一种苝二酰亚胺化合物,其合成方法及其在Fe3+检测中的应用与流程
本发明涉及化学合成及离子分析领域,具体地说是一种苝二酰亚胺化合物,其合成方法及其在fe3+检测中的应用。
背景技术:
铁是细胞中普遍存在的金属,它在许多生命过程中起着至关重要的作用,在很多生命活动中扮演着重要角色。例如电子转移过程、氧运输过程、dna和rna的合成、修复过程和光合作用进行固氮的过程等。fe3+在细胞中主要以与蛋白质结合的形式存在,然而过量或者不足都会导致严重的紊乱。生命体中fe3+含量不足会限制氧传递到细胞,导致疲劳、工作乏力以及免疫力低下;fe3+含量过高能够催化产生活性氧ros,活性氧过量能够破坏酯类、核酸和蛋白质,从而引起严重的疾病,如:阿尔茨海默氏病、慢性进行性舞蹈病和帕金森氏病。
近年来,fe3+对生物和环境系统的影响引起了人们越来越多的兴趣。目前,多种方法可以用于检测fe3+,比如原子吸收光谱法,电感耦合等离子体质谱法,伏安法等,这些技术具有较高的灵敏度和选择性,并常用于定量分析,但是这些方法所用仪器昂贵且样品预处理复杂。与上述技术相比,基于紫外的检测方法由于其简单,灵敏,快速的优势也得到了很好的发展,然而,目前报道的以紫外可见分光光度发检测fe3+的报道后期均用到了荧光光谱做辅助,如2017年zhang等人(fastresponseandhighsensitivityeuropiummetalorganicframeworkfluorescentprobewithchelatingterpyridinesitesforfe3+)利用金属有机框架结构螯合三联吡啶对fe3+进行检测,2013年,yang等人(ahighlyselectiveandsensitivefe3+fluorescentsensorbyassemblingthree1,8-naphthalimidefluorophoreswithatris(aminoethylamine)ligand)利用1,8-萘酰亚胺单体所制备的小分子对fe3+进行检测。这些方法除了紫外可见分析之外还用到了荧光光谱的分析。对仪器的要求相对还是较为昂贵。
技术实现要素:
本发明的技术任务是针对上述现有技术的不足,利用最为便捷的检测手段,提供一种化学和光学稳定性好,对fe3+进行检测有高的选择性和灵敏度的小分子苝二酰亚胺化合物。
本发明进一步的技术任务是提供上述化合物的合成方法。
本发明再进一步的技术任务是提供上述化合物在fe3+检测中的应用。
本发明的技术任务是按以下方式实现的:一种苝二酰亚胺化合物,其特点是具有与fe3+结合位点,可通过检测fe3+的紫外光谱变化实现fe3+的检测。
所述苝二酰亚胺化合物为1-对叔丁基酚氧基-7-(n-(8’-氨基喹啉)-乙酰氨基)哌嗪-n,n-二环己基3,4,9,10-苝二酰亚胺,其结构式为:
结构式(ⅰ)表示的苝二酰亚胺化合物的合成路线如下:
合成方法,包括以下步骤:
(1)双溴代苝二酰亚胺、对叔丁基苯酚、缚酸剂溶于有机溶剂中,在140~150℃下反应一定时间后,降至室温,调节ph值至2.5~3.5,继续反应至反应结束,经分离、提纯得到6-叔丁基苯酚-12-溴-环己基保护苝二酰亚胺;
(2)6-叔丁基苯酚-12-溴-环己基保护苝二酰亚胺、哌嗪、缚酸剂溶于有机溶剂中,在70~95℃下反应一定时间后,降至室温,经分离、提纯,得到6-叔丁基苯酚-12-哌嗪-环己基保护苝二酰亚胺;
(3)合成2-氯-n-(8-氨基喹啉)-乙酰胺;
(4)6-叔丁基苯酚-12-哌嗪-环己基保护苝二酰亚胺、2-氯-n-(8-氨基喹啉)-乙酰胺、缚酸剂、碘化钾溶于有机溶剂中,惰性气体保护下,在65~90℃下反应一定时间,经分离、提纯,得到目标化合物。
步骤(1)中双溴代苝二酰亚胺、对叔丁基苯酚、缚酸剂的摩尔比为1:0.6~1.4:1~1.8,优选为1:0.8~1.2:1.3~1.6;作为优选,在140~150℃下反应40~60min,调节ph值至2.5~3.5后继续反应30~40min。
双溴代苝二酰亚胺、对叔丁基苯酚、缚酸剂优选一次性加入到反应容器中,再向反应容器中加入有机溶剂。所述有机溶剂优选n-甲基吡咯烷酮、甲苯或吡啶,其用量以能够充分溶解固定反应物为宜,优选为固体反应物的8-15倍。
作为优选,以1-2m的盐酸溶液调节ph值。
步骤(2)中6-叔丁基苯酚-12-溴-环己基保护苝二酰亚胺、哌嗪、缚酸剂的摩尔比为1:2~4:2~5,优选为1:2.5~3.5:3~4;反应时间优选为3.5~4.5h。
6-叔丁基苯酚-12-溴-环己基保护苝二酰亚胺、哌嗪、缚酸剂优选一次性加入到反应容器中,再向反应容器中加入有机溶剂。所述有机溶剂优选dmf(二甲基甲酰胺)或吡啶,其用量以能够充分溶解固定反应物为宜,优选为固体反应物的8-15倍。
步骤(3)2-氯-n-(8-氨基喹啉)-乙酰胺的合成方法:
8-氨基喹啉、二氯甲烷溶液、三乙胺混合,混合体系降至0℃以下,滴加氯乙酰氯的二氯甲烷溶液,在5℃以下反应1.5~2h,经分离、提纯得到2-氯-n-(8-氨基喹啉)-乙酰胺。
8-氨基喹啉、三乙胺、氯乙酰氯的摩尔比优选为1:1~1.2:0.8~1.2,优选为1:1.05~1.15:1.0~1.1。
步骤(4)中6-叔丁基苯酚-12-哌嗪-环己基保护苝二酰亚胺、2-氯-n-(8-氨基喹啉)-乙酰胺、缚酸剂、碘化钾的摩尔比为1:1~3:2~5:0.3~0.5,优选为1:1.5~2.5:3~4:0.3~0.4。所述有机溶剂优选dmf(二甲基甲酰胺)或吡啶,其用量以能够充分溶解固定反应物为宜,优选为固体反应物的8-15倍。
所述碘化钾为催化剂。由于哌嗪中氮的亲核性较弱,需要更加容易的离去基团,碘甲烷的碘可以取代原料中的溴原子,使之更加容易反应。
本发明合成方法中,所述缚酸剂用于与反应体系中产生的hcl反应,优选为碳酸钾、碳酸铯、三乙醇胺或三乙胺。
各中间体(包括6-叔丁基苯酚-12-溴-环己基保护苝二酰亚胺、6-叔丁基苯酚-12-哌嗪-环己基保护苝二酰亚胺)及目标产物苝二酰亚胺化合物均可通过过硅胶柱提纯,展开剂可以是二氯甲烷、二氯甲烷与甲醇或乙醇的混合液、或者三氯甲烷与甲醇或乙醇的混合液、或者乙酸乙酯和石油醚的混合溶液,优选为二氯甲烷、二氯甲烷与甲醇或乙醇的混合液。
申请人发现,式(ⅰ)所示苝二酰亚胺化合物具有与fe3+结合的位点,与fe3+结合后光谱发生明显的改变,而与其他离子结合光谱没有任何变化,因此,可将该化合物用于fe3+检测中。
利用式(ⅰ)所示苝二酰亚胺化合物进行fe3+检测时,优选将苝二酰亚胺化合物配制在dmso和去离子水的混合液中,苝二酰亚胺化合物的浓度为1.0×10-5-1.2×10-5mol/l,最佳浓度为1.1×10-5mol/l。
dmso和去离子水的配比以能溶解苝二酰亚胺化合物为宜。
本发明的苝二酰亚胺化合物,其合成方法及其在fe3+检测中的应用与现有技术相比具有以下突出地有益效果:
(一)所述苝二酰亚胺化合物与fe3+结合后光谱发生明显的改变,而与其他离子结合光谱没有任何变化,灵敏度高、检测限低;
(二)化合物合成简便,并且易于提纯;
(三)测试用到的仪器常见、易得,能够快速检测。
附图说明
附图1是不同离子溶液与苝二酰亚胺衍生物混合溶液的紫外光谱图;
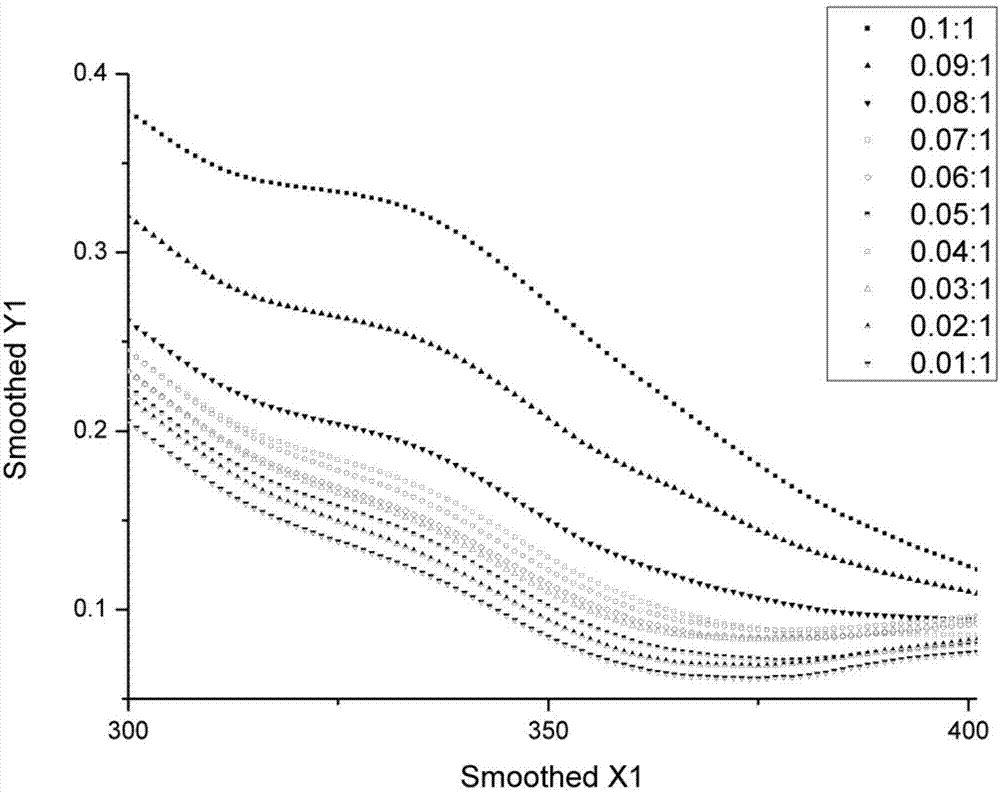
附图2是不同比例fe3+与苝二酰亚胺衍生物混合溶液的紫外光谱图。
具体实施方式
以具体实施例对本发明的锂萃取的功能化离子液体,其合成方法及其应用作以下详细地说明。应当理解,此处所描述的具体实施实例仅仅用以解释本发明,并不用于限定本发明。
如无特别说明,下述所用各成分的含量为重量百分比含量。
实施实例1:
(1)中间体1-溴-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺的合成
将1.065g(0.0015mol)双溴代苝二酰亚胺,0.269g(0.00195mol)无水碳酸钾,0.181g(0.0012mol)对叔丁基苯酚一次性加入到三口烧瓶中,再向三口烧瓶中加入15mln-甲基吡咯烷酮,145℃反应30min,停止反应,降温,将反应物倒入1m盐酸溶液中,调节ph值至3,搅拌35min过滤,晾干。柱层析提纯,二氯甲烷作为洗脱剂收集第二带产物。氯仿/甲醇重结晶得红色固体0.571g。nmr(300mhz,cdcl3)δ:9.41(d,1h),8.59(d,1h),8.50(s,1h),9.14(d,1h),8.28(d,1h),7.92(s,1h),4.80(m,2h),2.59(m,8h),1.99(m,8h),1.90(m,4h),0.94(s,9h).
(2)中间体1-哌嗪-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺的合成
将0.779g(1mmol)1-溴-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺,0.215g(2.5mmol)无水哌嗪,0.416g无水碳酸钾加入到20ml无水dmf中,氮气保护下80℃反应4h,减压蒸干溶剂得粗品。柱层析提纯,二氯甲烷/乙醇作为洗脱剂,得到暗红色固体0.641g。nmr(300mhz,cdcl3)δ:9.48(d,1h),8.64(d,1h),8.53(s,1h),9.21(d,1h),8.32(d,1h),7.96(s,1h),4.80(m,2h),3.63(t,2h),3.01(t,2h),2.59(m,8h),1.99(m,8h),1.90(m,4h),0.94(s,9h).
(3)2-氯-n-(8-氨基喹啉)-乙酰胺的合成
将8-氨基喹啉1.44g(0.01mol)加入到三口烧瓶中,加入干燥的二氯甲烷溶液50ml、1.06g(0.0105mol)三乙胺,体系降至0℃以下,滴加氯乙酰氯1.12g(0.01mol)的二氯甲烷溶液70ml,整个过程控制体系温度低于5℃,反应1.5h,经过滤、酸洗、水洗、干燥得到2-氯-n-(8-氨基喹啉)-乙酰胺1.98g。1hnmr(cdcl3):10.94(br,1h),8.89(dd,1h),8.78(dd,1h),8.21(dd,1h;),7.59(m,2h),7.51(q,1h),4.34(s,2h).
(4)目标化合物1-对叔丁基酚氧基-7-(n-(8’-氨基喹啉)-乙酰氨基)哌嗪-n,n-二环己基3,4,9,10-苝二酰亚胺的合成
将0.785g(1mmol)1-哌嗪-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺,0.332g(1.5mmol)2-氯-n-(8-氨基喹啉)-乙酰胺,无水碳酸钾0.420g(3mmol)和碘化钾49.8mg(0.3mmol)加入到10ml无水dmf中,氮气保护下80℃反应4h,减压蒸干溶剂得粗品。柱层析提纯,二氯甲烷/乙醇作为洗脱剂,得到暗红色固体0.724g。nmr(300mhz,cdcl3)δ:10.94(d,1h),9.48(d,1h),9.21(d,1h),8.89(dd,1h),8.78(dd,1h),8.59(d,1h),8.47(s,1h),8.29(d,1h),8.19(dd,1h;),7.87(s,1h),7.53(m,2h),7.46(q,1h),4.73(m,2h),4.39(s,2h).3.57(t,2h),2.99(t,2h),2.51(m,8h),1.92(m,8h),1.87(m,4h),0.95(s,9h).
目标化合物1-对叔丁基酚氧基-7-(n-(8’-氨基喹啉)-乙酰氨基)哌嗪-n,n-二环己基3,4,9,10-苝二酰亚胺化合物的结构式如下:
该化合物分子制备完成后,可以利用该分子检测fe3+。
该化合物分子在dmso-h20溶液中以自由单体的形式存在,当加入fe3+后,由于分子与fe3+的相互作用,使得紫外光谱在325nm处出现新的峰,而该化合物与其他金属离子混合后在该波长处都不会出现该峰,又将fe3+与其他离子共存时检测其吸光度,发现其他离子(包括fe2+)都不会影响fe3+的检测。
fe3+的检测:将所得苝二酰亚胺衍生物溶解到dmso溶液中,配制成10-5mol/l的溶液;分别配制不同金属离子的水溶液10-4mol/l。采用1ml苝二酰亚胺衍生物分别+(0.1~1ml)不同金属离子溶液的方式配制其混合溶液,测紫外光谱,如图1所示,再改变fe3+与苝二酰亚胺衍生物的比例,测紫外,如图2所示。
实施实例2
(1)中间体1-溴-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺的合成
将1.061g(0.0015mol)双溴代苝二酰亚胺,0.2898g(0.0021mol)无水碳酸钾、0.225g(0.0015mol)对叔丁基苯酚、12mln-甲基吡咯烷酮加入50ml三口烧瓶中,150℃反应45min,停止反应,降温,将反应物倒入1m盐酸溶液中,调节ph值至2.8,搅拌40min过滤,晾干。柱层析提纯,二氯甲烷作为洗脱剂收集第二带产物。氯仿/甲醇重结晶得红色固体0.593g。nmr(300mhz,cdcl3)δ:9.41(d,1h),8.59(d,1h),8.50(s,1h),9.14(d,1h),8.28(d,1h),7.92(s,1h),4.80(m,2h),2.59(m,8h),1.99(m,8h),1.90(m,4h),0.94(s,9h).
(2)中间体1-哌嗪-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺的合成
将0.781g(1mmol)1-溴-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺,0.258g(3mmol)无水哌嗪,0.495g无水碳酸钾加入到15ml无水dmf中,氮气保护下75℃反应4.0h,减压蒸干溶剂得粗品。柱层析提纯,二氯甲烷/乙醇作为洗脱剂,得到暗红色固体0.653g。nmr(300mhz,cdcl3)δ:9.48(d,1h),8.64(d,1h),8.53(s,1h),9.21(d,1h),8.32(d,1h),7.96(s,1h),4.80(m,2h),3.63(t,2h),3.01(t,2h),2.59(m,8h),1.99(m,8h),1.90(m,4h),0.94(s,9h).
(3)2-氯-n-(8-氨基喹啉)-乙酰胺的合成
将8-氨基喹啉1.43g(0.01mol)加入到三口烧瓶中,加入干燥的二氯甲烷溶液42ml、三乙胺1.11g(0.011mol),体系降至0℃以下,滴加氯乙酰氯1.18g(0.0105mol)的二氯甲烷溶液55ml,整个过程控制体系温度低于5℃,反应2h,经过滤、酸洗、水洗、干燥得到2-氯-n-(8-氨基喹啉)-乙酰胺1.87g。1hnmr(cdcl3):10.94(br,1h),8.89(dd,1h),8.78(dd,1h),8.21(dd,1h;),7.59(m,2h),7.51(q,1h),4.34(s,2h).
(4)目标化合物1-对叔丁基酚氧基-7-(n-(8’-氨基喹啉)-乙酰氨基)哌嗪-n,n-二环己基3,4,9,10-苝二酰亚胺的合成
将0.794g(1mmol)1-哌嗪-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺,0.44g(2mmol)2-氯-n-(8-氨基喹啉)-乙酰胺,无水碳酸钾0.491g(3.5mmol)和碘化钾(60mg,0.36mmol)加入到10ml无水dmf中,氮气保护下75℃反应4h,减压蒸干溶剂得粗品。柱层析提纯,二氯甲烷/乙醇作为洗脱剂,得到暗红色固体0.737g。nmr(300mhz,cdcl3)δ:10.94(d,1h),9.48(d,1h),9.21(d,1h),8.89(dd,1h),8.78(dd,1h),8.59(d,1h),8.47(s,1h),8.29(d,1h),8.19(dd,1h;),7.87(s,1h),7.53(m,2h),7.46(q,1h),4.73(m,2h),4.39(s,2h).3.57(t,2h),2.99(t,2h),2.51(m,8h),1.92(m,8h),1.87(m,4h),0.95(s,9h).
实施实例3:
(1)中间体1-溴-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺的合成
将1.072g(0.0015mol)双溴代苝二酰亚胺,0.33g(0.0024mol)无水碳酸钾,0.27g(0.0018mol)对叔丁基苯酚一次性加入到三口烧瓶中,再向三口烧瓶中加入20mln-甲基吡咯烷酮,140℃反应45min,停止反应,降温,将反应物倒入1m盐酸溶液中,调节ph值至3.1,搅拌30min过滤,晾干。柱层析提纯,二氯甲烷作为洗脱剂收集第二带产物。氯仿/甲醇重结晶得红色固体0.561g。nmr(300mhz,cdcl3)δ:9.41(d,1h),8.59(d,1h),8.50(s,1h),9.14(d,1h),8.28(d,1h),7.92(s,1h),4.80(m,2h),2.59(m,8h),1.99(m,8h),1.90(m,4h),0.94(s,9h).
(2)中间体1-哌嗪-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺的合成
将0.773g(1mmol)1-溴-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺,0.301g(3.5mmol)无水哌嗪,0.552g(4mmol)无水碳酸钾加入到20ml无水dmf中,氮气保护下85℃反应3.5h,减压蒸干溶剂得粗品。柱层析提纯,二氯甲烷/乙醇作为洗脱剂,得到暗红色固体0.649g。nmr(300mhz,cdcl3)δ:9.48(d,1h),8.64(d,1h),8.53(s,1h),9.21(d,1h),8.32(d,1h),7.96(s,1h),4.80(m,2h),3.63(t,2h),3.01(t,2h),2.59(m,8h),1.99(m,8h),1.90(m,4h),0.94(s,9h).
(3)2-氯-n-(8-氨基喹啉)-乙酰胺的合成
将8-氨基喹啉1.45(0.01mol)加入到三口烧瓶中,加入干燥的二氯甲烷溶液50ml、三乙胺1.162g(0.0115mol),体系降至0℃以下,滴加氯乙酰氯1.23g(0.011mol)的二氯甲烷溶液70ml,整个过程控制体系温度低于5℃,反应2h,经过滤、酸洗、水洗、干燥得到2-氯-n-(8-氨基喹啉)-乙酰胺2.01g。1hnmr(cdcl3):10.94(br,1h),8.89(dd,1h),8.78(dd,1h),8.21(dd,1h;),7.59(m,2h),7.51(q,1h),4.34(s,2h).
(4)目标化合物1-对叔丁基酚氧基-7-(n-(8’-氨基喹啉)-乙酰氨基)哌嗪-n,n-二环己基3,4,9,10-苝二酰亚胺的合成
将0.781g(1mmol)1-哌嗪-7-对叔丁基苯酚-n,n-二环己基-3,4,9,10-苝二酰亚胺,0.55g(2.5mmol)2-氯-n-(8-氨基喹啉)-乙酰胺,无水碳酸钾0.55g(4mmol)和碘化钾67mg(0.4mmol)加入到10ml无水dmf中,氮气保护下85℃反应3.5h,减压蒸干溶剂得粗品。柱层析提纯,二氯甲烷/乙醇作为洗脱剂,得到暗红色固体0.708g。nmr(300mhz,cdcl3)δ:10.94(d,1h),9.48(d,1h),9.21(d,1h),8.89(dd,1h),8.78(dd,1h),8.59(d,1h),8.47(s,1h),8.29(d,1h),8.19(dd,1h;),7.87(s,1h),7.53(m,2h),7.46(q,1h),4.73(m,2h),4.39(s,2h).3.57(t,2h),2.99(t,2h),2.51(m,8h),1.92(m,8h),1.87(m,4h),0.95(s,9h).
尽管这里参照本发明的多个解释性实施实例对本发明进行了描述,但是,应该理解,本领域技术人员可以设计出很多其他的修改和实施方式,这些修改和实施方式将落在本申请公开的原则范围和精神之内。更具体地说,在本申请公开、附图和权利要求的范围内,可以对主题组合布局的组成部件或布局进行多种变型和改进。
- 还没有人留言评论。精彩留言会获得点赞!