导致亲核性降低,不利于缩合主反应的顺利进行;并且会腐蚀实验设备、污染环境。因此应不断滴加本发明缚酸剂,中和反应产生的hcl,以保证缩合反应的顺利进行。
对本发明实例1-5所得蛋白基表面活性剂及其水解液利用乌氏粘度计进行特性粘数测定,其结果如表6所示。
表6表面活性剂及其水解液的特性粘数
由表6可以看出,实例1-5所得蛋白基表面活性剂的特性粘数均比与其对应的水解液的特性粘数要大,说明缩合产物的分子量均得到不同程度的增加,即已成功将油酰基的长碳链引入胶原多肽合成目标产物——蛋白基表面活性剂。
对本发明实例1-5所得蛋白基表面活性剂及其水解液进行红外光谱分析,其结果分别如图1-5所示。由图1-5可以看出实例所得5种蛋白基表面活性剂在1470~1450cm-1、1690~1620cm-1、2943~2854cm-1、3400~3300cm-1附近均有吸收峰出现。1470~1450cm-1附近出现的对称伸缩振动峰为多肽coo-的振动峰,1690~1620cm-1附近强烈的吸收峰为c=o的振动吸收峰,2943~2854cm-1处强烈的吸收峰为c-h的伸缩振动峰,3400~3300cm-1处强且宽的吸收峰为-nh的振动吸收峰。与水解液对比,发现表面活性剂在1690~1620cm-1,2943~2854cm-1附近的吸收峰明显增强,说明已经成功地向水解液多肽链上引入油酰基的碳链和羰基,即已合成目标产物油酰基复合多肽表面活性剂。
对实例1-5所得5种蛋白基表面活性剂(pbs1-5)进行性能测定:按照《表面活性剂hlb值的分析测定与计算》测定hlb值;按照gb/t7462-1994进行起泡能力测试;采用dsa-10mkz液滴形状分析仪测试接触角;按照《表面张力测量方法》测定表面张力、临界胶束浓度(cmc),并以市场上常用的表面活性剂油酸钠作为对比,其结果如表7所示:
表7表面活性剂的性能参数
由表7可知:合成的5种表面活性剂的hlb值均在7左右,适宜用作润湿剂、乳化剂;与油酸钠相比,合成的表面活性剂3、表面活性4、表面活性剂5降低水的表面张力的能力更强,说明合成的产品性能优异;合成的5种表面活性剂起泡性比油酸钠稍弱,但是稳泡性却比油酸钠要好;5种表面活性剂的润湿性能均优于油酸钠。
对实例1-5所得5种蛋白基表面活性剂(pbs1-5)进行乳化能力及乳化稳定性测定,并以市场上常用的表面活性剂油酸钠作为对比,其结果如图6-7所示:
图6-7可知,实例1-5合成的5种表面活性剂的析水率均比油酸钠低,乳化稳定指数均比油酸钠高,即合成的5种表面活性剂的乳化能力、乳化稳定性均明显优于油酸钠。
以铬革屑为原料通过加入自制脱铬剂壳聚糖4-(二乙氨基)水杨醛希夫碱、缚酸剂、亲核催化剂得到了5种铬含量极低、产品纯度更高的蛋白基表面活性剂产品。合成的5种表面活性剂均适宜用作乳化剂、润湿剂、渗透剂;表面活性剂1、表面活性剂2还适宜用作稳泡剂;表面活性剂3、表面活性剂4、表面活性剂5还适宜用作洗涤剂、发泡剂。该发明解决了制革废弃物环境污染的问题,在将蛋白质资源回收利用的同时也得到了性能优良、生物降解性好的表面活性剂产品,因此具有极好的应用及发展前景。
- 还没有人留言评论。精彩留言会获得点赞!