一种苯并菲双硼酸酯关键中间体关键中间体的制备方法与流程
本发明涉及oled领域中一种重要电子传输材料苯并菲双硼酸酯关键中间体关键中间体的制备。
背景技术:
oled是自主发光器件,无需背光源,具有响应速度快、驱动电压低、发光效率和分辨率高、对比度高、视角广等特点,此外它能以廉价的玻璃、金属甚至柔性的塑料为基板,因此还具有成本低、生产工艺简单、可进行大面积生产等优点,已成为新一代的全彩显示和照明技术。自1987年柯达公司的邓青云和vanslyke制备出第一个有机发光二极管(oled)以来(appl.phys.lett.1987,51,913),由于其在全彩显示和固态照明领域中巨大的潜在应用,无论是在学术界还是工业界,oled都吸引着越来越多的关注(chem.soc.rev.2015,44,8484;j.mater.chem.c.2015,3,913;sidsymposiumdigestoftechnicalpapers,2015,46,411;adv.mater.2017,29,1601861;adv.mater.2017,29,1603253)。尤其是近十年的快速发展,使其在智能手机、电视、显示器、数码相机、gps、可弯曲和折叠等电子产品等方面得到了越来越广泛的应用,并且在平面固态照明领域也有着巨大的市场空间,预计2020年仅在照明领域就有数十亿美金的市场份额。
一般情况下,oleds器件的阳极和阴极之间至少要有一层有机层。而空穴传输层和电子传输层在多层先进oleds器件中都是重要的组成部分。由于在通常的oleds器件中,电子迁移速率比空穴传输速率低一到两个数量级,这会导致发光层电荷不平衡,影响器件的寿命。因此相对于空穴传输材料而言,电子传输材料的发展显得更加重要。2012年,adachi等研究人员发展了一系列基于苯并菲结构的电子传输材料,例如bpytp-1,bpytp-2,2,3'-bpytpand2,4'-bpytp(图1),由于这些分子可以在基底上进行定向排列,使其可以采用更低的电压驱动,同时这些分子在绿光oleds中展现了很好的电化学稳定性(j.mater.chem.2012,22,20689;chem.lett.2013,42,651;acsappl.mater.interfaces2015,7,16240;adv.mater.2017,29,1605002;adv.funct.mater.2016,26,6703)。最近研究表明bpytp-2亦可以在红光(acsappl.mater.interfaces2015,7,16240;adv.mater.2017,29,1605002)和蓝光(j.chem.mater.2016,28,3276;(b)li,g.;appl.phys.lett.2017,110,113301)磷光oleds、荧光oleds(adv.funct.mater.2016,26,6703)以及热辅助延迟荧光等材料中(j.mater.chem.c.2015,3,1700;thinsolidfilms2016,619,120)用作高效而且稳定的电子传输材料。然而关于该类传输层物质以及苯并菲结构前体的合成路径研究却非常少。
2012年,adachi课题组发展了四步合成重要的苯并菲双硼酸酯关键中间体4的路径,见合成技术路线1(j.mater.chem.2012,22,20689;patentus9685612,2017)。此合成路线以昂贵的1,2-二碘苯($461/25g,tci)和3-(三甲基硅基)苯硼酸($257/5g,tci)作为起始原料,通过pd(0)催化铃木偶联反应,合成化合物1。接着通过溴化反应,mocl5促进的氧化偶联反应以及pd(ii)催化miyaura硼化反应依次得到中间体化合物2,3,4,最终苯并菲双硼酸酯关键中间体4的总收率为17.6%。进一步,通过采用偶联反应将所得关键中间体4与相应的溴代化合偶联,以11.6%的总收率得到了电子传输材料bpytp-2。同时关键中间体4与其它相应的芳基溴化物偶联可以制备一系列的电子传输材料(bpytp-1,bpytp-2,2,3'-bpytp和2,4'-bpytp)以及主体材料(bdbf-tp和bdbt-tp),参见技术路线2。
但是此关键中间体4的合成及电子传输层材料的制备路线存在以下问题:
(1)起始原料1,2-二碘苯($461/25g,tci)和3-(三甲基硅基)苯硼酸($257/5g,tci)价格昂贵;且1,2-二碘苯分子量大,需要消耗更多质量的原料;
(2)步骤2中要使用大量的易造成环境污染的液态溴;
(3)步骤3中mocl5促进的氧化偶联收率低;
(4)苯并菲双硼酸酯关键中间体4的合成总收率低;
(5)电子传输材料bpytp-2的合成总收率低。
技术路线1
技术路线2
综上所述,采用便宜易得的起始原料,发展一种新型高效实用的苯并菲双硼酸酯关键中间体4及传输材料bpytp-2的制备路径对oleds中电子传输层的发展具有重要意义。
技术实现要素:
本发明针对现有合成技术中存在的式ⅴ所示的苯并菲双硼酸酯关键中间体5中存在的起始原料价格昂贵、需要使用液态溴、关键步骤mocl5促进的氧化偶联收率低以及式ⅴ所示的关键中间体5的合成总收率低等问题,提供一种式ⅴ所示的苯并菲双硼酸酯关键中间体5的高效制备方法,其制备成本低,收率高,工艺稳定,质量好,操作筒单,适合克级以及更大量的制备。
为达到上述目的,本发明采用如下技术方案:
技术路线3
一种式ⅴ所示的苯并菲双硼酸酯关键中间体的制备方法,所述的方法按照如下步骤进行:
(1)以1,2-二溴苯和3-甲氧基苯硼酸为原料,通过偶联反应得到式ⅰ所示的化合物1;
(2)将步骤(1)所得式ⅰ所示的化合物1中加入五氯化钼进行氧化偶联得到式ⅱ所示的化合物2;
(3)将步骤(2)所得式ⅱ所示的化合物2加酸脱去甲基得到式ⅲ所示的化合物3;
(4)将步骤(3)所得式ⅲ所示的化合物3和三氟甲磺酸酐反应,得到式ⅳ所示的化合物4;
(5)将步骤(4)所得式ⅳ所示的化合物4与pd(ii)催化的硼化反应得到式ⅴ所示的苯并菲双硼酸酯关键中间体5;
进一步,步骤(1)所述的方法为:以1,2-二溴苯和3-甲氧基苯硼酸为原料,加入四(三苯基膦)钯和碱性物质,溶于有机溶剂a中,在氮气气氛下,在90-110℃下回流1-4天,所得反应液经后处理得到式ⅰ所示的化合物1;所述的碱性物质为碳酸钠或碳酸钾;所述的1,2-二溴苯,3-甲氧基苯硼酸与四(三苯基膦)钯及碱性物质的物质的量之比为1:2.0~3.0:0.01~0.05:5.0~7.0;
再进一步,步骤(1)所述的方法为:以1,2-二溴苯和3-甲氧基苯硼酸为原料,加入四(三苯基膦)钯和碱性物质,溶于有机溶剂a中,在氮气气氛下,在90-110℃下回流1-4天,所得反应液经后处理得到式ⅰ所示的化合物1;所述的碱性物质为碳酸钠或碳酸钾;所述的1,2-二溴苯,3-甲氧基苯硼酸与四(三苯基膦)钯及碱性物质的物质的量之比为1:2.0~3.0:0.01~0.05:5.0~7.0;所述的溶剂a为甲苯、去离子水和乙醇的混合溶液,所述的甲苯、去离子水与乙醇的体积之比为1:1:1;所述的溶剂a的加入量以1,2-二溴苯的物质的量计为1.5~4.5ml/mmol。
进一步,步骤(2)所述的方法为:在氮气气氛下,将式ⅰ所示的化合物1溶于有机溶剂b中,加入五氯化钼,在常温下搅拌反应2-3天,所得反应液b经后处理得到式ⅱ所示的化合物2;所述的五氯化钼分两次等量加入,间隔时间为24小时;所述的式ⅰ所示的化合物1与五氯化钼加入总量的物质的量之比为1.0:2.0~3.5。
进一步,步骤(3)所述的方法为:在氮气气氛下,向式ⅱ所示的化合物2中加入浓度为48%的浓氢溴酸,并溶于有机溶剂醋酸中,在110-120℃下回流2-4天,所得反应液c经后处理得到式ⅲ所示的化合物3;所述的浓氢溴酸的加入量以式ⅱ所示的化合物2的物质的量计为2~4ml/mmol。
再进一步,步骤(3)中,所述的醋酸的加入量以式ⅱ所示的化合物2的物质的量计为10~20ml/mmol。
进一步,步骤(4)所述的的方法为:将式ⅲ所示的化合物3溶于有机溶剂c中,加入吡啶和三氟甲磺酸酐,在0-5℃下,搅拌反应2-4小时,所得反应液d经后处理得到式ⅳ所示的化合物4;所述的式ⅲ所示的化合物3与吡啶、三氟甲磺酸酐的物质的量之比为1.0:7.0~9.0:2.5~3.0。
再进一步,步骤(4)中,所述的有机溶剂c为二氯甲烷,所述的有机溶剂c的加入量以式ⅲ所示的化合物3的物质量计为15.0~20.0ml/mmol。
进一步,步骤(5)所述的方法为:将式ⅳ所示的化合物4中、双(频哪醇合)二硼、醋酸钾和[1,1'-双(二苯基膦基)二茂铁]二氯化钯溶于有机溶剂d中,在氮气气氛下,置于80-100℃下搅拌反应2-4天,所得反应液e经后处理得到式ⅴ所示的苯并菲双硼酸酯关键中间体5;所述的式ⅳ所示的化合物4中、双(频哪醇合)二硼、醋酸钾和[1,1'-双(二苯基膦基)二茂铁]二氯化钯的物质的量之比为1.0:3.0~5.0:5.0~7.0:0.03~0.07。
再进一步,步骤(5)所述的方法为:将式ⅳ所示的化合物4中、双(频哪醇合)二硼、醋酸钾和[1,1'-双(二苯基膦基)二茂铁]二氯化钯溶于有机溶剂d中,在氮气气氛下,置于80-100℃下搅拌反应2-4天,所得反应液e经后处理得到式ⅴ所示的苯并菲双硼酸酯关键中间体5;所述的式ⅳ所示的化合物4中、双(频哪醇合)二硼、醋酸钾和[1,1'-双(二苯基膦基)二茂铁]二氯化钯的物质的量之比为1.0:3.0~5.0:5.0~7.0:0.03~0.07;所述的有机溶剂d为二氧六环;所述的有机溶剂d的加入量以式ⅳ所示的化合物4的物质量计为7.0~9.0ml/mmol。
更进一步,较为具体的,本发明所述的方法具体按照如下步骤进行:
(1)以1,2-二溴苯和3-甲氧基苯硼酸为原料,加入四(三苯基膦)钯和碱性物质,溶于有机溶剂a中,在氮气气氛下,在90-110℃下回流1-4天,所得反应液经后处理得到式ⅰ所示的化合物1;所述的碱性物质为碳酸钠或碳酸钾;所述的1,2-二溴苯,3-甲氧基苯硼酸与四(三苯基膦)钯及碱性物质的物质的量之比为1:2.0~3.0:0.01~0.05:5.0~7.0;所述的溶剂a为甲苯、去离子水和乙醇的混合溶液,所述的甲苯、去离子水与乙醇的体积之比为1:1:1;所述的溶剂a的加入量以1,2-二溴苯的物质的量计为1.5~4.5ml/mmol;
(2)在氮气气氛下,将式ⅰ所示的化合物1溶于有机溶剂b中,加入五氯化钼,在常温下搅拌反应2-3天,所得反应液b经后处理得到式ⅱ所示的化合物2;所述的五氯化钼分两次等量加入,间隔时间为24小时;所述的式ⅰ所示的化合物1与五氯化钼总加入量的物质的量之比为1.0:2.5~3.5;所述的有机溶剂b为二氯甲烷;所述的有机溶剂b的加入量以式ⅰ所示的化合物1的物质的量计为15.0~20.0ml/mmol;
(3)在氮气气氛下,向式ⅱ所示的化合物2中加入浓度为48%的浓氢溴酸,并溶于有机溶剂醋酸中,在110-120℃下回流2-4天,所得反应液c经后处理得到式ⅲ所示的化合物3;所述的浓氢溴酸的加入量以式ⅱ所示的化合物2的物质的量计为2~4ml/mmol;所述的醋酸的加入量以式ⅱ所示的化合物2的物质的量计为10~20ml/mmol;
(4)将式ⅲ所示的化合物3溶于有机溶剂c中,加入吡啶和三氟甲磺酸酐,在0-5℃下,搅拌反应2-4小时,所得反应液d经后处理得到式ⅳ所示的化合物4;所述的式ⅲ所示的化合物3与吡啶、三氟甲磺酸酐的物质的量之比为1.0:7.0~9.0:2.5~3.0;所述的有机溶剂c为二氯甲烷;所述的有机溶剂c的加入量以式ⅲ所示的化合物3的物质量计为15.0~20.0ml/mmol;
(5)将式ⅳ所示的化合物4中、双(频哪醇合)二硼、醋酸钾和[1,1'-双(二苯基膦基)二茂铁]二氯化钯溶于有机溶剂d中,在氮气气氛下,置于80-100℃下搅拌反应2-4天,所得反应液e经后处理得到式ⅴ所示的苯并菲双硼酸酯关键中间体5;所述的式ⅳ所示的化合物4中、双(频哪醇合)二硼、醋酸钾和[1,1'-双(二苯基膦基)二茂铁]二氯化钯的物质的量之比为1.0:3.0~5.0:5.0~7.0:0.03~0.07;所述的有机溶剂d为二氧六环;所述的有机溶剂d的加入量以式ⅳ所示的化合物4的物质量计为7.0~9.0ml/mmol。
更进一步,步骤(1)中,反应结束后,将反应液a冷却至室温,过滤,所得滤饼采用乙酸乙酯清洗,浓缩并用大量乙酸乙酯稀释,随后采用盐酸清洗,分离有机相,采用nahco3清洗,直到不产生气泡,分离有机相,采用na2so4干燥,过滤,浓缩,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=20:1—10:1),继续打浆纯化(石油醚/乙酸乙酯=5:1),得式ⅰ所示的化合物1。
更进一步,步骤(2)中,反应结束后,向所得反应液b中加入甲醇搅拌1小时淬灭反应,所得产物过滤,所得滤饼经二氯甲烷洗涤,滤液浓缩,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=20:1-10:1-5:1-3:1),再通过石油醚/乙酸乙酯(10:1)打浆纯化得式ⅱ所示的化合物2。
更进一步,步骤(3)中,反应结束后,待反应液c冷却至室温,减压蒸馏除去有机溶剂,所得混合物用水稀释,经k2co3溶液中和,过滤,将得到的沉淀物用水洗涤,干燥,得式ⅲ所示的化合物3。
更进一步,步骤(4)中,反应结束后,向所得反应液d中加入水淬灭反应,然后加入二氯甲烷直至反应体系中固体全部溶解,分液,合并有机相经干燥、过滤、浓缩,再加入环己烷稀释,过滤,所得滤饼经洗涤、干燥,得式ⅳ所示的化合物4。
更进一步,步骤(5)中,反应结束后,将反应液e冷却至室温,滤液浓缩,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=60:1-40:1)得含有目标产物的混合物,进一步通过石油醚/乙酸乙酯(20:1)常温搅拌4-6小时打浆纯化,过滤干燥,得式ⅴ所示的苯并菲双硼酸酯关键中间体5。
与现有技术相比,本发明的的有益效果在于:
(1)起始原料1,2-二溴苯和3-甲氧基苯硼酸便宜易得,且其偶联反应收率高达96%;
(2)反应过程无需采用易造成环境污染的液态溴;
(3)通过分批加入氧化剂五氯化钼构建了苯并菲结构单元,收率大大提高,可达82%;
(4)苯并菲双硼酸酯关键中间体5的制备总收率可高达48.6%,远高于现有技术的总收率。
附图说明
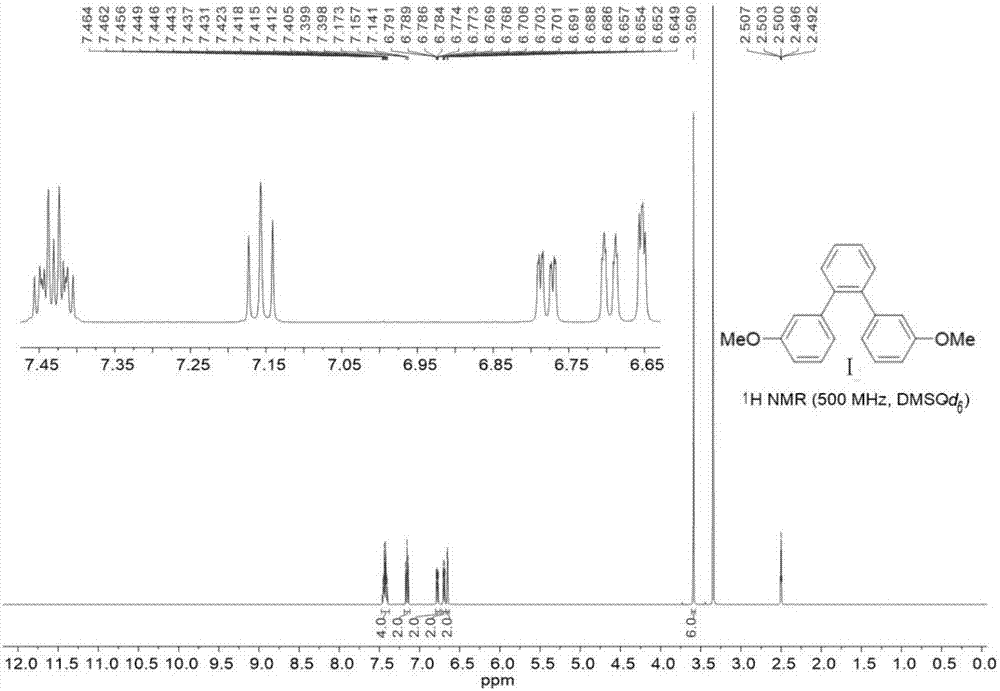
图1为化合物1的核磁共振氢谱图;
图2为化合物1的核磁共振碳谱图;
图3为化合物1的质谱图;
图4为化合物2的核磁共振氢谱图;
图5为化合物2的核磁共振碳谱图;
图6为化合物2的质谱图;
图7为化合物3的核磁共振氢谱图;
图8为化合物3的核磁共振碳谱图;
图9为化合物3的质谱图;
图10为化合物4的核磁共振氢谱图;
图11为化合物4的核磁共振碳谱图;
图12为化合物4的质谱图;
图13为化合物5的核磁共振氢谱图;
图14为化合物5的核磁共振碳谱图;
图15为化合物5的质谱图。
具体实施方式
下面通过具体实施例对本发明作进一步说明,但本发明的保护范围并不仅限于此。
步骤1:1,2-二(3-甲氧基苯基)苯(化合物1)的制备
向带有磁力转子和冷凝管的三口烧瓶依次加入3-甲氧基苯硼酸(28.80g,189mmol,3.00当量),pd(pph3)4(2.91g,2.52mmol,0.05eq)和na2co3(40.10g,378.00mmol,6.00当量)。抽换氮气三次,然后加入1,2-二溴苯(7.50ml,63.00mmol,1.00当量),甲苯(150ml),去离子水(150ml)和乙醇(150ml)。然后鼓氮气泡30分钟,反应混合物回流2天。然后冷却至室温,过滤,采用乙酸乙酯清洗滤柄(100ml×3),浓缩,采用大量乙酸乙酯稀释,随后采用3m的盐酸清洗,分离有机相,采用nahco3清洗,直到不产生气泡,分离有机相,采用na2so4干燥,过滤,浓缩。将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=20:1—10:1),得白色液体12.97g,收率50%。为分离干净的部分通过打浆纯化(10ml,石油醚/乙酸乙酯=5:1),得到4.66g白色固体。产物总重17.63g,收率96%。1hnmr(500mhz,dmso-d6):δ3.59(s,6h),6.65(dd,j=2.5,1.5hz,2h),6.70(ddd,j=7.5,1.5,1.0hz,2h),6.78(ddd,j=8.5,2.5,1.0hz,2h),7.16(t,j=8.0hz,2h),7.40-7.46(m,4h)(参见附图1).13cnmr(126mhz,dmso-d6):δ54.8,112.4,115.0,121.7,127.7,129.0,130.2,139.8,142.4,158.7(参见附图2).hrms(tofmsei+)forc20h18o2[m]+:calcd290.1307,found290.1303(参见附图3).
步骤2:2,7-二甲氧基三苯基苯(化合物2)的制备
在氮气保护下,向带有磁力转子和冷凝管的三口烧瓶依次加入化合物5(8.31g,28.60mmol,1.00当量)的二氯甲烷溶液(500ml),mocl5(7.82g,28.60mmol,1.00当量),混合物常温搅拌反应24小时,然后再加入mocl5(7.82g,28.60mmol,1.00当量),随后混合物再常温搅拌反应24小时后,加入甲醇(100ml)搅拌1小时淬灭反应。过滤,用二氯甲烷(500ml)清洗滤柄。滤液浓缩,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=20:1-10:1-5:1-3:1),得白色固体7.52g,进一步通过石油醚/乙酸乙酯(22ml,10:1)打浆纯化,得得白色固体6.82g,收率82%。1hnmr(500mhz,dmso-d6):δ3.99(s,6h),7.28(dd,j=9.0,2.5hz,2h),7.70(dd,j=6.0,3.0hz,2h),8.17(d,j=2.5hz,2h),8.59(d,j=9.5hz,2h),8.80(dd,j=6.0,3.0hz,2h)(参见附图4).13cnmr(126mhz,dmso-d6):δ55.4,105.7,116.5,123.3,123.9,124.7,127.5,129.3,129.6,158.1(参见附图5).hrms(tofmsei+)forc20h16o2[m]+:calcd,288.1150,found288.1154(参见附图6).
步骤3:2,7-二羟基三苯基苯(化合物3)的制备
在氮气保护下,将上一步得到的化合物6(4.30g,14.90mmol),浓氢溴酸((30ml,浓度48%)和醋酸(220ml)在装有磁力搅拌子和冷凝管的单口瓶中回流2天。然后让混合物自然冷却至室温,减压蒸馏除去有机溶剂,所得混合物用水稀释(30ml),随后用k2co3溶液中和至无气泡产生为止。过滤所产生的沉淀物,用水洗涤3次(50ml×3).,干燥,得目标产物棕色固体化合物7,定量收率。1hnmr(500mhz,dmso-d6):δ7.14(dd,j=9.0,2.5hz,2h),7.66(dd,j=6.0,3.0hz,2h),7.97(d,j=2.0hz,2h),8.46(d,j=9.0hz,2h),8.54(dd,j=6.0,3.0hz,2h),9.76(s,2h)(参见附图7).13cnmr(126mhz,dmso-d6):δ107.6,117.2,122.4,123.4,124.4,127.3,129.2,129.3,156.0(参见附图8).hrms(dartpositiveionmode)forc18h13o2[m+h]+:calcd261.0910,found261.0909(参见附图9).
步骤4:2,7-二羟基三苯基苯(化合物4)的制备
向带有磁力转子三口烧瓶加入化合物7(3.94g,14.90mmol,1.00当量)的二氯甲烷溶液(250ml),将其置于0-5℃低温浴槽中,随后加入吡啶(9.60ml,119.16mmol,8.00当量),滴加tf2o(6.60ml,39.25mmol,2.60当量)。随后混合物在该温度下搅拌反应2小时后,加入水(25ml)淬灭反应。接着加入二氯甲烷直至反应体系中固体全部溶解。分液,有机相采用na2so4干燥,过滤,浓缩至40ml。再加入环己烷(200ml)稀释,过滤,采用环己烷(100ml)清洗滤柄,干燥,得白色固体6.77g,收率87%。1hnmr(500mhz,dmso-d6):δ7.83-7.86(m,4h),8.92(dd,j=6.5,3.5hz,2h),8.99(d,j=2.5hz,2h),9.03(d,j=9.5hz,2h)(参见附图10).13cnmr(126mhz,dmso-d6):δ116.70,118.35(q,j=320hz),120.7,124.4,127.3,128.1,128.6,129.1,131.5,149.2(参见附图11).hrms(dartpositiveionmode)forc20h10f6o6s2[m]+:calcdfor523.9818,found523.9814(参见附图12).
步骤5:苯并菲双硼酸酯关键中间体5的制备
向带有磁力转子和冷凝管的三口烧瓶依次加入化合物8(9.00g,17.16mmol,1.00当量),双(频哪醇合)二硼(17.43g,68.65mmol,4.00当量),koac(10.10g,102.96mmol,6.00当量)和pd(dppf)cl2(0.84g,1.03mmol,0.06当量)。抽换氮气三次,然后加入二氧六环(136ml)。然后鼓氮气泡20分钟,反应混合物置于80℃油浴搅拌反应2天。然后冷却至室温,滤液浓缩,将所得粗品通过硅胶柱色谱分离纯化,淋洗剂(石油醚/乙酸乙酯=60:1-40:1)得含有少量双(频哪醇合)二硼的目标产物混合物。进一步通过石油醚/乙酸乙酯(20:1)常温搅拌4-6小时打浆纯化,过滤干燥,得白色固体5.86g,收率71%。1hnmr(500mhz,dmso-d6):δ1.39(s,24h),7.77(dd,j=6.5,3.5hz,2h),7.98(dd,j=8.0,1.0hz,2h),8.78(dd,j=6.0,3.5hz,2h),8.84(d,j=8.0hz,2h),9.03(s,2h)(参见附图13).13cnmr(126mhz,dmso-d6):δ24.7,84.0,123.33,123.4,128.0,128.8,128.9,129.7,131.1,132.8(参见附图14).hrms(dartpositiveionmode)forc30h3410b2o4[m]+:calcd478.2710,found478.2710(参见附图15).
显然,上述实施例仅仅是为清楚地说明本发明的内容所做的举例,而非对实施方式的限定。对于所属领域的技术人员来说,在上述说明的基础之上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以一一举例。而由此所引申的显而易见的变化或变动仍处于本发明的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!