阿立哌唑十二烷酸酯的制备方法与流程
本发明涉及医药化工领域,尤其涉及一种工艺简单、产品收率和纯度有显著提高且可以重现的阿立哌唑十二烷酸酯的制备方法。
背景技术:
阿立哌唑十二烷酸酯的化学名为7-{4-[4-(2,3-二氯苯基)-1-哌嗪基]丁氧基}-2-氧代-3,4-二氢喹啉-1(2h)-甲基十二烷酸酯(cas:1259305-29-7),由爱尔兰alkermes公司研发,于2015年10月5日获得美国食品药品监督管理局(fda)批准上市,商品名为aristada。该药为缓释注射混悬剂,为每月注射一次或每6周注射一次的长效治疗非典型精神分裂症药物,用于成人精神分裂症的治疗。阿立哌唑十二烷酸酯结构如下式i:
第wo2010151689a1号专利文献中公开了阿立哌唑十二烷酸酯的制备方法和治疗用途,其方法为:以阿立哌唑为起始物料与37%的甲醛溶液反应得到阿立哌唑n-亚甲基醇,n-亚甲基醇再与十二烷酸酐反应,柱层析得到阿立哌唑十二烷酸酯(i),合成路线如下:
然而,该方法存在如下缺点:(1)其后处理纯化需要柱层析,操作繁琐,不方便工业化生产;(2):收率低,两步总收率仅13.65%。
另,第wo2011140183a1专利文献页公开了另一种阿立哌唑十二烷酸酯的制备方法,以阿立哌唑为起始物料与37%甲醛反应得到阿立哌唑n-亚甲基醇,n-亚甲基醇再与十二烷酰氯反应,柱层析得到阿立哌唑十二烷酸酯(i),合成路线如下:
该方法是对第wo2010151689a1专利文献的延伸,用十二烷酰氯代替十二烷酸酐进行反应,虽然其未公开收率,但理论上与第wo2010151689a1专利文献的收率接近,且该制备方法仍然存在后处理纯化需要柱层析,操作繁琐,不方便工业化生产。
有鉴于此,有必要对现有的阿立哌唑十二烷酸酯的制备方法予以改进,以解决上述问题。
技术实现要素:
本发明的目的在于提供一种工艺简单、产品收率和纯度有显著提高且可以重现的阿立哌唑十二烷酸酯的制备方法。
为实现上述发明目的,本发明提供了阿立哌唑十二烷酸酯的制备方法,其特征在于:包括如下步骤:s1于酰胺类溶剂中,化合物(ii)与卤代硅烷在缚酸剂的存在下反应保护酚羟基得到化合物(iii),
其中r1,r2,r3为相同或不同的烷基;
s2在醇类溶剂中,化合物(iii)在缚酸剂存在下与多聚甲醛反应,处理后的中间产物在呋喃或酰胺溶剂中,在缚酸剂存在下与十二烷酰氯反应,生成化合物(iv),
其中r1,r2,r3为相同或不同的烷基;
s3化合物(iv)在呋喃溶剂中用四丁基氟化铵脱去硅醚得化合物(v)
s4在丙酮溶剂、酰胺溶剂、腈类溶剂中的一种溶剂多种溶剂混合的混合溶剂中,化合物(v)与1-溴-4-氯丁烷及1-(2,3-二氯苯基)哌嗪在缚酸剂、碘化钠存在下反应,制备化合物(i)阿立哌唑十二烷酸酯
作为本发明的进一步改进,所述卤代硅烷为叔丁基二甲基氯硅烷。
作为本发明的进一步改进,步骤s1中,化合物(ii)与卤代硅烷的摩尔比为1:1~1:2。
作为本发明的进一步改进,步骤s1中,化合物(ii)与卤代硅烷、缚酸剂的摩尔比为1:1:2~1:2:2。
作为本发明的进一步改进,步骤s2中,式(iii)与多聚甲醛的摩尔比为1:1~1:20,式(iii)与缚酸剂的摩尔比为1:0.2~1:1。
作为本发明的进一步改进,步骤s2中,式(iii)与多聚甲醛反应生成的醇与十二烷酰氯摩尔比为1:1~1:5。
作为本发明的进一步改进,步骤s3中,化合物(iv)与tbaf的摩尔比为1:1~1:5。
作为本发明的进一步改进,步骤s4中,化合物(v)与1-溴-4-氯丁烷及1-(2,3-二氯苯基)哌嗪的摩尔比为1:1:1~1:3:3。
作为本发明的进一步改进,步骤s1~步骤s4中,反应温度介于0℃~100℃之间。
作为本发明的进一步改进,所述缚酸剂为有机弱碱或无机碱,所述有机弱碱可以是三乙胺,三乙烯二胺,1,5-二氮杂二环[5.4.0]十一烯-5,1,5-二氮杂二环[4.3.0]壬烯-5,4-二甲氨基吡啶,吡啶,n-甲基吗啉,四甲基乙二胺中的一种或多种的混合;所述无机碱为氢氧化钠、氢氧化钾、氢氧化锂、氢氧化铯、碳酸钾、碳酸钠、碳酸铯、碳酸锂、碳酸氢钾、碳酸氢钾、碳酸氢锂中的一种或多种。
本发明的有益效果是:本发明的阿立哌唑十二烷酸酯的制备方法,后期处理无需柱层析等繁琐工艺、产品收率高达78%、纯度高达99.8%,降低了生产成本,便于工业生产。
附图说明
图1是本发明化合物(iii)的核磁共振图谱。
图2是本发明化合物(iv)的核磁共振图谱。
图3是本发明化合物(v)的核磁共振图谱。
图4是本发明化合物(i)的核磁共振图谱。
具体实施方式
为了使本发明的目的、技术方案和优点更加清楚,下面结合附图和具体实施例对本发明进行详细描述。
本发明描述中用到的缩写的含义为:ea:乙酸乙酯;thf:四氢呋喃;dmf:n,n-二甲基甲酰胺;tlc:薄层色谱法;tbaf:四丁基氟化铵。
本发明的阿立哌唑十二烷酸酯的制备方法,包括如下步骤:
s1于酰胺类溶剂中,化合物(ii)7-羟基喹啉酮与卤代硅烷在缚酸剂的存在下反应保护酚羟基得到化合物(iii),
其中r1,r2,r3为相同或不同的烷基。
s2在醇类溶剂中,化合物(iii)在缚酸剂存在下与多聚甲醛反应,处理后的中间产物在呋喃或酰胺溶剂中,在缚酸剂存在下与十二烷酰氯反应,生成化合物(iv),
其中r1,r2,r3为相同或不同的烷基。
s3在呋喃溶剂中,将化合物(iv)用tbaf脱去硅醚得化合物(v)
s4在丙酮溶剂、酰胺溶剂、腈类溶剂中的一种溶剂多种溶剂混合的混合溶剂中,化合物(v)与1-溴-4-氯丁烷及1-(2,3-二氯苯基)哌嗪在缚酸剂、碘化钠存在下反应,制备化合物(i)阿立哌唑十二烷酸酯
其合成路线为:
其中r1,r2,r3为相同或不同的烷基。
具体地,步骤s1中,化合物(ii)与卤代硅烷的摩尔比为1:1~1:2,更优选1:1.2~1:1.5;化合物(ii)与卤代硅烷、缚酸剂的摩尔比为1:1:2~1:2:2;能够保证化合物(ii)的酚羟基得到有效的保护;反应温度介于0℃~100℃之间;所用酰胺类溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺等中的一种或多种的混合。
步骤s2中,式(iii)与多聚甲醛的摩尔比为1:1~1:20,更优选1:10~1:5;式(iii)与缚酸剂的摩尔比为1:0.2~1:1,更优选1:0.4~1:0.6;式(iii)与多聚甲醛反应生成的醇与十二烷酰氯摩尔比为1:1~1:5,更优选1:1.1~1:1.3,反应生成的醇与十二烷酰氯反应生成化合物(iv);该步骤中,其反应温度介于0℃~100℃之间;所用醇类溶剂包括乙醇、甲醇、异丙醇等。
步骤s3中,化合物(iv)与tbaf的摩尔比为1:1~1:5,更优选1:1~1:1.2;反应温度介于0℃~100℃之间;呋喃溶剂包括四氢呋喃、二氧六环等。
步骤s4中,将化合物(v)与1-溴-4-氯丁烷及1-(2,3-二氯苯基)哌嗪在丙酮、或酰胺、或腈类溶剂中的一种或多种的组合溶剂中,在无机碱存在下反应,制备阿立哌唑十二烷酸酯。化合物(v)与1-溴-4-氯丁烷及1-(2,3-二氯苯基)哌嗪的摩尔比为1:1:1~1:3:3,更优选1:1.2:1.2~1:1.5:2;反应温度介于0℃~100℃之间;酮类溶剂包括丙酮、1-丁酮甲乙酮、甲基异丁酮、环己酮、二异丁酮等;
上述制备方法中用到的缚酸剂为有机弱碱或无机碱,所述有机弱碱可以是三乙胺,三乙烯二胺(dabco),1,5-二氮杂二环[5.4.0]十一烯-5(dbu),1,5-二氮杂二环[4.3.0]壬烯-5(dbn),4-二甲氨基吡啶(dmap),吡啶,n-甲基吗啉,四甲基乙二胺等中的一种或多种的混合。所述无机碱为氢氧化钠、氢氧化钾、氢氧化锂、氢氧化铯、碳酸钾、碳酸钠、碳酸铯、碳酸锂、碳酸氢钾、碳酸氢钾、碳酸氢锂等中的一种或多种。优选地,步骤s1和步骤s2中的所述缚酸剂为有机弱碱。
上述步骤中,各优选条件可任意组合即得本发明的各优选实施例;且本发明所用的试剂和原料均市售可得。
在一具体实施例中,化合物ii为7-羟基喹啉酮;所述卤代硅烷为叔丁基二甲基氯硅烷,化合物iii为7-叔丁基二甲基硅氧基-3,4-二氢-2(1h)-喹啉酮;化合物iv为7-叔丁基二甲基硅氧基-2-氧代-3,4-二氢-1(2h)-喹啉酮甲基十二烷酸酯;化合物v为7-羟基-2-氧代-3,4-二氢-1(2h)-喹啉酮甲基十二烷酸酯;化合物ⅰ为目标产物阿立哌唑十二烷酸酯。
其具体步骤为:
s1:7-叔丁基二甲基硅氧基-3,4-二氢-2(1h)-喹啉酮的制备
于500ml三口瓶中,加入20.0g7-羟基喹啉酮,100mldmf,25.0g三乙胺,降温至0℃,保温滴加22.2g叔丁基二甲基氯硅烷溶于150mldmf中的溶液,溶液由澄清变浑浊。滴毕转至室温反应,呈淡黄色。反应30~45min,将反应液倒入750ml冰水中,加入300ml正己烷,萃取分液,水相再用300ml正己烷萃取,己烷相合并,纯水洗涤,饱和食盐水洗,有机相用无水硫酸钠干燥,过滤,滤液蒸干得淡黄色固体33.8g。收率99.4%。ms(+1):278.5。1h-nmr(cdcl3,400mhz)δ0.219(s,6h),1.000(s,9h),2.634(t,2h),2.908(t,2h),6.307(s,1h),6.470(d,1h),7.012(d,1h),8.236(s,1h)。(见图1)
s2:7-叔丁基二甲基硅氧基-2-氧代-3,4-二氢-1(2h)-喹啉酮甲基十二烷酸酯的制备
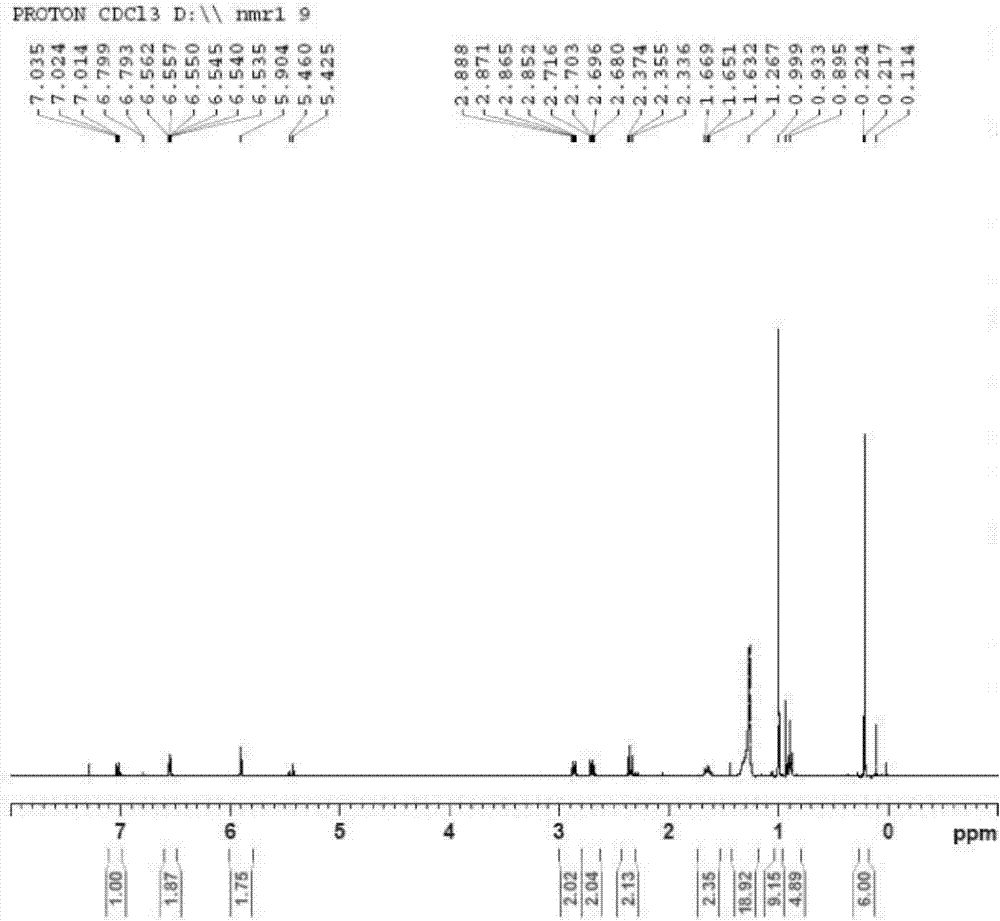
于500ml单口瓶中,加入28.0g7-叔丁基二甲基硅氧基-3,4-二氢-2(1h)-喹啉酮,300ml异丙醇,60.7g多聚甲醛,7ml三乙胺,搅拌,升温至80℃回流反应,呈白色浑浊,反应过夜。tlc点板,转化60-70%,停止反应,减压蒸去部分异丙醇,加500ml纯水及300mlea,萃取分液,水相用100mlea萃取,合并ea相,用150ml5%碳酸氢钠水溶液洗涤,再用纯水及饱和食盐水洗涤,无水硫酸镁干燥,过滤,滤液减压蒸去溶剂得27.4g固体。固体用180mlthf溶解,加入23.5g十二烷酰氯,氮气保护,降温至0℃左右搅拌加入三乙胺的thf溶液(20ml三乙胺溶于20mlthf中),滴加完毕转至室温,反应过夜。tlc点板,原料基本反应完全(未与多聚甲醛反应的原料不变),停止反应,减压蒸去部分thf,加250ml水及200mlea,萃取分液,水相用100mlea萃取,合并有机相,用100ml5%碳酸氢钠水溶液洗涤,再用纯水及饱和食盐水洗涤,无水硫酸镁干燥,过滤,蒸干溶剂得黄色油状物,硅胶柱层析纯化得27.0g油状产物,收率:54.0%,纯度:98.5%,回收原料7-叔丁基二甲基硅氧基-3,4-二氢-2(1h)-喹啉酮12g。ms(+1):376.5。1h-nmr(cdcl3,400mhz)δ0.24(s,6h),0.90(t,3h),1.00(s,9h),1.26(s,16h),1.65(m,2h),2.35(t,2h),2.70(t,2h),2.86(t,2h),5.90(s,2h),6.55(m,2h),7.02(d.2h)。(见图2)
s3:7-羟基-2-氧代-3,4-二氢-1(2h)-喹啉酮甲基十二烷酸酯的制备
于100ml单口瓶中,加入1.80g-叔丁基二甲基硅氧基-2-氧代-3,4-二氢-1(2h)-喹啉酮甲基十二烷酸酯,30mlthf溶解,室温缓慢滴加4.4ml1mol/l的tbaf溶液,搅拌30min,tlc点板,原料反应完全,停止反应,加入ph4~5的稀盐酸水溶液淬灭反应,ea萃取,ea相用水及饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压蒸去溶剂得油状物1.4g,甲醇结晶得到1.25g白色固体,收率:90%,纯度:99.0%。ms(+1):491。1h-nmr(cdcl3,400mhz)δ0.947(t,3h),1.27(s,17h),1.63(m,2h),2.36(t,2h),2.70(t,2h),2.86(t,2h),5.928(s,2h),6.60(t,2h),7.02(d,1h)。(见图3)
s4:阿立哌唑十二烷酸酯的制备
于250ml单口瓶中,加入2.00g7-羟基-2-氧代-3,4-二氢-1(2h)-喹啉酮甲基十二烷酸酯,2.80g粉碎后的碳酸钾,1.10g1-溴-4-氯丁烷,50ml丙酮,氮气保护,回流反应过夜。tlc点板,原料剩余很少,加入1.20g碘化钠,反应2h,继续加入1.95g1-(2,3-二氯苯基)哌嗪,回流反应过夜。tlc点板中间态剩余较少,停止反应,降温,过滤,滤液加水及乙酸乙酯,萃取分液,乙酸乙酯相分别用纯水及饱和食盐水洗涤,无水硫酸镁干燥,过滤,滤液减压蒸去大部分的乙酸乙酯,缓慢加入乙醇结晶,过滤,得阿立哌唑十二烷酸酯粗品。粗品经重结晶得阿立哌唑十二烷酸酯纯品2.7g,纯度:99.8%,收率:78%。ms(+1):661.7。
1h-nmr(cdcl3,400mhz)δ0.895(t,3h),1.295(s,16h),1.64(m,2h),1.735(m,2h),1.84(m,2h),2.373(t,2h),2.512(t,2h),2.70(m,6h),2.882(t,2h),3.094(s,4h),3.994(t,2h),5.937(s,2h),6.62(m,2h),6.98(m,1h),7.08(d,1h),7.16(m,2h)。(见图4)
综上所述,本发明的阿立哌唑十二烷酸酯的制备方法,后期处理无需柱层析等繁琐工艺、产品收率高达78%、纯度高达99.8%,降低了生产成本,便于工业生产。
以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!