一种1‑甲基‑4‑硝基吡唑的制备方法与流程
本发明涉及有机化合物的合成领域,尤其涉及一种1-甲基-4-硝基吡唑的制备方法。
背景技术:
1-甲基-4-硝基吡唑是一种典型的氮杂环类化合物,该结构含有高焓c=c双键和n-n单键,具有较高生成焓、高密度以及对环境友好的特征,具有非常可观的能量,在高能炸药、安全钝感、低感添加剂等领域有很好的前景,是新型含能材料1-甲基-3,4,5-三硝基吡唑等的中间体。
吡唑或甲基吡唑的硝化一般采用发烟硝酸-乙酸酐或发烟硝酸-浓硫酸作为硝化试剂,吡唑环的n-硝化采用发烟硝酸-乙酸酐为硝化试剂,吡唑环的c-硝化采用发烟硝酸-浓硫酸为硝化试剂。
现有技术中,1-甲基-4-硝基吡唑的制备一般采用1-甲基吡唑与发烟硝酸-浓硫酸硝化体系反应。但是这种方法使用的硝化剂发烟硝酸-浓硫酸会产生大量的氮氧化物气体以及过多的废酸而造成环境污染。
技术实现要素:
有鉴于此,本发明的目的在于提供一种1-甲基-4-硝基吡唑的制备方法,该方法合成工艺简单,避免了使用发烟硝酸为硝化剂,环境污染小。
为了实现上述发明目的,本发明提供以下技术方案:
一种1-甲基-4-硝基吡唑的制备方法,包括以下步骤:
将1-甲基吡唑、n-硝基吡唑和硫酸溶液混合进行硝化反应,得到1-甲基-4-硝基吡唑。
优选地,所述1-甲基吡唑、n-硝基吡唑和硫酸溶液的物质的量比为1:1~3:7.5~12.5。
优选地,所述硝化反应的温度为20~80℃。
优选地,所述硝化反应的温度为40~60℃。
优选地,所述硝化反应的时间为2~8h。
优选地,所述硝化反应的时间为4~6h。
优选地,所述硫酸溶液的质量浓度为40%~98%。
优选地,所述1-甲基吡唑、n-硝基吡唑和硫酸溶液的混合具体为:
将1-甲基吡唑和硫酸溶液混合后加热至20~80℃,得到1-甲基吡唑和硫酸溶液的热混合液;
分8~12次向所述1-甲基吡唑和硫酸溶液的热混合液中加入n-硝基吡唑。
优选地,所述硝化反应后还包括提纯过程,包括以下步骤:
将硝化反应后所得硝化反应液冷却后过滤,得到滤液和第一固体;
将所述滤液依次进行萃取和蒸馏,得到第二固体;
第一固体和第二固体混合后进行重结晶,得到1-甲基-4-硝基吡唑。
优选地,所述冷却温度为0~5℃。
有益技术效果:
本发明提供了一种1-甲基-4-硝基吡唑的制备方法。本发明的制备方法以n-硝基吡唑和硫酸为硝化试剂,直接将1-甲基吡唑进行硝化即可得到1-甲基-4-硝基吡唑,原料来源丰富,成本低,且避免了使用硝酸和硫酸的混酸为硝化剂,反应过程无污染气体氮氧化物的产生,不产生废酸,对环境污染小;进一步的,本发明提供的制备方法在20~80℃温度下进行硝化反应,反应条件温和,不需要在高温下长时间反应,对反应设备要求较低,易于提纯,且所得产物纯度高。实施例结果表明,以本发明提供的制备方法制备1-甲基-4-硝基吡唑,所得纯化后的1-甲基-4-硝基吡唑的纯度可以达到99%以上,收率为50%~75%。
附图说明
图1是实施例1制备得到的目标产物的高效液相色谱图;
图2是实施例1制备得到的目标产物的红外光谱图;
图3是实施例1制备得到的目标产物的质谱图;
图4是实施例1制备得到的目标产物的核磁共振氢谱;
图5是实施例1制备得到的目标产物的核磁共振碳谱。
具体实施方式
本发明提供了一种1-甲基-4-硝基吡唑的制备方法,包括以下步骤:
将1-甲基吡唑、n-硝基吡唑和硫酸溶液混合进行硝化反应,得到1-甲基-4-硝基吡唑。
在本发明中,所述1-甲基吡唑、n-硝基吡唑和硫酸溶液中硫酸的物质的量比优选为1:1~3:7.5~12.5,更优选为1:1.5~2.5:9~11;所述硫酸溶液的质量浓度优选为40%~98%,更优选为75%~90%,最优选为85~90%。在本发明中,硫酸能将n-硝基吡唑的未取代氮原子质子化,使得其邻氮原子上的硝基活化而产生硝酰氧离子(no2+),硝酰氧离子(no2+)进一步与n-甲基吡唑发生c-硝化反应。
在本发明中,所述硝化反应的温度优选为20~80℃,更优选为40~60℃;所述硝化反应的时间优选为2~8h,更优选为4~6h。
在本发明中,所述1-甲基吡唑、n-硝基吡唑和硫酸溶液的混合优选具体为:
将1-甲基吡唑和硫酸溶液混合后加热至20~80℃,得到1-甲基吡唑和硫酸溶液的热混合液;
分8~12次向所述1-甲基吡唑和硫酸溶液的热混合液中加入n-硝基吡唑。
本发明将1-甲基吡唑和硫酸溶液混合后加热至20~80℃,得到1-甲基吡唑和硫酸溶液的热混合液。本发明优选在搅拌条件下将1-甲基吡唑滴加到硫酸溶液中,得到1-甲基吡唑和硫酸溶液的混合液,所述滴加速度优选为1~2滴/秒;所述搅拌速度优选为300~400转/分,更优选为320~380转/分,最优选为350转/分;本发明对搅拌方法没有特殊限制,选用本领域常规的搅拌方法即可,具体的如机械搅拌。
得到1-甲基吡唑和硫酸的混合液后,本发明优选将1-甲基吡唑和硫酸的混合液加热至20~80℃,得到1-甲基吡唑和硫酸的热混合液。在本发明中,所述加热的速率优选为1~5℃/分,更优选为2~3℃/分;所述热混合液更优选为加热至40~60℃。
得到热混合液后,本发明优选将n-硝基吡唑分8~12次加入热混合液中。在本发明中,所述n-硝基吡唑的加入批次优选为8~12次,更优选为10次,每批次加入的n-硝基吡唑的质量为所需n-硝基吡唑的总质量与相应加入批次的比值。在本发明的具体实施例中,每次加入n-硝基吡唑后会出现升温现象,本发明优选在温度降至初始加料温度时再次加入n-硝基吡唑。
得到1-甲基吡唑、n-硝基吡唑和硫酸的混合液后,本发明优选将混合液在20~80℃保温2~8h,进行硝化反应;本发明的硝化反应时间自n-硝基吡唑加料完成后开始计算。
在本发明中,所述硝化反应的方程式如式i所示:
硝化反应完成后,本发明优选还包括:将得到的硝化反应液进行提纯,得到1-甲基-4-硝基吡唑纯品,所述提纯优选包括以下步骤:
将硝化反应液冷却后过滤,得到滤液和第一固体;
将所述滤液依次进行萃取和蒸馏,得到第二固体;
将所述第一固体和所述第二固体混合后进行重结晶,得到1-甲基-4-硝基吡唑。
本发明将硝化反应液冷却后过滤,得到滤液和第一固体。在本发明中,所述冷却的温度优选为0~5℃,更优选为2~4℃;所述冷却的降温速率优选为5~10℃/分钟,更优选为7~9℃/分钟;本发明对所述冷却的方法没有特殊限制,能够冷却至要求温度即可;在本发明的具体实施例中,优选将硝化反应液与冰块混合进行冷却。本发明对所述过滤没有特殊要求,使用本领域技术人员熟知的过滤方法即可,具体的如抽滤。
过滤得到滤液和第一固体后,本发明优选将所述滤液依次进行萃取和蒸馏,得到第二固体。在本发明中,所述萃取的萃取剂优选为乙醚、乙醇或乙酸乙酯,更优选为乙醚或乙醇;本发明对所述萃取的具体条件没有特殊限制,选用本领域常规萃取条件,能够将滤液中的1-甲基-4-硝基吡唑完全萃取到有机相中即可。
萃取后,本发明优选将所述萃取得到的有机相进行蒸馏,得到第二固体。在本发明中,所述蒸馏温度优选为35~80℃,更优选为45~65℃,最优选为50~60℃,所述蒸馏程度为将有机相蒸馏至原来三分之一时停止蒸馏,倒出液体自然晾干得到第二固体。
在本发明中,萃取后的萃余液中主要成分为硫酸,本发明优选将萃余液进行浓缩回收硫酸,本发明对浓缩的方法没有特殊限定,采用本领域技术人员熟知的浓缩方法即可,如蒸发浓缩。
在本发明中,硝化反应后n-硝基吡唑脱掉硝基转变为副产物吡唑,吡唑存在于萃取后的水相,可通过苯萃取吡唑进行回收,可为其它药物合成的前体。本发明优选将产生的副产物吡唑回收,本发明对吡唑的回收方法没有特殊限制,采用本领域技术人员熟知的回收方法即可,如将萃取后的水相中加入苯溶剂萃取出吡唑,蒸发苯溶剂回收吡唑。
得到第一固体和第二固体后,本发明优选将所述第一固体和第二固体混合后进行重结晶,得到1-甲基-4-硝基吡唑纯品。在本发明中,所述重结晶用溶剂优选为醇类溶剂,更优选为乙醇。本发明对所述重结晶的方法没有特殊的限制,选用本领域常规的重结晶方法即可。
下面结合实施例对本发明提供的对1-甲基-4-硝基吡唑的制备方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
分别取1.4g(0.012mol)n-硝基吡唑,1g(0.012mol)1-甲基吡唑备用,将8ml98%的浓硫酸加入装有机械搅拌、温度计、加料漏斗的100ml四口瓶中,于20℃下用加料漏斗向浓硫酸中滴加1-甲基吡唑,加料完成水浴升温至60℃,于此温度下分10次加入n-硝基吡唑,每次加入量为0.0012mol,加料完成,保持搅拌恒温反应6h,反应完成后,趁热将反应液倒入装有冰块的烧杯中搅拌,有白色细小晶状固体析出,冰块全融,抽滤,得白色固体i和滤液;滤液用乙醚萃取,合并溶剂,减压旋蒸除去溶剂,得到白色固体ii,将白色固体i与白色固体ii合并后用乙醇进行重结晶,得到目标产物。产品的纯度为99%,产品熔点为:90~93℃;收率为72.53%。
表1对实施例1中目标产物的元素分析结果
由表1的元素分析可知,所得目标产物n、c、h的元素含量与1-甲基-4-硝基吡唑理论值基本一致。
图1是测试所得目标产物纯度的高效液相色谱图
色谱条件:采用色谱甲醇配制含目标物的溶液,以水和甲醇为流动相,水和甲醇的体积比为45:55为流动相,紫外吸收波长237nm,流速为1ml·min-1,进样量10μl。测试结果如图1可知,1-甲基-4-硝基吡唑的保留时间为4.37min,用面积归一法计算产物纯度为99.1%。
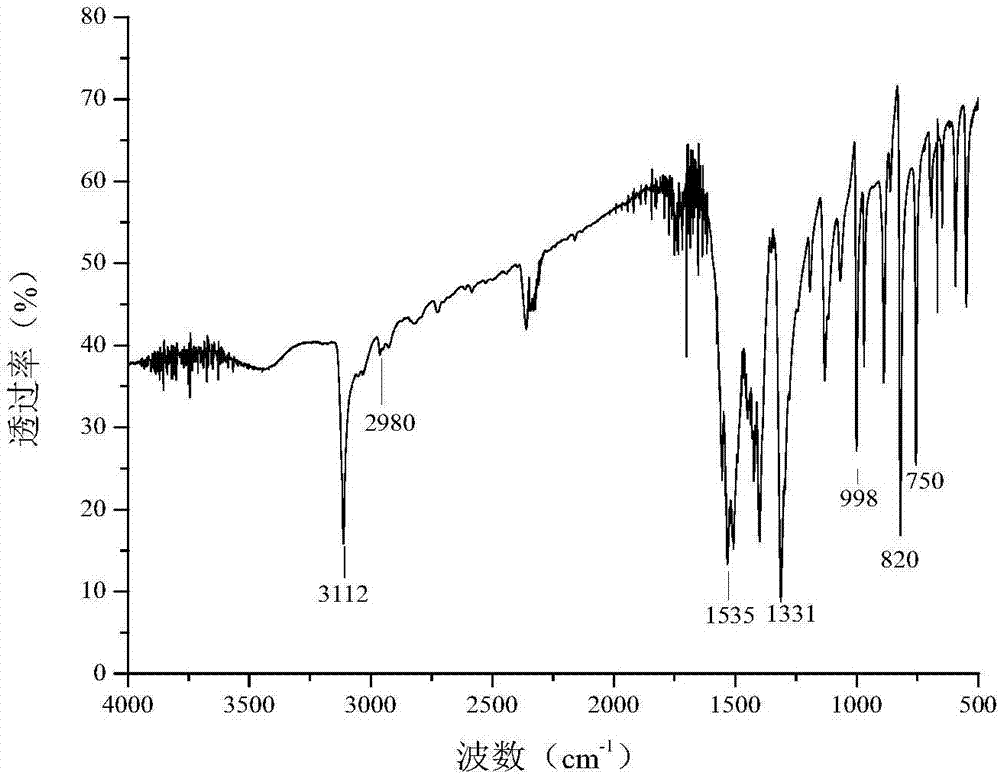
图2是实施例1制备得到的目标产物的红外光谱图,所得数据为:
ir(kbr,vcm-1):3112(吡唑环c-h),2980(甲基c-h),1535,1331(c-no2)。由图2可知被测目标产物结构中存在硝基,甲基,吡唑环。
图3是实施例1制备得到的目标产物的质谱图,图3中分子离子峰为127,与1-甲基-4-硝基吡唑分子量127.10相近。
图4是实施例1制备得到的目标产物的核磁共振氢谱图,所得数据如下:
1hnmr(acetone-d6)δ:8.55(1h),8.05(1h),3.99(3h)。由上述数据可知,被测目标产物有3种氢原子存在。
图5为所得目标产物核磁共振碳谱图,所得数据如下:
13cnmr(acetone-d6):39.34(t,ch3),135.59(t,c3),130.09(t,c5),135.05(t,c4)。由上述数据可知,被测目标产物有4种碳原子存在。
综上所述,所测目标产物的分子结构与1-甲基-4-硝基吡唑分子结构相符,即本发明实施例1中所得目标产物为1-甲基-4-硝基吡唑,产品的纯度为99.1%。
实施例2
分别取4.1g(0.036mol)n-硝基吡唑,1g(0.012mol)1-甲基吡唑备用,将6ml98%的浓硫酸加入装有机械搅拌、温度计、加料漏斗的100ml的四口瓶中,于20℃下用恒压滴液漏斗向浓硫酸中滴加1-甲基吡唑,加料完成水浴升温至80℃,于此温度下分12次加入n-硝基吡唑,每次加入0.003mol,加料完成后,在20℃下恒温反应4h,趁热将反应液倒入装有冰块的烧杯中搅拌,有白色细小晶状固体析出,冰块全融后,抽滤,得白色固体i;滤液用乙醚萃取,合并溶剂,减压旋蒸除去溶剂,得到白色固体ii;将白色固体i与白色固体ii合并后用乙醇进行重结晶得目标产物。
采用实施例1中的测试方法对该目标产物的结构进行表征,所得测试结果与实施例1相似,证明实施例2中所得目标产物为1-甲基-4-硝基吡唑,产品的纯度为99.1%,产品熔点为:89~92℃;收率为69.52%。
实施例3
分别取2.7g(0.024mol)n-硝基吡唑,1g(0.012mol)1-甲基吡唑备用,将8ml98%的浓硫酸加入装有机械搅拌、温度计、加料漏斗的100ml四口瓶,于20℃下用恒压滴液漏斗向浓硫酸中滴加1-甲基吡唑,加料完成水浴升温至35℃,于此温度下分10次加入n-硝基吡唑,每次加入0.0024mol,恒温反应6h,趁热将反应液倒入装有冰块的烧杯中搅拌,有白色细小晶状固体析出,冰块全融后,抽滤,得白色固体i和滤液;滤液用乙醚萃取,合并溶剂,减压旋蒸除去溶剂,得到白色固体ii,将白色固体i和白色固体ii合并后用乙醇重结晶得目标产物。
采用实施例1中的测试方法对该目标产物的结构进行表征,所得测试结果与实施例1相似,证明实施例3中所得目标产物为1-甲基-4-硝基吡唑,产品的纯度为99.2%,产品熔点为:90~91℃;收率为74.01%。
实施例4
分别取2.7g(0.024mol)n-硝基吡唑,1g(0.012mol)1-甲基吡唑备用,将8ml60%的浓硫酸加入装有机械搅拌、温度计、加料漏斗的100ml四口瓶,于20℃下用恒压滴液漏斗向浓硫酸中滴加1-甲基吡唑,加料完成水浴升温至30℃,于此温度下分8次加入n-硝基吡唑,每次加入0.003mol,恒温反应4h,趁热将反应液倒入装有冰块的烧杯中搅拌,有白色细小晶状固体析出,冰块全融后,抽滤得白色固体i和滤液;所得滤液用乙醚萃取,合并溶剂,减压旋蒸除去溶剂,得到白色固体ii,将白色固体i和白色固体ii合并后用乙醇重结晶得目标产物。
采用实施例1的测试方法对该目标产物的结构进行表征,所得测试结果与实施例1相似,证明实施例4中所得目标产物为1-甲基-4-硝基吡唑,产品的纯度为99.1%,产品熔点为:90~92℃;收率为53.81%。
实施例5
分别取4.1g(0.036mol)n-硝基吡唑,1g(0.012mol)1-甲基吡唑备用,将8ml40%的浓硫酸加入装有机械搅拌、温度计、加料漏斗的100ml四口瓶,于20℃下用恒压滴液漏斗向浓硫酸中滴加1-甲基吡唑,加料完成水浴升温至50℃,于此温度下分12次加入n-硝基吡唑,每次加入量为0.003mol,恒温反应3h,趁热将反应液倒入装有冰块的烧杯中搅拌,有白色细小晶状固体析出,冰块全融后,抽滤得白色固体i和滤液;所得滤液用乙醚萃取,合并溶剂,减压旋蒸除去溶剂,得到白色固体ii,将白色固体i和白色固体ii合并后用乙醇重结晶得目标产物。
采用实施例1中的测试方法对该目标产物的结构进行表征,所得测试结果与实施例1相似,证明实施例5中所得目标产物为1-甲基-4-硝基吡唑,产品的纯度为99.1%,产品熔点为:89~92℃;收率为51.26%。
由以上实施例可知本发明的制备方法以n-硝基吡唑和硫酸为硝化试剂,直接将1-甲基吡唑进行硝化即可得到1-甲基-4-硝基吡唑。本发明的制备方法原料来源丰富,成本低,且避免了使用硝酸和硫酸的混酸为硝化剂,反应过程无污染气体氮氧化物的产生,对环境污染小;并且本发明提供的制备方法反应条件温和,不需要在高温下长时间反应,对反应设备要求较低,且所得产物纯度可达99%,收率为50%~75%。
本发明以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种含虫酰肼和吡唑茚满基甲酰胺的杀虫组合物的制作方法
- 5-(4-氟苯基)-N–(4-氯苯乙基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 制备1-烷基-3-二氟甲基-5-氟-1h-吡唑-4-甲醛和1-烷基-3-二氟甲基-5-氟-1h-吡唑-4 ...的制作方法
- 含有色酮结构的吡唑类n‐对硝基苯基马来酰亚胺衍生物及其制备方法与应用
- 含有色酮结构的吡唑类n-苯基马来酰亚胺衍生物及其制备方法与应用
- 三氟甲基吡唑基胍F<sub>1</sub>F<sub>0</sub>-ATP酶抑制剂及其治疗用图
- 5-(4-氟苯基)-7-三氟甲基吡唑并[1,5-a] 嘧啶-3-羧酸乙酯的合成方法
- 5-(4-氟苯基)-7-三氟甲基吡唑并[1,5-a] 嘧啶-3-羧酸的合成方法
- 5-(4-氟苯基)-N–1-(3-羟基金刚烷基)-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 一种胺苯吡唑酮残留量的gc-ei-ms快速测定方法
- 5-(4-氟苯基)-N,N-二甲基-7-三氟甲基吡唑并[1,5-a]嘧啶-3-酰胺的合成方法
- 制备1-烷基-3-二氟甲基-5-氟-1h-吡唑-4-甲醛和1-烷基-3-二氟甲基-5-氟-1h-吡唑-4 ...的制作方法
- 三氟甲基吡唑基胍F<sub>1</sub>F<sub>0</sub>-ATP酶抑制剂及其治疗用图
- 3,5-二甲基-1h-吡唑-1-甲脒盐酸盐的制备方法
- 取代吡唑基吡唑衍生物及其作为除草剂的用图
- 取代吡唑基吡唑衍生物及其作为除草剂的用图
- 封端异氰酸酯、涂料组合物、粘接剂组合物和物品的制作方法
- 一种1-(3-甲基-1-苯基-1h-吡唑-5-基)哌嗪的制备方法
- 一种3-二氟甲基-1-甲基-1h-吡唑-4-羧酸乙酯的合成方法
- 一种1,3,5-三甲基-4-吡唑甲酸甲酯的合成方法