一种合成1‑(2,4‑二氯苯基)‑3‑甲基‑4‑二氟甲基‑1,2,4‑三唑‑5‑酮的方法与流程
本发明涉及除草剂制备技术领域,具体为一种合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法。
背景技术:
甲磺草胺是一种属于二氟甲基三唑啉酮类的除草剂,具有高效、低毒、广谱的特点,1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮是合成甲磺草胺的重要中间体,化学结构式如下:
目前大部分厂家生产采用以苯肼为起始原料,先合成1-苯基-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮,再在苯环上两个氯生成中间体,但此种方式需要经过二次氯化,苯环上两个氯比较困难,杂质高,收率低;此外也有以2,4-二氯苯胺为起原料,但由于苯环上有两个氯,杂环成环阻力大,收率低,同时氟甲化反应转化率也低于目前水平。目前国内现有技术公开了关于制备1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的工艺路线主要有以下两种。
张元元等人公开了除草剂甲磺草胺的合成(除草剂甲磺草胺的合成,张元元等,农药研究与应用,第1期,2008年)工艺,该合成方法以2,4-二氯苯胺为初始原料,经重氮化、缩合、n-烷基化得到甲磺草胺中间体二氟甲基三唑啉酮,本发明的有益效果体现在反应原料易得,条件温和,操作简便,适合在工业上推广利用;但该合成存在一定的缺点,以2,4-二氯苯胺为原料,中间产物易出现位阻现象,且产物转化率低,同时对位上氯在氟甲化时高温下容易脱卤,形成杂质。
中国专利cn106478532a公开了一种合成二氟甲基三唑啉酮的方法:以邻氯苯肼盐酸盐为原料,经过碱将邻氯苯肼盐酸盐游离邻氯苯肼,邻氯苯肼经二氯甲烷萃取后,与原乙酸三甲酯和氰酸钾反应得到1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮,1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮再经n-烷基化、氯化得到1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮。本发明的有益效果体现在采用邻氯苯肼盐酸盐,使中间产物的位阻小,收率高,但该方法存在一定的缺点,游离邻氯苯肼需要用二氯甲烷萃取,二氯甲烷沸点低,回收损耗大,增加成本,环合时使用原乙酸三甲酯和氰酸钾价格比较高,并且氯化反应需要复合型催化剂,催化剂回收操作复杂。
技术实现要素:
本发明的目的在于提供一种合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,以邻氯苯胺为原料,通过重氮化、还原、成盐合成邻氯苯肼盐,经碱中和成游离邻氯苯肼,邻氯苯肼和乙醛、氰酸钠、次氯酸钠经过缩合、环化和氧化反应生成1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮,1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮再经过成盐、氟钾化和氯化反应得到1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮(甲磺草胺中间体),以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:一种合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,以邻氯苯胺为原料,通过重氮化、还原、成盐合成邻氯苯肼盐,经碱中和成游离邻氯苯肼,邻氯苯肼和乙醛、氰酸钠、次氯酸钠经过缩合、环化和氧化反应生成1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮,1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮再经过成盐、氟钾化和氯化反应得到1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮(甲磺草胺中间体)。
本发明更进一步的技术方案,包括以下步骤:
(1)邻氯苯肼盐酸盐合成
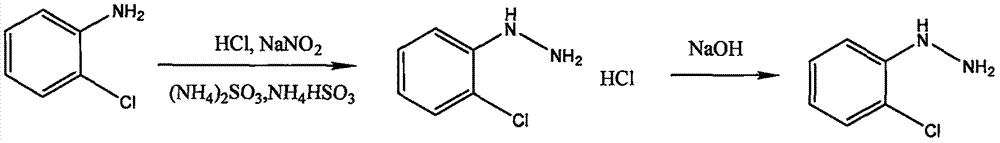
将邻氯苯胺加入装有定量盐酸的反应釜中,保持反应温度在-30-5℃,然后向反应釜中加入亚硝酸钠溶液,邻氯苯胺与亚硝酸钠发生重氮化反应,之后加入亚硫酸铵和亚硫酸氢铵溶液发生还原反应,生成邻氯苯肼盐酸盐,反应结束得到中间体1,滴加定量氢氧化钠溶液中和,整个重氮化、还原、成盐和中和反应均在常压下进行。
其中,所述中间体1为c6h7cln2,产物为c6h7cln2和水h2o;c6h7cln2和水h2o的质量比为142∶18。
(2)成腙反应
常温下在成腙反应釜内依次加入定量邻氯苯肼,定量溶剂叔丁醇,控制温度在-5-10℃滴加定量乙醛,-5-10℃保温搅拌15分钟,,生成中间体2待下一步反应。
其中,所述中间体2为c8h9cln2,产物为c8h9cln2、水h2o;c8h9cln2、水h2o的质量比为168∶18。
(3)环化反应
上述中间体2加入定量氰酸钠和定量盐酸,在酸性条件下控制温度为-5-10℃保温反应2.5小时,反应结束得到中间体3,待下步反应。
其中,所述中间体3为c9h10cln3o,产物为c9h10cln3o和氯化钠nacl;c9h10cln3o和氯化钠nacl的质量比为422∶117。
(4)氧化反应
升温到5-30℃,于环化反应产物中间体3中滴加定量配好的次氯酸钠溶液,滴加过程中控制温度在5-30℃。滴加完次氯酸钠,5-30℃保温30分钟,反应结束得到中间体4,待下步反应。
其中,所述中间体4为c9h8cln3o,产物为c9h8cln3o和氯化钠nacl、水h2o;c9h8cln3o和氯化钠nacl、水h2o的质量比为418∶117∶36。
(5)成盐反应
向反应釜内投入定量dmf和定量中间体4,搅拌0.5小时,而后在常温条件下投入定量碳酸钾,升温至160-165℃,保温反应0.5小时,生成中间体5,待下一步反应。
其中,所述中间体5为kc9h7cln3o,产物为kc9h7cln3o和二氧化碳co2、水h2o;kc9h7cln3o和二氧化碳co2、水h2o的质量比为247∶44∶18。
(6)氟钾化反应
将脱水后反应釜升温至180-190℃,通入定量一氯二氟甲烷,保温反应15分钟,降温至80℃,转料至结晶釜降温,离心过滤钾盐后,脱除溶剂,得到中间体6,待下一步反应。
其中,所述中间体6为c10h8cln3of2,产物为c10h8cln3of2和氯化钾kcl;c10h8cln3of2和氯化钾kcl的质量比为518∶149。
(7)氯化反应
反应釜内投入定量dmf和定量中间体6,打开引风通入定量氯气,温度控制30-70℃,通完氯气,保温1小时,得到1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮(甲磺草胺中间体)。
优选的,在步骤(1)中,所述邻氯苯胺c6h6ncl、盐酸hcl、亚硝酸钠nano2、亚硫酸铵(nh4)2so3、亚硫酸氢铵nh4hso3、氢氧化钠naoh的质量比为254∶73∶138∶232∶198∶80。
优选的,在步骤(2)中,所述中间体1邻氯苯肼c6h7cln2、乙醛c2h4o的质量比为142∶44。
优选的,在步骤(3)中,所述中间体2c8h9cln2、氰酸钠naocn、盐酸hcl的质量比为336∶130∶73。
优选的,在步骤(4)中,所述中间体3c9h10cln3o、次氯酸钠naclo的质量比为422∶149。
优选的,在步骤(5)中,所述中间体4c9h8cln3o、碳酸钾k2co3的质量比为209∶138。
优选的,在步骤(6)中,所述中间体5kc9h7cln3o、一氯二氟甲烷hcf2cl的质量比为494∶173。
优选的,在步骤(7)中,所述中间体6c10h8cln3of2、氯气cl2的质量比为259∶71。
优选的,在步骤(1-7)中,邻氯苯肼盐酸盐合成、成腙反应、环化反应、氧化反应、成盐反应、氟钾化反应和氯化反应的转化率均大于97%。
与现有技术相比,本发明的有益效果是:
1、本发明合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,以邻氯苯胺为起始原料,只需一步氯化反应,能够使合成中间体的位阻小,杂质低,从而提高了收率。
2、本发明合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,邻氯苯肼盐酸盐用氢氧化钠溶液中和后得到的游离邻氯苯肼,不需要有机溶剂萃取,直接与乙醛进行下一步反应,操作简便。
3、本发明合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,反应原料乙醛和氰酸钠价格低且易得,一定程度上降低了生产成本。
4、本发明合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,氯化反应不需要复合型催化剂,不存在催化剂回收问题。
5、本发明合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,氯化反应采用酸性dmf溶剂,经过简单脱溶即可直接回收套用,操作简便。
6、本发明合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,反应步数少,且各步工艺条件温和,安全性高,操作简便,原料廉价易得,反应温和,操作简便,有利于实现规模化生产。
附图说明
图1为本发明的甲磺草胺中间体合成反应方程式;
图2为本发明的邻氯苯肼盐酸盐合成方程式;
图3为本发明的成腙反应方程式;
图4为本发明的环化反应方程式;
图5为本发明的氧化反应方程式;
图6为本发明的成盐反应方程式;
图7为本发明的氟钾化反应方程式;
图8为本发明的氯化反应方程式;
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
请参阅图1,一种合成1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮的方法,以邻氯苯胺为原料,通过重氮化、还原、成盐合成邻氯苯肼盐,经碱中和成游离邻氯苯肼,邻氯苯肼和乙醛、氰酸钠、次氯酸钠经过缩合、环化和氧化反应生成1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮,1-邻氯苯基-3-甲基-1h-1,2,4-三唑-5-酮再经过成盐、氟钾化和氯化反应得到1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮(甲磺草胺中间体)。
本发明更进一步的技术方案,包括以下步骤:
(1)邻氯苯肼盐酸盐合成
将邻氯苯胺加入装有定量盐酸的反应釜中,保持反应温度在-30-5℃,然后向反应釜中加入亚硝酸钠溶液,邻氯苯胺与亚硝酸钠发生重氮化反应,之后加入亚硫酸铵和亚硫酸氢铵溶液发生还原反应,生成邻氯苯肼盐酸盐,反应结束得到中间体1,滴加定量氢氧化钠溶液中和,整个重氮化、还原、成盐和中和反应均在常压下进行。
其中,所述中间体1为c6h7cln2,产物为c6h7cln2和水h2o;c6h7cln2和水h2o的质量比为142∶18。
其中,所述邻氯苯胺c6h6ncl、盐酸hcl、亚硝酸钠nano2、亚硫酸铵(nh4)2so3、亚硫酸氢铵nh4hso3、氢氧化钠naoh的质量比为254∶73∶138∶232∶198∶80。
(2)成腙反应
常温下在成腙反应釜内依次加入定量邻氯苯肼,定量溶剂叔丁醇,控制温度在-5-10℃滴加定量乙醛,-5-10℃保温搅拌15分钟,,生成中间体2待下一步反应。
其中,所述中间体2为c8h9cln2,产物为c8h9cln2、水h2o;c8h9cln2、水h2o的质量比为168∶18。
其中,所述中间体1邻氯苯肼c6h7cln2、乙醛c2h4o的质量比为142∶44。
(3)环化反应
上述中间体2加入定量氰酸钠和定量盐酸,在酸性条件下控制温度为-5-10℃保温反应2.5小时,反应结束得到中间体3,待下步反应。
其中,所述中间体3为c9h10cln3o,产物为c9h10cln3o和氯化钠nacl;c9h10cln3o和氯化钠nacl的质量比为422∶117。
其中,所述中间体2c8h9cln2、氰酸钠naocn、盐酸hcl的质量比为336∶130∶73。
(4)氧化反应
升温到5-30℃,于环化反应产物中间体3中滴加定量配好的次氯酸钠溶液,滴加过程中控制温度在5-30℃。滴加完次氯酸钠,5-30℃保温30分钟,反应结束得到中间体4,待下步反应。
其中,所述中间体4为c9h8cln3o,产物为c9h8cln3o和氯化钠nacl、水h2o;c9h8cln3o和氯化钠nacl、水h2o的质量比为418∶117∶36。
其中,所述中间体3c9h10cln3o、次氯酸钠naclo的质量比为422∶149。
(5)成盐反应
向反应釜内投入定量dmf和定量中间体4,搅拌0.5小时,而后在常温条件下投入定量碳酸钾,升温至160-165℃,保温反应0.5小时,生成中间体5,待下一步反应。
其中,所述中间体5为kc9h7cln3o,产物为kc9h7cln3o和二氧化碳co2、水h2o;kc9h7cln3o和二氧化碳co2、水h2o的质量比为247∶44∶18。
其中,所述中间体4c9h8cln3o、碳酸钾k2co3的质量比为209∶138。
(6)氟钾化反应
将脱水后反应釜升温至180-190℃,通入定量一氯二氟甲烷,保温反应15分钟,降温至80℃,转料至结晶釜降温,离心过滤钾盐后,脱除溶剂,得到中间体6,待下一步反应。
其中,所述中间体6为c10h8cln3of2,产物为c10h8cln3of2和氯化钾kcl;c10h8cln3of2和氯化钾kcl的质量比为518∶149。
其中,所述中间体5kc9h7cln3o、一氯二氟甲烷hcf2cl的质量比为494∶173。
(7)氯化反应
反应釜内投入定量dmf和定量中间体6,打开引风通入定量氯气,温度控制30-70℃,通完氯气,保温1小时,得到1-(2,4-二氯苯基)-3-甲基-4-二氟甲基-1,2,4-三唑-5-酮(甲磺草胺中间体)。
其中,所述中间体6c10h8cln3of2、氯气cl2的质量比为259∶71。
在步骤(1-7)中,邻氯苯肼盐酸盐合成、成腙反应、环化反应、氧化反应、成盐反应、氟钾化反应和氯化反应的转化率均大于97%。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种2-氯-n-(2,4-二氯苯基)-亚胺代乙酰氯的合成方法
- 制备l-(4-(4-(3,4-二氯-2-氟苯基氨基)-7-甲氧基喹唑啉-6-基氧)哌啶-l-基)丙-2-烯 ...的制作方法
- 一种1-(4-氯苯基)-2-环丙基-1-丙酮的合成方法
- 一种2,4-二氯-5-氟苯乙酮酰化反应尾气处理装置的制造方法
- 1-(5′-(5-(3,5-二氯-4-氟苯基)-5-(三氟甲基)-4,5-二氢异噁唑-3-基)-3′h-螺[氮杂 ...的制作方法
- 一种2-[(4-氯苯基)(4-哌啶基氧基)甲基]吡啶及其衍生物的合成方法
- 一种药物中间体4-(4-氯苯基)环己基-1-甲酸的合成方法
- 一种3-(4-氯苯基)戊二酸镉配位聚合物及其制备方法
- 1-苯甲酰基-3-取代苯基硫脲的微波合成法的制作方法
- 负载型催化剂在合成n-苯基马来酰亚胺的应用的制作方法