阳离子脂质的制作方法
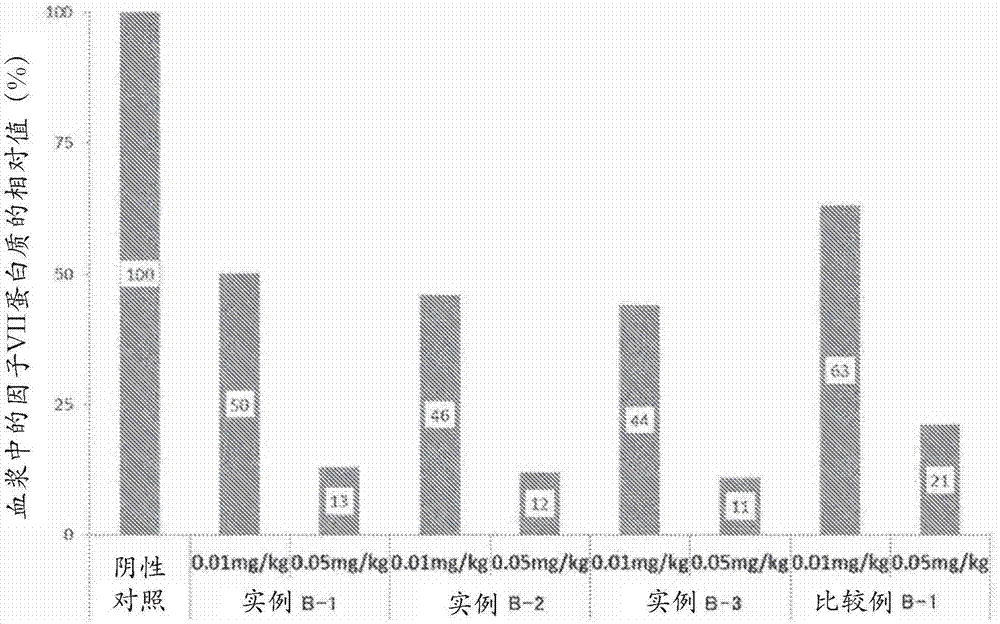
本发明是关于一种新颖阳离子脂质。
先前技术
sirna(smallinterferingrna,小干扰rna)、mirna(microrna,微rna)、shrna(短发夹rna(shorthairpinrna)、或小发夹rna(smallhairpinrna))表达载体、反义寡核苷酸等核酸是在活体内诱导序列特异性的基因表达抑制的核酸,作为核酸药物而为人所知。
这些核酸药物中,尤其是sirna受到关注。sirna是包含19~23个碱基对的双链rna,其诱导被称为rna干扰(rnainterference,rnai)的序列特异性的基因表达抑制。
虽然sirna在化学上稳定,但就容易被血浆中的rnase(ribonuclease,核糖核酸酶)所分解的方面、及不易单独通过细胞膜的方面等而言,在治疗用途方面存在问题(例如参照专利文献1)。
针对上述问题,已知借由在含有阳离子脂质的微粒内封入sirna,可保护所封入的sirna免在在血浆中被分解,且使其通过脂溶性的细胞膜(例如参照专利文献1)。
此外,专利文献2~5中记载有一种用在sirna等核酸药物的传递的阳离子脂质,其会提高生物分解能力。
此外,含有阳离子脂质的微粒具有在保管期间容易凝聚的稳定性问题,已知有在该微粒中含有聚乙二醇修饰脂质(peg脂质)而抑制凝聚的方法。进而,专利文献6中记载有如下方法,即,借由将作为特定peg脂质的peg-dpg(dipalmitoylglycerol,二棕榈酰甘油)设为微粒的构成成分、或制成包含该微粒与经去离子化的溶剂的制剂,而改善凝聚抑制及核酸的传递效率。
先前技术文献
专利文献
[专利文献1]国际公开第2010/144740号说明书
[专利文献2]国际公开第2011/153493号说明书
[专利文献3]国际公开第2013/086354号说明书
[专利文献4]国际公开第2013/158579号说明书
[专利文献5]国际公开第2015/095346号说明书
[专利文献6]国际公开第2014/089239号说明书
技术实现要素:
发明所欲解决的问题
然而,尽管近来有所进展,但业界仍在谋求可用于对细胞质的核酸传递的阳离子脂质。
解决问题的技术手段
本发明是关于以下的〔1〕至〔15〕。
〔1〕一种化合物或其药学上可接受的盐,该化合物是以下述式(1a)表示,
〔化1〕
〔式中,l1及l2各自独立地表示碳数3~10的亚烷基,r1及r2各自独立地表示碳数4~22的烷基或碳数4~22的烯基,x1表示单键或-co-o-,环p表示下述式(p-1)~(p-5)的任一者〕
〔化2〕
〔式中,r3表示碳数1~3的烷基〕。
〔2〕如〔1〕所记载的化合物或其药学上可接受的盐,其中化合物以下述式(1)表示,
〔化3〕
〔式中,l1及l2各自独立地表示碳数3~10的亚烷基,r1及r2各自独立地表示碳数4~22的烷基或碳数4~22的烯基,x1表示单键或-co-o-〕。
〔3〕如〔1〕或〔2〕所记载的化合物或其药学上可接受的盐,其中化合物是选自由下述式(a1)~(a22)所表示的化合物所组成的组,
〔化4〕
〔3a〕如〔3〕所记载的化合物或其药学上可接受的盐,其中化合物是选自由上述式(a1)、(a2)、(a3)、(a4)、(a5)、(a9)、(a12)、(a15)、(a16)、(a17)、(a19)、(a20)、(a22)所表示的化合物所组成的组。
〔3b〕如〔3〕所记载的化合物或其药学上可接受的盐,其中化合物是选自由上述式(a1)、(a2)、(a3)、(a4)、(a5)、(a9)、(a12)、(a15)、(a20)所表示的化合物所组成的组。
〔3c〕如〔3〕所记载的化合物或其药学上可接受的盐,其中化合物是选自由上述式(a1)~(a5)所表示的化合物所组成的组。
〔3d〕如〔3〕所记载的化合物或其药学上可接受的盐,其中化合物是选自由上述式(a6)~(a8)所表示的化合物所组成的组。
〔3〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a1)表示,
〔化5〕
〔4〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a2)表示,
〔化6〕
〔5〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a3)表示,
〔化7〕
〔6〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a4)表示,
〔化8〕
〔7〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a5)表示,
〔化9〕
〔8〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a9)表示,
〔化10〕
〔9〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a12)表示,
〔化11〕
〔10〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a15)表示,
〔化12〕
〔11〕如〔1〕至〔3〕中任一项所记载的化合物或其药学上可接受的盐,其中化合物是以下述式(a20)表示,
〔化13〕
〔13〕一种脂质复合体,其含有(i)如〔1〕至〔12〕中任一项所记载的化合物或其药学上可接受的盐、及(ii)选自由中性脂质、聚乙二醇修饰脂质、及固醇所组成的组中的至少一种脂质。
〔14〕一种组合物,其含有(i)如〔1〕至〔12〕中任一项所记载的化合物或其药学上可接受的盐、(ii)选自由中性脂质、聚乙二醇修饰脂质、及固醇所组成的组中的至少一种脂质、及(iii)核酸。
〔15〕一种组合物的制造方法,其包括:将含有(i)如〔1〕至〔12〕中任一项所记载的化合物或其药学上可接受的盐、及(ii)选自由中性脂质、聚乙二醇修饰脂质、及固醇所组成的组中的至少一种脂质的含极性有机溶剂的水溶液与含有(iii)核酸的水溶液加以混合而获得混合液的步骤;及减少混合液中的极性有机溶剂的含有率的步骤。
发明的效果
本发明的阳离子脂质具有以下中的一种以上的效果。
(1)根据本发明的阳离子脂质,能够将核酸高效率地释放至细胞质中。
(2)根据本发明的阳离子脂质,能够在保管一定时间的情形时抑制脂质复合体的粒径增大。
因此,本发明的阳离子脂质具有作为对细胞质的核酸传递用的脂质的可利用性。
附图说明
〔图1〕是表示试验例1的结果的图表。
〔图2〕是表示试验例2的结果的图表。
具体实施方式
以下,示出实施例及示例等而对本发明进行详细说明,但本发明并不限于以下所示的实施例及示例等,可在不脱离本发明的主旨的范围内任意变更而实施。再者,本说明书所记载的全部文献及刊物不论其目的为何,均借由参照而将其全部内容并入本说明书中。
<阳离子脂质>
在一实施例中,本发明是下述式(1a)所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
〔化14〕
式(1a)中,环p表示下述式(p-1)~(p-5)的任一者。
〔化15〕
上述式(p-1)~(p-5)中,r3表示碳数1~3的烷基。
在一实施例中,环p表示下述式(p-1)、式(p-2)、(p-4)、(p-5)的任一者。
在一实施例中,环p表示下述式(p-1)。
在一实施例中,本发明是下述式(1)所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
〔化16〕
式(1a)及式(1)中,l1及l2各自独立地表示碳数3~10的亚烷基,r1及r2各自独立地表示碳数4~22的烷基或碳数4~22的烯基,x1表示单键或-co-o-。
在本说明书中,所谓“烷基”意指具有指定数量的碳原子的直链状、环状或支链状的饱和脂肪族烃基。
在本说明书中,所谓“烯基”意指具有指定数量的碳原子及至少1个碳-碳双键的直链或支链的烃基。例如可列举单烯、二烯、三烯及四烯等,但并不限于这些。
在本说明书中,所谓“亚烷基”意指具有指定数量的碳原子的直链状、环状或支链状的二价饱和脂肪族烃基。
在本说明书中,“卤素”意指f、cl、br、i。
本发明之一实施例是上述式(1a)或式(1)中,l1及l2各自独立为碳数3~10(例如,碳数5~10或碳数3~8)的亚烷基,r1及r2各自独立为碳数4~18的烷基或碳数4~18的烯基,x1为单键或-co-o-的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
本发明的一实施例是上述式(1a)或式(1)中,l1及l2各自独立为碳数3~10(例如,碳数5~10或碳数3~8)的直链状的亚烷基,r1及r2各自独立为碳数4~18的直链状或支链状的烷基或者碳数4~18的直链状的烯基,x1为-co-o-的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
因此,本发明的一实施例是下述式(1b)所表示的化合物或其药学上可接受的盐。
〔化17〕
式(1b)中,r1及r2各自独立为碳数4~22的烷基或碳数4~22的烯基,优选为碳数4~18的直链状或支链状的烷基或者碳数4~18的直链状的烯基,n1及n2各自独立地表示3~10(例如5~10或3~8)的整数。
在本发明的一实施例中,本发明是上述式(1a)或式(1)中,x1为-co-o-,l1与l2相同,r1与r2相同的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
在本发明的一实施例中,本发明是上述式(1a)或式(1)中,l1及l2各自独立为碳数5~10的直链状的亚烷基,r1为碳数4~18的直链状或支链状的烷基或者碳数4~18的直链状的烯基,r2为碳数4~18的直链状的烷基,x1为单键的化合物或其药学上可接受的盐。在本实施例中,优选为l2及r2的合计的碳数为9~12。本实施例的化合物可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
因此,本发明的一实施例是下述式(1c)所表示的化合物或其药学上可接受的盐。
〔化18〕
式(1c)中,r1为碳数4~22的烷基或碳数4~22的烯基,优选为碳数4~18的直链状或支链状的烷基或者碳数4~18的直链状的烯基,n1表示3~10(例如5~10或3~8)的整数,n2表示8~25、优选为8~11的整数。
以下,示出实施例的化合物的一例。
〔化19〕
本发明的一实施例是上述式(a1)~(a22)的任一者所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
本发明的一实施例是上述式(a1)~(a5)、(a9)~(a22)的任一者所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
本发明的一实施例是上述式(a1)、(a2)、(a3)、(a4)、(a5)、(a9)、(a12)、(a15)、(a16)、(a17)、(a19)、(a20)、(a22)的任一者所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
本发明的一实施例是上述式(a1)、(a2)、(a3)、(a4)、(a5)、(a9)、(a12)、(a15)、(a20)的任一者所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
本发明的一实施例是上述式(a1)、(a2)、(a3)、(a4)、(a5)的任一者所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
本发明的一实施例是上述式(a6)、(a7)、(a8)的任一者所表示的化合物或其药学上可接受的盐,其可用作阳离子脂质。阳离子脂质亦可为上述盐的水合物或上述盐的溶剂合物。
在本说明书中,所谓阳离子脂质是具有含有一个或多个的烃基的脂质亲和性区域、与含有在生理性ph值下进行质子化的极性基的亲水性区域的两亲媒性分子。即,本发明的阳离子脂质可进行质子化而形成阳离子。例如上述式(1)所表示的化合物包含对哌啶环的氮原子上的孤电子对配位有氢离子的下述式(1)′所表示的化合物(阳离子)。
〔化20〕
作为与上述阳离子成对而本实施例的阳离子脂质可含有的阴离子,只要为药学上可接受的那些,则无特别限定,例如可列举:氯化物离子、溴化物离子、硝酸根离子、硫酸根离子、磷酸根离子等无机离子;乙酸根离子、草酸根离子、顺丁烯二酸根离子、反丁烯二酸根离子、柠檬酸根离子、苯甲酸根离子、甲磺酸根离子等有机酸根离子等。
本发明的阳离子脂质可存在几何异构物、光学异构物等立体异构物、互变异构物等,本发明的阳离子脂质包含包括这些异构物在内的全部可能的异构物及这些的混合物。
<阳离子脂质的制造方法>
对本发明的阳离子脂质的制造方法进行说明。下述式(10)及(11)表示阳离子脂质的合成流程的一实施例。本说明书所记载的化合物全部以化合物的形式包含在本发明中。本发明的化合物可依照以下的流程所记载的方法的至少1种而合成。
〔化21〕
(式中,l1及r1分别与上述含义相同,r为式(1)中的-l2-x1-r2--(l2、x1及r2分别与上述含义相同))
式(1)的阳离子脂质(x1为单键的化合物)例如可依照上述式(10)所示的流程1而合成。
(步骤1-1:酯化)
首先,在缩合剂的存在下使醇(a1)与卤化烷基羧酸x-l1-cooh(x为卤素原子,l1与上述含义相同)(优选为溴化烷基羧酸)进行反应,而获得酯的卤化物(a2)。作为缩合剂,例如可列举:1-[3-(二甲胺基)丙基]-3-乙基碳二酰亚胺(edc)盐酸盐、n,n′-二环己基碳二酰亚胺(dcc)等。视需要亦可添加碱。作为碱,例如可列举:nmm(n-methylmorpholine,n-甲基味啉)、tea(triethylamine,三乙胺)、dipea(n,n-diisopropylethylamine,n,n-二异丙基乙基胺)、dmap(4-dimethylaminopyridine,4-二甲胺基吡啶)、吡啶、甲基吡啶、二甲基吡啶等。作为溶剂,例如可列举:四氢呋喃(thf)、二氯甲烷、氯仿、苯、己烷、乙酸乙酯等。
(步骤1-2:导入烷基链)
继而,在碱的存在下使酯卤化物(a2)与丙二酸二叔丁酯进行反应。借此,夺去丙二酸二酯的活性亚甲基的氢原子,并导入烷基酯链,而获得化合物(a3)。作为碱,例如可列举nah。作为溶剂,例如可列举:二噁烷、四氢呋喃、环戊基甲基醚、1,2-二甲氧基乙烷等醚类等。
(步骤1-3:导入烷基链)
继而,在碱的存在下使化合物(a3)与卤化烷基(优选为碘化物)进行反应,而导入烷基链,获得化合物(a4)。碱及溶剂可使用与上述步骤1-2相同的那些。
(步骤1-4:去保护)
继而,在酸性水解条件下将化合物(a4)的叔丁基去保护,而获得化合物(a5)。作为去保护所使用的酸,例如可列举三氟乙酸(tfa)、盐酸等。作为溶剂,例如可列举二氯甲烷等。
(步骤1-5:脱羧)
继而,借由化合物(a5)的脱羧而获得羧酸(a6)。脱羧反应例如可借由在溶剂中进行加热而进行。作为溶剂,例如可使用苯、甲苯、二甲苯等芳香族烃类等。
(步骤1-6:还原步骤)
在还原剂的存在下将化合物(a6)的羧基还原为羟基,而获得化合物(a7)。作为还原剂,例如可列举硼烷(bh3)-四氢呋喃络合物、硼烷-二甲基硫醚络合物等硼烷络合物等。作为溶剂,例如可使用:二乙醚、四氢呋喃、二噁烷等醚类;氯仿、二氯甲烷、二氯乙烷等卤化烃类;己烷、苯、甲苯等烃类;及这些的混合溶剂等。
(步骤1-7:酯化)
在缩合剂、碱的存在下使所获得的醇(a7)与1-甲基-哌啶-4-羧酸或其衍生物(氢卤酸盐等)进行反应,而获得作为最终物的化合物(a8)(r=l2-x1-r2)(相当在式(1)的阳离子脂质的化合物)。作为缩合剂及碱,可使用与步骤1-1相同的那些。
再者,在合成x1为-co-o-的化合物时,在上述流程1的步骤1-3中,按照下文所述的步骤2-1制备羧基经保护的化合物,使其与化合物(a3)进行反应,最后进行去保护及酯化即可。
〔化22〕
(式中,l1及r1分别与上述含义相同,r为式(1)中的-l2-x1-r2(l2、x1及r2分别与上述含义相同))
上述流程2表示从业者借由不同方法而合成式(1)的阳离子脂质(x1为单键的化合物)的方法。
(步骤2-1:酯化)
首先,借由使卤化烷基羧酸与苄醇进行缩合,而获得酯的卤化物(b1)。酯化条件与步骤1-1相同。
(步骤2-2:导入烷基链)
继而,与步骤1-2同样地,在碱的存在下使酯的卤化物(b1)与丙二酸二叔丁酯进行反应,而获得化合物(b2)。
(步骤2-3:导入烷基链)
继而,与步骤1-3同样地,在碱的存在下使化合物(b2)与卤化烷基进行反应,而获得化合物(b3)。
(步骤2-4:去保护)
继而,与步骤1-4同样地,在酸性水解条件下将化合物(b3)的叔丁氧基羰基进行去保护,而获得化合物(b4)。
(步骤2-5:脱羧)
继而,与步骤1-5同样地,获得羧酸(b5)。
(步骤2-6:还原步骤)
进而,与步骤1-6同样地,在还原剂的存在下将其还原而获得化合物(b6)。
(步骤2-7:酯化)
在缩合剂、碱的存在下使所获得的化合物(b6)与1-甲基-哌啶-4-羧酸或其衍生物(氢卤酸盐等)进行酯化反应,而获得化合物(b7)。
(步骤2-8:去保护)
继而,在还原条件下将苄基保护基进行去保护,而获得化合物(b8)。去保护例如借由在钯/碳等金属触媒的存在下进行催化氢化反应而进行即可。
(步骤2-9:酯化)
最后,借由使化合物(b8)与醇(r1oh)进行反应,可获得化合物(b9)(r=l2-x1-r2)(相当于式(1)的阳离子脂质的化合物)。
再者,在合成x1为-co-o-的化合物时,在上述流程2的步骤2-3中,按照步骤2-1使羧基经保护的化合物与化合物(b2)进行反应,将其在步骤2-8中进行去保护即可。
关于上述式(1a)的化合物,亦可按照上述流程1或2而合成。具体而言,在环p为式(p-1)、(p-4)、(p-5)的结构的情形时,例如在上述步骤1-7或步骤2-7中,使用与式(p-1)、(p-4)、(p-5)的结构相对应的羧酸代替1-甲基-哌啶-4-羧酸进行酯化反应即可。此外,在环p为式(p-2)、(p-3)的结构的情形时,代替步骤1-7,而在碱的存在下,使步骤1-6中所获得的化合物a7、羰基化试剂(例如氯甲酸4-硝基苯酯等氯甲酸酯)、n-烷基哌嗪或n-烷基高哌嗪(具有式(p-2)、(p-3)的结构的化合物)进行反应,借此可获得式(1a)所表示的化合物(参照下文所述的实例a-8)。
在本发明的化合物的合成中,只要未特别记载起始原料的制造,则这些化合物为公知,或者可借由该技术领域中公知的类似的方法,或如以下的实例中所记载般进行制备。从业者应当理解上述的流程仅为本发明化合物的代表性的制备方法,亦可同样地使用其他周知的方法。
在本发明的化合物的制备中,有时存在对分子的官能基加以保护的需要和/或愿望。该保护可利用从业者所周知的先前的保护基而进行。保护基可在其后合适的阶段,使用该技术领域周知的方法而去除。再者,上述流程中所示的保护基(叔丁基保护基、苄基保护基)亦可换为从业者所周知的其他保护基。
<脂质复合体>
本发明提供一种含有(i)上述的阳离子脂质、及(ii)选自由中性脂质、聚乙二醇修饰脂质及固醇所组成的组中的至少一种脂质的脂质复合体。作为本发明的一实施例,脂质复合体含有(i)上述的阳离子脂质、(ii)选自由中性脂质、聚乙二醇修饰脂质及固醇所组成的组中的至少一种脂质、及(iii)核酸。因此,本发明的脂质复合体可含有核酸,亦可不含核酸。本实施例的脂质复合体能够将核酸高效率地释放至细胞质中。此外,本实施例的脂质复合体在保管一定时间(例如1个月或3个月)的情形时,粒径的增大受到抑制,而可显示出优异的物理稳定性。
作为由含有阳离子脂质的脂质与核酸所形成的复合体的形态,例如可列举核酸与含有脂质单(单分子)层的膜(反胶束)的复合体、核酸与脂质体的复合体、核酸与胶束的复合体等。在本发明的一实施例的脂质复合体中,将核酸封入至包含含有阳离子脂质的脂质的微粒内。
本实施例的脂质复合体以脂质复合体所含的全部脂质为基准,而含有例如10~100摩尔%、例如20~90摩尔%、例如40~80摩尔%的上述的阳离子脂质。阳离子脂质可单独使用1种,或者亦可将2种以上混合而使用。
作为核酸,可列举:sirna、mirna、shrna表达载体、反义寡核苷酸、mrna、核酶等。在一实施例中,核酸亦可为sirna、mirna、mrna。
本实施例的脂质复合体相对在脂质复合体的总重量而含有例如0.01~50重量%、例如0.1~30重量%、例如1~10重量%的核酸。
本实施例的脂质复合体含有(i)上述的阳离子脂质、及(ii)选自由中性脂质、聚乙二醇修饰脂质、及固醇所组成的组中的至少一种脂质作为脂质成分。本实施例的脂质复合体相对在脂质复合体的总重量,而含有例如50~100重量%、例如70~99.99重量%、例如90~99重量%的脂质成分。
所谓中性脂质意指在生理性ph值下以不带电或中性的两性离子的任一种形式存在的脂质。作为中性脂质,可列举:二油酰磷脂酰乙醇胺(dope)、棕榈酰油酰磷脂酰胆碱(popc)、卵磷脂酰胆碱(epc)、二肉豆蔻酰磷脂酰胆碱(dmpc)、二棕榈酰磷脂酰胆碱(dppc)、二硬脂酰磷脂酰胆碱(dspc)、二花生酰磷脂酰胆碱(dapc)、二山萮酰磷脂酰胆碱(dbpc)、二(木蜡酰)磷脂酰胆碱(dlpc)、二油酰磷脂酰胆碱(dopc)、神经鞘磷脂、脑酰胺、二油酰磷脂酰甘油(dopg)、二棕榈酰磷脂酰甘油(dppg)、磷脂酰乙醇胺(pope)、二油酰磷脂酰乙醇胺4-(n-顺丁烯二酰亚胺甲基)-环己烷-1-羧酸酯(dope-mal)等。中性脂质可单独使用1种,或者亦可将2种以上混合而使用。
本实施例的脂质复合体以脂质复合体所含的全部脂质为基准,而可含有例如0~50摩尔%、例如0~40摩尔%、例如0~30摩尔%的中性脂质。
作为聚乙二醇修饰脂质,可列举:peg2000-dmg(peg2000-二肉豆蔻基甘油)、peg2000-dpg(peg2000-二棕榈酰甘油)、peg2000-dsg(peg2000-二硬脂酰甘油)、peg5000-dmg(peg5000-二肉豆蔻基甘油)、peg5000-dpg(peg5000-二棕榈酰甘油)、peg5000-dsg(peg5000-二硬脂酰甘油)、peg-cdma(n-[(甲氧基聚(乙二醇)2000)胺甲酰基]-1,2-二肉豆蔻氧基丙基-3-胺)、peg-c-domg(r-3-[(ω-甲氧基-聚(乙二醇)2000)胺甲酰基]-1,2-二肉豆蔻氧基丙基-3-胺)、聚乙二醇(peg)-二酰甘油(dag)、peg-二烷基氧基丙基(daa)、peg-磷脂质、peg-脑酰胺(cer)等。
作为peg-二烷基氧基丙基,可列举:peg-二月桂基氧基丙基、peg-二肉豆蔻基氧基丙基、peg-二棕榈基氧基丙基、peg-二硬脂基氧基丙基等。聚乙二醇修饰脂质可单独使用1种,或者亦可将2种以上混合而使用。
本实施例的脂质复合体以脂质复合体所含的全部脂质为基准,而可含有例如0~30摩尔%、例如0~20摩尔%、例如0~10摩尔%的聚乙二醇修饰脂质。
固醇是具有类固醇骨架的醇。作为固醇,可列举:胆固醇、二氢胆固醇、羊毛固醇、β-植固醇、菜油固醇、豆固醇、菜籽固醇、麦角固醇、海藻固醇、3β-[n-(n′,n′-二甲胺基乙基)胺甲酰基]胆固醇(dc-chol)等。固醇可单独使用1种,或者亦可将2种以上混合而使用。
本实施例的脂质复合体以脂质复合体所含的全部脂质为基准,而可含有例如0~90摩尔%、例如10~80摩尔%、例如20~50摩尔%的固醇。
本实施例的脂质复合体中的脂质成分的组合并无特别限定,例如可列举:上述的阳离子脂质、中性脂质及固醇的组合;上述的阳离子脂质、中性脂质、聚乙二醇修饰脂质及固醇的组合等。
<组合物>
在一实施例中,本发明提供一种含有(i)上述的阳离子脂质、(ii)选自由中性脂质、聚乙二醇修饰脂质及固醇所组成的组中的至少一种脂质、及(iii)核酸的组合物。本实施例的组合物能够将核酸高效率地释放至细胞质中。本实施例的组合物亦可为含有上述的脂质复合体、药学上可接受的介质、及视情形的其他添加剂者。以下,对药学上可接受的介质、其他添加剂进行说明。
本实施例的组合物以组合物所含的全部脂质为基准,而含有例如10~100摩尔%、例如20~90摩尔%、例如40~70摩尔%的上述的阳离子脂质。阳离子脂质可单独使用1种,或者亦可将2种以上混合而使用。
作为核酸,可列举与上述者相同的那些。本实施例的组合物相对于组合物的总重量,而含有例如0.01~50重量%、例如0.1~30重量%、例如1~10重量%的核酸。
本实施例的组合物含有(i)上述的阳离子脂质、及(ii)选自由中性脂质、聚乙二醇修饰脂质、及固醇所组成的组中的至少一种脂质作为脂质成分。
作为中性脂质,可列举与上述者相同的那些。本实施例的组合物以组合物所含的全部脂质为基准,而可含有例如0~50摩尔%、例如0~40摩尔%、例如0~30摩尔%的中性脂质。
作为聚乙二醇修饰脂质,可列举与上述者相同的那些。本实施例的组合物以组合物所含的全部脂质为基准,而可含有例如0~30摩尔%、例如0~20摩尔%、例如0~10摩尔%的聚乙二醇修饰脂质。
作为固醇,可列举与上述者相同的那些。本实施例的组合物以组合物所含的全部脂质为基准,而可含有例如0~90摩尔%、例如10~80摩尔%、例如20~50摩尔%的固醇。
本实施例的组合物中的脂质成分的组合并无特别限定,例如可列举:上述的阳离子脂质、中性脂质及固醇的组合;上述的阳离子脂质、中性脂质、聚乙二醇修饰脂质及固醇的组合等。
本实施例的组合物除了上述的成分以外,作为其他添加剂,亦可含有:蔗糖、葡萄糖、山梨糖醇、乳糖等糖;谷酰胺、谷胺酸、谷胺酸钠、组胺酸等氨基酸;柠檬酸、磷酸、乙酸、乳酸、碳酸、酒石酸等酸的盐等。
本实施例的组合物亦可配制为药物组合物。作为药物组合物的剂型,例如可列举注射剂。
本实施例的组合物例如可为借由冷冻干燥等而去除溶剂的粉末状态,亦可为液体状态。本发明的一实施例的组合物为含有上述的实施例的脂质复合体的粉末组合物。粉末组合物可由液体状态的组合物(分散液)借由例如过滤、离心分离等去除溶剂而制备,亦可将该分散液加以冷冻干燥而制备。在组合物为粉末状态的情形时,可在使用前使其悬浮或溶解在药学上可接受的介质中,制成注射剂而使用。本发明的一实施例的组合物为含有上述的实施例的脂质复合体及药学上可接受的介质的液体组合物。在组合物为液体状态的情形时,可直接、或者使其悬浮或溶解在药学上可接受的介质中制成注射剂而使用。
作为药学上可接受的介质,可列举:杀菌水;生理盐水;含有葡萄糖、d-山梨糖醇、d-甘露糖、d-甘露糖醇、氯化钠等佐药的等渗液;磷酸缓冲液、柠檬酸缓冲液、乙酸缓冲液等缓冲液等。本实施例的组合物亦可进而含有:乙醇、丙二醇、聚乙二醇等醇等增溶剂、稳定剂、抗氧化剂、防腐剂、制造药剂上通常使用的赋形剂、填充剂、增量剂、结合剂、湿润剂、崩解剂、润滑剂、表面活性剂、分散剂、保存剂、调味剂、舒缓剂等添加剂。
组合物对患者的给予例如可借由动脉内注射、静脉内注射、皮下注射等非经口用法而进行。组合物的给予量根据给予对象、对象器官、症状、给予方法而异。组合物的给予对象并无限定,可应用于各种动物,尤其可应用于哺乳动物、优选为人类及临床试验、筛选、实验的实验动物。
本实施例的组合物是形成在包含含有阳离子脂质的脂质的微粒内封入有核酸的脂质复合体。脂质复合体的“平均粒径”可借由体积平均、数量平均、z-平均的任一方法而算出。在本实施例的组合物中,脂质复合体的平均粒径(z-平均)可为例如10~1000nm、例如30~500nm、例如30~200nm。
本实施例的组合物优选为在保管期间,脂质复合体的粒径与保管前相比尽量不增大。例如,在4℃下保管3个月的情形时的平均粒径(z-平均)的大小优选为相对于保管前的粒径的大小而为1.3倍以下,更佳为1.2倍以下,尤佳为1.1倍以下。
就抑制非特异性的吸附或免疫反应的观点而言,本实施例的组合物优选为在例如血液中等ph值约为7.4的环境下,表面几乎无电荷。此外,在借由内噬作用而被吸收至细胞内时,就提高与内体膜的融合效率的观点而言,优选为在低ph值(例如3.5~7.0)环境下带正电。
<组合物的制造方法>
在一实施例中,本发明提供一种组合物的制造方法,该制造方法包括:将含有(i)上述的阳离子脂质、及(ii)选自由中性脂质、聚乙二醇修饰脂质及固醇所组成的组中的至少一种脂质的含极性有机溶剂的水溶液与含有(iii)核酸的水溶液加以混合而获得混合液的步骤(a);及减少混合液中的极性有机溶剂的含有率的步骤(b)。根据本实施例的制造方法,可制造能够将核酸高效率地释放至细胞质中的组合物。
借由水溶性的核酸与上述的阳离子脂质的静电性相互作用、及脂质间的疏水性相互作用,可形成在包含脂质的微粒内封入有核酸的脂质复合体。例如,借由减少混合液中的极性有机溶剂的含有率,而改变含有(i)上述的阳离子脂质、及(ii)选自由中性脂质、聚乙二醇修饰脂质及固醇所组成的组中的至少一种脂质的脂质成分相对于含极性有机溶剂的水溶液的溶解度,借此可形成脂质复合体。作为极性有机溶剂,可列举乙醇等醇。
首先,在步骤(a)中,将溶解有(i)上述的阳离子脂质、及(ii)选自由中性脂质、聚乙二醇修饰脂质及固醇所组成的组中的至少一种脂质的含极性有机溶剂的水溶液与含有(iii)核酸的水溶液加以混合,而获得混合液。关于含极性有机溶剂的水溶液中的极性有机溶剂的浓度,只要与核酸的水溶液混合后亦满足溶解脂质分子的条件,则无特别限定。若列举一例,则步骤(a)的含极性有机溶剂的水溶液中的极性有机溶剂的浓度可为0~60重量%。
其次,在步骤(b)中,借由向上述的混合液中添加水等,而减少极性有机溶剂的含有率。借此,可形成脂质复合体。为了高效率地形成脂质复合体,优选为快速降低极性有机溶剂的含有率。若列举一例,则步骤(b)中的最终的含极性有机溶剂的水溶液中的极性有机溶剂的浓度可为0~5重量%。
此外,对在步骤(a)所获得的上述混合液,亦可借由透析而去除极性有机溶剂,将溶剂换为药学上可接受的介质。由于在透析过程中溶液中的极性有机溶剂的含有率会减少,因此,借此可形成脂质复合体。
根据本实施例的组合物的制造方法,可获得将核酸高效率地封入至微粒内部的脂质复合体。该脂质复合体可具有优异的物理稳定性。例如,在保管一定时间(例如1个月、例如3个月)的情形时,可抑制粒径的增大。
在封入至组合物中的核酸为核酸药物的情形时,可将组合物作为药物组合物而利用。例如,本发明的组合物可用于在体内(invivo)或体外(invitro)对成为靶标的细胞质(例如成为各种疾病的原因的细胞质)导入所需的核酸的治疗(例如基因治疗)。因此,本发明的一实施例提供一种使用含有上述的脂质复合体的药物组合物的各种疾病的治疗方法(尤其是基因治疗方法)。再者,给予的对象、给予的方法及条件与上述相同。
本发明的一实施例可为含有上述阴离子性脂质的核酸药物传递用试剂盒。此外,该试剂盒可优选地用于针对各种靶细胞的治疗(例如基因治疗)等。在本实施例的试剂盒中,阴离子性脂质的保存状态并无特别限定,可考虑各自的稳定性(保存性)及使用容易性等而设定为溶液状或粉末状等任意的状态。本实施例的试剂盒除了上述阴离子性脂质以外,亦可包含例如各种核酸、各种介质(药学上可接受的介质、缓冲剂)、及使用说明书(使用手册)等。本实施例的试剂盒可用于制备包含导入至靶细胞内的所需的核酸及含有上述阴离子性脂质的脂质的组合物或脂质复合体。所制备的组合物或脂质复合体可有效地用于对靶细胞的核酸传递。进而,本发明的一实施例可为包含含有上述阴离子性脂质的药物组合物的核酸药物传递用试剂盒。本实施例的试剂盒除了上述药物组合物以外,亦可包含例如各种介质(药学上可接受的介质)、及使用说明书(使用手册)等。
实例
以下,列举实例、制造例及试验例对本发明进一步进行详细描述,但本发明并不限于这些实例。实例、制造例中的化合物名称是使用软件(商品名“chemdrawultraver.12.0”,perkinelmer公司制造)进行命名。
为了合成本发明的化合物而利用的起始原料、试剂、酸、碱、脱水剂、溶剂、及触媒全部为市售,或可借由从业者所公知的有机合成法而生成。进而,本发明的化合物如以下的实例所示般,可借由从业者所公知的有机合成法而制造。
实例中所使用的缩写为从业者所周知的惯用缩写。将若干缩写示在以下。
dipea:n,n-二异丙基乙基胺
dmap:4-(二甲胺基)吡啶
dmf:n,n-二甲基甲酰胺
edc:1-乙基-3-(3-二甲胺基丙基)碳二酰亚胺盐酸盐
n-:正
tert-:叔
etoac:乙酸乙酯
tfa:三氟乙酸
thf:四氢呋喃
1h-nmr:质子核磁共振谱分析
以下的实例及制造例中的“室温”通常表示约10℃至约35℃。只要未特别记载,则%表示重量百分比。
质子核磁共振谱的化学位移是以相对于四甲基硅烷的δ单位(ppm)进行记录。图案的缩写如下所述。
s:单峰、d:双峰、t:三重峰、q:四重峰、quin:五重峰、m:多重峰、br:宽峰。
关于层析法,使用yamazen公司制造的parallelprep{柱:yamazen公司制造的hi-flashtmcolumn(硅胶(silicagel))、尺寸:s(16×60mm)、m(20×75mm)、l(26×100mm)、2l(26×150mm)}、或biotage公司制造的快速自动精制系统isoleratm{柱:snapcartridgekp-sil(10g、25g、50g、100g、340g)}。
a.阳离子脂质的合成
〔制造例1〕
9-溴壬酸2-丁基辛酯的合成
〔化23〕
将9-溴壬酸(1.1g,4.7mmol)、dipea(0.91ml,5.3mmol)及dmap(76mg,0.63mmol)溶解在二氯甲烷(13ml)中,在冰浴冷却下添加edc(1.0g,5.3mmol)。添加2-丁基-1-辛醇(0.58g,3.1mmol),在室温下搅拌2小时。在反应混合物中添加饱和碳酸氢钠水溶液,利用乙酸乙酯进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(1.2g,2.96mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.82-0.95(m,6h),1.17-1.49(m,24h),1.52-1.70(m,3h),1.75-1.93(m,2h),2.23-2.36(m,2h),3.34-3.47(m,2h),3.91-4.02(m,2h)。
〔制造例2〕
9-溴壬酸苄酯的合成
〔化24〕
将苄醇(0.50ml,4.84mmol)、9-溴壬酸(1.38g,5.80mmol)及dmap(59mg,0.48mmol)溶解在二氯甲烷(9.6ml)中,在冰浴冷却下添加edc(1.16g,6.05mmol),在室温下搅拌2小时。在反应混合物中添加饱和碳酸氢钠水溶液,利用二乙醚进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(1.49g,4.55mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.22-1.48(m,8h),1.58-1.71(m,2h),1.78-1.90(m,2h),2.30-2.40(m,2h),3.35-3.44(m,2h),5.12(s,2h),7.26-7.45(m,5h)。
〔制造例3〕
8-溴辛酸苄酯的合成
〔化25〕
按照制造例2的方法,由苄醇(1.85ml,17.9mmol)、8-溴辛酸(4.0g,17.9mmol)、edc(3.78g,19.7mmol)、dmap(0.22g,1.79mmol)、及二氯甲烷(36ml)获得标题化合物(4.8g,15.3mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.22-1.48(m,6h),1.58-1.71(m,2h),1.78-1.90(m,2h),2.30-2.38(m,2h),3.33-3.45(m,2h),5.12(s,2h),7.28-7.44(m,5h)。
〔制造例4〕
8-戊基十三烷-1-醇的合成
〔化26〕
(1)(6-羧基己基)三苯基溴化膦的合成
〔化27〕
将7-溴庚酸(2.0g,9.57mmol)与三苯基膦(2.5g,9.57mmol)悬浮在乙腈(20ml)中,加热回流18小时。放置冷却至室温后,在减压下进行浓缩而获得标题化合物(4.1g,8.66mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.22-1.59(m,8h),2.12-2.25(m,2h),3.47-3.62(m,2h),7.19-7.28(m,2h),7.34-7.44(m,2h),7.71-7.85(m,10h),7.85-7.94(m,3h),11.97(brs,1h)。
(2)8-戊基-7-十三碳烯酸的合成
〔化28〕
将制造例4-(1)中所获得的化合物(4.65g,9.87mmol)溶解在thf(15ml)中,在氮气环境、室温下添加双(三甲基硅烷基)酰胺钠/thf溶液(1m,19.7ml,19.7mmol)。将反应液加热至45℃后搅拌30分钟。滴加十一烷-6-酮(1.7ml,8.22mmol)的thf(5ml)溶液后进行加热回流。将反应混合物搅拌22小时后,仔细小心地添加盐酸(2m)直至ph值成为2为止。添加水(100ml),利用二乙醚进行萃取后,利用无水硫酸镁将有机相加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(乙酸乙酯/环己烷)对残渣进行精制,而获得标题化合物与十一烷-6-酮的1∶1的混合物(1.36g)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.80-0.89(m,6h),1.16-1.38(m,18h),1.43-1.53(m,2h),1.89-1.99(m,4h),2.17(t,j=7.43hz,2h),5.07(t,j=6.90hz,1h),11.83(brs,1h)。
(3)8-戊基十三烷酸的合成
〔化29〕
将制造例4-(2)中所获得的化合物(1.36g)与10%钯-碳(0.06g,0.060mmol)悬浮在乙醇(5ml)中,在氢气环境、室温常压下搅拌18小时。利用硅藻土将反应混合物加以过滤,并利用乙醇洗净。在减压下将滤液浓缩,而获得标题化合物(0.60g,2.18mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.85(t,j=7.1hz,6h),1.14-1.32(m,25h),1.42-1.52(m,2h),2.18(t,j=7.43hz,2h)。
(4)8-戊基十三烷-1-醇的合成
〔化30〕
在氮气环境下在冰浴冷却下向氢化锂铝/thf溶液(2.4m,1.3ml,3.15mmol)中滴加制造例4-(3)中所获得的化合物(1.36g)的thf(5ml)溶液。在室温下搅拌2小时后,滴加盐酸(1m,8ml)并搅拌1小时。利用二乙醚对有机相进行萃取后,依序利用饱和碳酸氢钠水溶液、饱和食盐水将其洗净,利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(乙酸乙酯/环己烷)对残渣进行精制,而获得标题化合物(0.28g,1.02mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=7.24hz,6h),1.13-1.41(m,28h),1.54-1.60(m,2h),3.59-3.68(m,2h)。
〔制造例5〕
4-壬基十三烷-1-醇的合成
〔化31〕
(1)(3-溴丙氧基)(叔丁基)二甲基硅烷的合成
〔化32〕
将3-溴-1-丙醇(6.5ml,71.9mmol)溶解在二氯甲烷(250ml)中,在室温下添加咪唑(7.84g,115.0mmol)并完全溶解。添加叔丁基二甲基氯硅烷(13.0g,86.3mmol),在室温下搅拌3天。将反应液过滤后在减压下进行浓缩。在残渣中添加二乙醚(300ml),依序利用饱和碳酸氢钠水溶液、饱和食盐水将其洗净,利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(环己烷)对残渣进行精制,而获得标题化合物(16.2g,64.0mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.08(s,6h),0.91(s,9h),2.05(quin,j=6.05hz,2h),3.53(t,j=6.42hz,2h),3.75(t,j=5.69hz,2h)。
(2){3-[(叔丁基二甲基硅烷基)氧基]丙基}三苯基溴化膦的合成
〔化33〕
按照制造例4-(1)的方法,由制造例5-(1)中所获得的化合物(16.2g,63.9mmol)、三苯基膦(16.8g,63.9mmol)及乙腈(125ml)获得标题化合物(21.2g,41.1mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.01(s,6h),0.84(s,9h),1.65-1.75(m,2h),3.48-3.57(m,2h),3.69(t,j=6.14hz,2h),7.73-7.84(m,12h),7.87-7.93(m,3h)。
(3)叔丁基二甲基[(4-壬基-3-十三碳烯-1-基)氧基]硅烷的合成
〔化34〕
将制造例5-(2)中所获得的化合物(4.38g,8.50mmol)溶解在thf(15ml)中,在氮气环境、室温下添加双(三甲基硅烷基)酰胺钠/thf溶液(1m,8.5ml,8.5mmol)。将反应液加热至45℃后搅拌30分钟。滴加十九烷-10-酮(2.0g,7.08mmol)的thf(10ml)溶液后加热回流21小时。将反应混合物冷却至室温后,添加二乙醚,依序利用水、饱和食盐水将其洗净,利用无水硫酸镁将有机相加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(环己烷)对残渣进行精制,而获得标题化合物(1.16g)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.07(s,6h),0.90(m,15h),1.21-1.41(m,28h),1.92-2.04(m,4h),2.24(q,j=7.34hz,2h),3.58(t,j=7.24hz,2h),5.09(t,j=7.15hz,1h)。
(4)4-壬基十三烷-1-醇的合成
〔化35〕
将制造例5-(3)中所获得的化合物(1.16g,2.64mmol)及10%钯-碳(0.28g,0.26mmol)悬浮在乙酸乙酯(10ml)及乙酸(0.15ml,2.64mmol)中,在氢气环境下在室温、常压下搅拌18小时。利用硅藻土将反应混合物加以过滤,并利用乙醇洗净后,在减压下将滤液浓缩。借由硅胶柱层析法(乙酸乙酯/环己烷)对残渣进行精制,而获得标题化合物(0.35g,1.08mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.80-0.90(m,6h),1.13-1.30(m,35h),1.32-1.39(m,2h),3.32-3.38(m,2h),4.30(t,j=5.23hz,1h)。
〔制造例6〕
4-庚基十一烷-1-醇的合成
〔化36〕
(1)叔丁基[(4-庚基-3-十一碳烯-1-基)氧基]二甲基硅烷的合成
〔化37〕
按照制造例5-(3)的方法,由{3-[(叔丁基二甲基硅烷基)氧基]丙基}三苯基溴化膦(3.0g,5.83mmol)、thf(10ml)、双(三甲基硅烷基)酰胺钠/thf溶液(1m,5.8ml,5.8mmol)、及十五烷-8-酮(1.1g,4.86mmol)获得标题化合物(0.83g,2.17mmol)。
1h-nmr(600hz,cdcl3)δ(ppm):0.07(s,6h),0.84-0.95(m,15h),0.89(s,1h),1.21-1.41(m,20h),1.92-2.05(m,4h),2.24(q,j=7.34hz,2h),3.58(t,j=7.24hz,2h),5.09(t,j=7.24hz,1h)。
(2)4-庚基十一烷-1-醇的合成
〔化38〕
按照制造例5-(4)的方法,由制造例6-(1)中所获得的化合物(0.83g,2.17mmol)、10%钯-碳(0.23g,0.22mmol)、乙酸乙酯(10ml)、及乙酸(0.25ml,4.35mmol)获得标题化合物(0.42g,1.56mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.82-0.89(m,6h),1.14-1.31(m,27h),1.33-1.39(m,2h),3.32-3.38(m,2h),4.30(t,j=5.14hz,1h)。
〔制造例7〕
4-戊基壬烷-1-醇的合成
〔化39〕
(1)叔丁基二甲基硅烷基[(4-戊基-3-壬烯-1-基)氧基]硅烷的合成
〔化40〕
按照制造例5-(3)的方法,由{3-[(叔丁基二甲基硅烷基)氧基]丙基}三苯基溴化膦(5.0g,9.72mmol)、thf(4ml)、双(三甲基硅烷基)酰胺钠/thf溶液(1m,9.7ml,9.7mmol)、及十一烷-6-酮(1.7ml,8.10mmol)获得标题化合物(1.16g)。
1h-nmr(600hz,cdcl3)δ(ppm):0.07(s,6h),0.90(m,15h),1.19-1.43(m,13h),1.92-2.05(m,4h),2.21-2.29(m,2h),3.58(t,j=7.24hz,2h),5.10(t,j=7.15hz,1h)。
(2)4-戊基壬烷-1-醇的合成
〔化41〕
按照制造例5-(4)的方法,由制造例7-(1)中所获得的化合物(1.12g,3.43mmol)、10%钯-碳(0.37g,0.34mmol)、乙酸乙酯(15ml)、及乙酸(0.39ml,6.86mmol)获得标题化合物(0.22g,1.02mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.87-0.93(m,6h),1.18-1.36(m,19h),1.37-1.45(m,2h),3.36-3.42(m,2h),4.35(t,j=5.23hz,1h)。
〔制造例8〕
4-溴丁酸苄酯的合成
〔化42〕
将苄醇(2.84ml,27.3mmol)、4-溴丁酸(5.01g,30.0mmol)及dmap(0.33g,2.73mmol)溶解在二氯甲烷(50ml)中,在冰浴冷却下添加edc(6.5g,34.1mmol),在室温下搅拌2.5小时。在减压下将反应混合物浓缩后,溶解在二乙醚中。依序利用饱和碳酸氢钠水溶液、水、10%柠檬酸水溶液进行洗净,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除,而获得标题化合物(6.41g,24.9mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):2.20(quin,j=6.83hz,2h),2.56(t,j=7.15hz,2h),3.46(t,j=6.51hz,2h),5.13(s,2h),7.28-7.42(m,5h)。
〔制造例9〕
6-溴己酸苄酯的合成
〔化43〕
按照制造例8的方法,由苄醇(1.93ml,18.5mmol)、6-溴己酸(4.33g,22.2mmol)、dmap(0.23g,1.85mmol)、edc(4.43g,23.1mmol)及二氯甲烷(35ml)获得标题化合物(5.43g,19.0mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.44-1.51(m,2h),1.64-1.72(m,2h),1.83-1.90(m,2h),2.38(t,j=7.43hz,2h),3.39(t,j=6.69hz,2h),5.12(s,2h),7.30-7.40(m,5h)。
〔制造例10〕
(1r,5s,6r)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸盐酸盐的合成
〔化44〕
(1)(1r,5s,6r)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸乙酯的合成
将(1r,5s,6r)-3-氮杂双环[3.1.0]己烷-6-羧酸乙酯(cas174456-77-0)(0.89g,5.74mmol)溶解在thf(20ml)中,在室温下依序添加乙酸(0.49ml,8.6mmol)、甲醛溶液(11.7ml,161.5mol)并搅拌30分钟。添加三(乙酰氧基)硼氢化钠(2.43g,11.5mmol),并在室温下搅拌3小时。在减压下将反应混合物浓缩后,添加饱和碳酸氢钠水溶液,利用二氯甲烷进行萃取。利用无水硫酸钠加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(二氯甲烷/甲醇)对残渣进行精制,而获得标题化合物(0.70g,4.14mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.25(t,j=7.2hz,3h),1.94(brs,2h),2.01(brs,1h),2.29(s,3h),2.35(d,j=9.2hz,2h),3.05(d,j=9.2hz,2h),4.09(q,j=7.2hz,2h)。
(2)(1r,5s,6r)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸盐酸盐的合成
在制造例10-(1)中所获得的化合物(0.60g,3.55mmol)中添加浓盐酸(10ml),并在70℃下搅拌12小时。在减压下将反应混合物浓缩,而获得标题化合物(0.52g,3.05mmol)。
1h-nmr(400mhz,dmso-d6)δ(ppm):2.14(brs,2h),2.25(brs,1h),2.74(brs,3h),3.25-3.37(m,2h),3.56-3.67(m,2h),10.91(brs,1h),12.49(brs,1h)。
〔制造例11〕
(1r,5s,6s)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸盐酸盐的合成
〔化45〕
(1)(1r,5s,6s)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸乙酯的合成
按照制造例10-(1)的方法,由(1r,5s,6s)-3-氮杂双环[3.1.0]己烷-6-羧酸乙酯(cas1144099-54-6)(2.5g,16.1mmol)、乙酸(1.4ml,24.2mmol)、甲醛溶液(10.0ml,134.3mmol)、三(乙酰氧基)硼氢化钠(6.8g,32.2mmol)、及thf(50ml)获得标题化合物(2.5g,14.8mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.23(t,j=7.1hz,3h),1.48-1.68(m,3h),2.22(s,3h),2.33(brd,j=8.8hz,2h),3.02(d,j=8.8hz,2h),4.10(q,j=7.1hz,2h)。
(2)(1r,5s,6s)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸盐酸盐的合成
按照制造例10-(2)的方法,由制造例10-(2)中所获得的化合物(500mg,2.96mmol)与浓盐酸(10ml)获得标题化合物(350mg,1.97mmol)。
1h-nmr(400mhz,dmso-d6)δ(ppm):1.88-1.94(m,0.45h),1.94-2.00(m,0.55h),2.35-2.45(m,2h),2.68(brs,1.65h),2.67(brs,1.35h),3.08-3.18(m,1.1h),3.45-3.57(m,0.9h),3.73-3.88(m,2h),9.16(brs,1h),11.52(brs,1h)。
<阳离子脂质的合成(1)>
〔实例a-1〕
1-甲基哌啶-4-羧酸2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}十二烷基酯(阳离子脂质1)
〔化46〕
(1)壬烷-1,1,9-三羧酸1,1-二叔丁酯9-(2-丁基辛酯)的合成
使60%氢化钠(59mg,1.48mmol)悬浮在1,4-二噁烷(6.5ml)中,在室温下缓慢添加丙二酸二叔丁酯(0.30ml,1.35mmol),并搅拌20分钟。使用1,4-二噁烷(1ml)添加制造例1中所获得的化合物(573mg,1.41mmol),在70℃下搅拌3小时,并在95℃下搅拌15小时。将反应混合物在冰水浴中加以冷却,添加饱和氯化铵水溶液,并利用正庚烷进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(490mg,0.91mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-0.98(m,6h),1.18-1.38(m,24h),1.39-1.54(m,2h),1.46(s,18h),1.55-1.69(m,3h),1.71-1.87(m,2h),2.24-2.35(m,2h),3.06-3.15(m,1h),3.91-4.02(m,2h)。
(2)十九烷-1,9,9-三羧酸9,9-二叔丁酯1-(2-丁基辛酯)的合成
将实例a-1-(1)中所获得的化合物(490mg,0.91mmol)溶解在1,4-二噁烷(4ml)中,在水浴冷却下添加60%氢化钠(47mg,1.18mmol),并在室温下搅拌5分钟。添加1-碘癸烷(0.39ml,1.81mmol),在80℃下搅拌15小时。将反应混合物在冰水浴中加以冷却,添加饱和氯化铵水溶液,并利用正庚烷进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(450mg,0.66mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-0.96(m,9h),1.05-1.36(m,40h),1.39-1.49(m,2h),1.44(s,18h),1.53-1.68(m,3h),1.70-1.82(m,4h),2.23-2.34(m,2h),3.92-4.01(m,2h)。
(3)2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}十二烷酸的合成
将实例a-1-(2)中所获得的化合物(450mg,0.66mmol)溶解在二氯甲烷(2ml)中,在冰浴冷却下添加tfa(1ml),并在室温下搅拌1.5小时。在反应混合物中添加甲苯,在减压下将溶剂蒸馏去除。将甲苯的添加及蒸馏去除重复进行2次,借此进行干燥而获得2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}-2-癸基丙二酸的粗产物。将所获得的粗产物溶解在二甲苯(5ml)中,在150℃下搅拌8小时。将反应混合物恢复至室温,并在减压下进行浓缩。借由柱层析法(正庚烷/乙酸乙酯)对所获得的残渣进行精制,而获得标题化合物(240mg,0.46mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-0.97(m,9h),1.16-1.70(m,49h),2.22-2.41(m,3h),3.92-4.02(m,2h)。
(4)10-(羟基甲基)二十烷酸2-丁基辛酯的合成
将实例a-1-(3)中所获得的化合物(240mg,0.46mmol)溶解在thf(4ml)中,在-15℃下滴加0.92m硼烷-thf络合物(0.75ml,0.69mmol),并在0℃下搅拌2小时。添加饱和碳酸氢钠水溶液,在室温下搅拌5分钟,利用乙酸乙酯进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(197mg,0.39mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-0.98(m,9h),1.10-1.39(m,46h),1.39-1.50(m,1h),1.54-1.69(m,4h),2.23-2.36(m,2h),3.48-3.59(m,2h),3.92-4.02(m,2h)。
(5)1-甲基哌啶-4-羧酸2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}十二烷基酯的合成
将实例a-1-(4)中所获得的化合物(38mg,0.074mmol)、dipea(0.054ml,0.31mmol)、1-甲基-哌啶-4-羧酸盐酸盐(27mg,0.15mmol)及dmap(1.8mg,0.015mmol)溶解在二氯甲烷(0.8ml)中,在冰浴冷却下添加edc(31mg,0.16mmol),并在室温下搅拌15小时。在反应混合物中添加饱和碳酸氢钠水溶液,利用氯仿进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯/甲醇)对残渣进行精制,而获得标题化合物(38mg,0.060mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.77-0.97(m,9h),1.13-1.41(m,46h),1.50-1.69(m,4h),1.69-1.85(m,2h),1.85-2.09(m,4h),2.18-2.35(m,3h),2.27(s,3h),2.72-2.88(m,2h),3.90-4.03(m,4h)。
<阳离子脂质的合成(2)>
〔实例a-2〕
1-甲基哌啶-4-羧酸2-{9-侧氧基-9-[(3-戊基辛基)氧基]壬基}十二烷基酯(阳离子脂质2)
〔化47〕
(1)壬烷-1,1,9-三羧酸9-苄酯1,1-二叔丁酯的合成
使60%氢化钠(83mg,2.07mmol)悬浮在1,4-二噁烷(9.4ml)中,在室温下缓慢添加丙二酸二叔丁酯(0.42ml,1.88mmol),并搅拌10分钟。添加制造例2中所获得的化合物(650mg,1.99mmol),在95℃下搅拌13小时。将反应混合物在冰水浴中加以冷却,添加饱和氯化铵水溶液,并利用正庚烷进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(536mg,1.16mmol)。
(2)十九烷-1,9,9-三羧酸1-苄酯9,9-二叔丁酯的合成
按照实例a-1-(2)的方法,由实例a-2-(1)中所获得的化合物(590mg,1.28mmol)、1-碘癸烷(0.54ml,2.55mmol)、65%氢化钠(71mg,1.91mmol)及1,4-二噁烷(5ml)获得标题化合物(470mg,0.78mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.83-0.93(m,3h),1.05-1.36(m,26h),1.44(s,18h),1.57-1.68(m,2h),1.71-1.82(m,4h),2.29-2.39(m,2h),5.11(s,2h),7.28-7.42(m,5h)。
(3)2-[9-(苄氧基)-9-侧氧基壬基]十二烷酸的合成
按照实例a-1-(3)的方法,由实例a-2-(2)中所获得的化合物(470mg,0.78mmol)、二氯甲烷(2ml)、tfa(1ml)、及二甲苯(2ml)获得标题化合物(286mg,0.64mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.95(m,3h),1.18-1.37(m,26h),1.39-1.52(m,2h),1.54-1.69(m,4h),2.28-2.41(m,3h),5.11(s,2h),7.28-7.40(m,5h)。
(4)10-(羟基甲基)二十烷酸苄酯的合成
按照实例a-1-(4)的方法,由实例a-2-(3)中所获得的化合物(285mg,0.64mmol)、0.92m硼烷-thf络合物(1.0ml,0.96mmol)、及thf(3.2ml)获得标题化合物(223mg,0.52mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.95(m,3h),1.12-1.38(m,31h),1.39-1.50(m,1h),1.58-1.72(m,2h),2.23-2.36(m,2h),3.48-3.59(m,2h),5.11(s,2h),7.28-7.41(m,5h)。
(5)1-甲基哌啶-4-羧酸2-[9-(苄氧基)-9-侧氧基壬基]十二烷基酯的合成
按照实例a-1-(5)的方法,由实例a-2-(4)中所获得的化合物(114mg,0.26mmol)、1-甲基-哌啶-4-羧酸盐酸盐(95mg,0.53mmol)、edc(111mg,0.58mmol)、dipea(0.090ml,0.53mmol)、dmap(6.4mg,0.053mmol)及二氯甲烷(1.3ml)获得标题化合物(128mg,0.23mmol)。
1h-nmr(400mhz,cdgl3)δ(ppm):0.81-0.96(m,3h),1.15-1.41(m,31h),1.52-1.70(m,2h),1.70-1.84(m,2h),1.85-2.06(m,4h),2.18-2.40(m,3h),2.27(s,3h),2.73-2.89(m,2h),3.92-4.03(m,2h),5.12(s,2h),7.28-7.44(m,5h)。
(6)10-{[(1-甲基哌啶-4-羰基)氧基]甲基}二十烷酸的合成
将实例a-2-(5)中所获得的化合物(127mg,0.23mmol)溶解在乙酸乙酯(2ml)中,在室温下添加10%钯-碳(24mg,50%含水),在氢气环境、常压下搅拌3小时。将反应液过滤,利用乙酸乙酯将其洗净。在减压下将滤液浓缩,借由柱层析法(氯仿/甲醇)对所获得的残渣进行精制,而获得标题化合物(94mg,0.20mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.95(m,3h),1.12-1.44(m,31h),1.53-1.71(m,2h),1.74-1.95(m,2h),1.97-2.10(m,2h),2.11-2.37(m,5h),2.40(s,3h),3.11-3.28(m,2h),3.79-3.92(m,1h),4.14-4.25(m,1h)。
(7)1-甲基哌啶-4-羧酸2-{9-侧氧基-9-[(3-戊基辛基)氧基]壬基}十二烷基酯的合成
按照制造例2的方法,由实例a-2-(6)中所获得的化合物(93mg,0.20mmol)、3-戊基辛烷-1-醇(cas1443519-63-8)(60mg,0.30mmol)、edc(42mg,0.22mmol)、dmap(4.9mg,0.040mmol)及二氯甲烷(1.5ml)获得标题化合物(103mg,0.16mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.82-0.95(m,9h),1.14-1.46(m,46h),1.50-1.68(m,6h),1.70-1.84(m,2h),1.85-2.06(m,4h),2.19-2.33(m,3h),2.27(s,3h),2.74-2.87(m,2h),3.93-4.02(m,2h),4.03-4.14(m,2h)。
<阳离子脂质的合成(3)>
〔实例a-3〕
1-甲基哌啶-4-羧酸2-壬基-11-侧氧基-11-[(3-戊基辛基)氧基]十一烷基酯(阳离子脂质3)
〔化48〕
(1)十八烷-1,9,9-三羧酸1-苄酯9,9-二叔丁基酯的合成
将实例a-2-(1)中所获得的化合物(536mg,1.16mmol)溶解在thf(6ml)中,在水浴冷却下添加60%氢化钠(58mg,1.45mmol),并在室温下搅拌5分钟。添加1-碘壬烷(0.23ml,1.16mmol),在80℃下搅拌20小时。将反应混合物在冰水浴中加以冷却,添加饱和氯化铵水溶液,并利用正庚烷进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(340mg,0.577mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.82-0.92(m,3h),1.03-1.36(m,24h),1.44(s,18h),1.57-1.68(m,2h),1.71-1.81(m,4h),2.28-2.40(m,2h),5.11(s,2h),7.28-7.41(m,5h)。
(2)11-(苄氧基)-2-壬基-11-侧氧基十一烷酸的合成
按照实例a-1-(3)的方法,由实例a-3-(1)中所获得的化合物(340mg,0.58mmol)、二氯甲烷(2ml)、tfa(1ml)、及二甲苯(2ml)获得标题化合物(96mg,0.22mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-0.95(m,3h),1.12-1.74(m,30h),2.28-2.41(m,3h),5.11(s,2h),7.28-7.40(m,5h)。
(3)10-(羟基甲基)十九烷酸苄酯的合成
按照实例a-1-(4)的方法,由实例a-3-(2)中所获得的化合物(96mg,0.22mmol)、0.92m硼烷-thf络合物(0.36ml,0.33mmol)及thf(1.1ml)获得标题化合物(80mg,0.19mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.94(m,3h),1.09-1.38(m,29h),1.38-1.50(m,1h),1.58-1.71(m,2h),2.30-2.40(m,2h),3.48-3.58(m,2h),5.11(s,2h),7.29-7.42(m,5h)。
(4)1-甲基哌啶-4-羧酸11-(苄氧基)-2-壬基-11-侧氧基十一烷基酯的合成
按照实例a-1-(5)的方法,由实例a-3-(3)中所获得的化合物(80mg,0.19mmol)、1-甲基-哌啶-4-羧酸盐酸盐(69mg,0.38mmol)、edc(81mg,0.42mmol)、dipea(0.066ml,0.38mmol)、dmap(4.7mg,0.038mmol)及二氯甲烷(1.0ml)获得标题化合物(97mg,0.18mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.83-0.94(m,3h),1.15-1.39(m,29h),1.52-1.70(m,2h),1.70-1.85(m,2h),1.85-2.07(m,4h),2.19-2.40(m,3h),2.27(s,3h),2.73-2.88(m,2h),3.94-4.02(m,2h),5.12(s,2h),7.28-7.43(m,5h)。
(5)10-{[(1-甲基哌啶-4-羰基)氧基]甲基}十九烷酸的合成
按照实例a-2-(6)的方法,将实例a-3-(4)中所获得的化合物(96mg,0.18mmol)溶解在乙酸乙酯(2ml)中,在室温下添加10%钯-碳(19mg,50%含水),并在氢气环境、常压下搅拌3小时。将反应液过滤,利用乙酸乙酯将其洗净。在减压下将滤液浓缩,借由柱层析法(氯仿/甲醇)对所获得的残渣进行精制,而获得标题化合物(74mg,0.163mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.95(m,3h),1.12-1.44(m,29h),1.53-1.71(m,2h),1.74-1.94(m,2h),1.98-2.08(m,2h),2.08-2.38(m,5h),2.39(s,3h),3.10-3.30(m,2h),3.80-3.91(m,1h),4.14-4.25(m,1h)。
(6)1-甲基哌啶-4-羧酸2-壬基-11-侧氧基-11-[(3-戊基辛基)氧基]十一烷基酯的合成
按照制造例2的方法,由实例a-3-(5)中所获得的化合物(74mg,0.16mmol)、3-戊基辛烷-1-醇(cas1443519-63-8)(49mg,0.25mmol)、edc(38mg,0.20mmol)、dmap(4.0mg,0.033mmol)及二氯甲烷(1.2ml)获得标题化合物(81mg,0.13mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.95(m,9h),1.15-1.47(m,44h),1.50-1.68(m,6h),1.70-1.84(m,2h),1.84-2.06(m,4h),2.18-2.33(m,3h),2.27(s,3h),2.73-2.87(m,2h),3.94-4.01(m,2h),4.04-4.12(m,2h)。
<阳离子脂质的合成(4)>
〔实例a-4〕
9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸双(3-戊基辛基)酯(阳离子脂质4)
〔化49〕
(1)辛烷-1,1,8-三羧酸8-苄酯1,1-二叔丁酯的合成
使60%氢化钠(59mg,1.48mmol)悬浮在dmf(5.4ml)中,在冰浴冷却下缓慢添加丙二酸二叔丁酯(0.30ml,1.35mmol),并在0℃下搅拌5分钟后,撤去冰水浴,并搅拌20分钟。在冰浴冷却下添加碘化钠(61mg,0.40mmol)、制造例3中所获得的化合物(443mg,1.41mmol),并在室温下搅拌15小时。将反应混合物在冰水浴中加以冷却,添加水,利用二乙醚进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(456mg,1.02mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.23-1.36(m,8h),1.45(s,18h),1.58-1.69(m,2h),1.72-1.84(m,2h),2.30-2.40(m,2h),3.05-3.15(m,1h),5.11(s,2h),7.28-7.41(m,5h)。
(2)十五烷-1,8,8,15-四羧酸1,15-二苄酯8,8-二叔丁酯的合成
按照实例a-4-(1)的方法,由实例a-4-(1)中所获得的化合物(456mg,1.02mmol)、制造例3中所获得的化合物(478mg,1.53mmol)、碘化钠(46mg,0.31mmol)、60%氢化钠(49mg,1.22mmol)及dmf(3.3ml)获得标题化合物(380mg,0.56mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.03-1.17(m,4h),1.23-1.36(m,12h),1.43(s,18h),1.56-1.68(m,4h),1.70-1.80(m,4h),2.29-2.38(m,4h),5.11(s,4h),7.27-7.41(m,10h)。
(3)10-(苄氧基)-2-[8-(苄氧基)-8-侧氧基辛基]-10-侧氧基癸酸的合成
按照实例a-1-(3)的方法,由实例a-4-(2)中所获得的化合物(380mg,0.56mmol)、二氯甲烷(2ml)、tfa(1ml)、及二甲苯(2ml)获得标题化合物(234mg,0.45mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.18-1.37(m,16h),1.38-1.71(m,8h),2.26-2.41(m,5h),5.11(s,4h),7.28-7.43(m,10h)。
(4)9-(羟基甲基)十七烷二酸二苄酯的合成
按照实例a-1-(4)的方法,由实例a-4-(3)中所获得的化合物(234mg,0.45mmol)、0.92m硼烷-thf络合物(0.73ml,0.67mmol)及thf(1.8ml)获得标题化合物(188mg,0.37mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.12-1.51(m,22h),1.56-1.72(m,4h),2.29-2.41(m,4h),3.47-3.58(m,2h),5.11(s,4h),7.28-7.44(m,10h)。
(5)9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸二苄酯的合成
按照实例a-1-(5)的方法,由实例a-4-(4)中所获得的化合物(188mg,0.37mmol)、1-甲基-哌啶-4-羧酸盐酸盐(132mg,0.74mmol)、edc(155mg,0.81mmol)、dipea(0.126ml,0.74mmol)、dmap(9.0mg,0.074mmol)及二氯甲烷(1.9ml)获得标题化合物(217mg,0.34mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):1.16-1.38(m,21h),1.53-1.69(m,4h),1.70-1.84(m,2h),1.85-2.04(m,4h),2.19-2.40(m,5h),2.26(s,3h),2.74-2.86(m,2h),3.92-4.01(m,2h),5.11(s,4h),7.28-7.41(m,10h)。
(6)9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸双(3-戊基辛基)酯的合成
将实例a-4-(5)中所获得的化合物(217mg,0.34mmol)溶解在thf(4ml)及甲醇(2ml)中,在室温下添加10%钯-碳(19mg,50%含水),并在氢气环境、常压下搅拌2小时。将反应是以氮气加以置换后,过滤反应液,并利用甲醇将其洗净。在减压下将滤液浓缩,而获得9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸的粗产物(160mg)。
按照制造例2的方法,由所获得的粗产物(40mg)、3-戊基辛烷-1-醇(cas1443519-63-8)(44mg,0.22mmol)、edc(37mg,0.19mmol)、dmap(2.1mg,0.018mmol)及thf(1ml)获得标题化合物(34mg,0.041mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.83-0.94(m,12h),1.14-1.48(m,53h),1.51-1.67(m,10h),1.70-1.84(m,2h),1.85-2.07(m,4h),2.18-2.34(m,5h),2.27(s,3h),2.74-2.88(m,2h),3.93-4.01(m,2h),4.03-4.12(m,4h)。
<阳离子脂质的合成(5)>
〔实例a-5〕
9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸二[(z)-2-壬烯-1-基]酯(阳离子脂质5)
〔化50〕
按照制造例2的方法,由实例a-4-(6)中所获得的9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸的粗产物(60mg)、顺-2-壬烯-1-醇(0.066ml,0.40mmol)、edc(56mg,0.29mmol)、dmap(3.2mg,0.026mmol)及thf(1.3ml)获得标题化合物(76mg,0.11mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.96(m,6h),1.15-1.44(m,35h),1.52-1.68(m,6h),1.70-1.84(m,2h),1.85-2.05(m,4h),2.05-2.15(m,4h),2.21-2.35(m,5h),2.27(s,3h),2.74-2.88(m,2h),3.93-4.01(m,2h),4.57-4.67(m,4h),5.47-5.58(m,2h),5.59-5.70(m,2h)。
<阳离子脂质的合成(6)>
〔实例a-6〕
(1r,5s,6r)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}十二烷基酯(阳离子脂质6)
〔化51〕
按照实例a-1-(5)的方法,由实例a-1-(4)中所获得的化合物(40mg,0.078mmol)、制造例10-(2)中所获得的化合物(28mg,0.16mmol)、edc(33mg,0.17mmol)、dipea(0.027ml,0.16mmol)、dmap(1.9mg,0.016mmol)及二氯甲烷(1.0ml)获得标题化合物(40mg,0.063mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-1.00(m,9h),1.16-1.40(m,46h),1.52-1.70(m,4h),1.89-1.97(m,2h),1.98-2.06(m,1h),2.23-2.42(m,4h),2.30(s,3h),3.00-3.12(m,2h),3.87-4.03(m,4h)。
<阳离子脂质的合成(7)>
〔实例a-7〕
(1r,5s,6s)-3-甲基-3-氮杂双环[3.1.0]己烷-6-羧酸2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}十二烷基酯(阳离子脂质7)
〔化52〕
按照实例a-1-(5)的方法,由实例a-1-(4)中所获得的化合物(40mg,0.078mmol)、制造例11-(2)中所获得的化合物(27.8mg,0.16mmol)、edc(33mg,0.17mmol)、dipea(0.027ml,0.157mmol)、dmap(1.9mg,0.016mmol)及二氯甲烷(1.0ml)获得标题化合物(38mg,0.060mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.81-0.97(m,9h),1.15-1.40(m,46h),1.46-1.70(m,7h),2.21(s,3h),2.24-2.38(m,4h),2.96-3.07(m,2h),3.85-4.02(m,4h)。
<阳离子脂质的合成(8)>
〔实例a-8〕
4-甲基哌嗪-1-羧酸2-{9-[(2-丁基辛基)氧基]-9-侧氧基壬基}十二烷基酯(阳离子脂质8)
〔化53〕
将实例a-1-(4)中所获得的化合物(11mg,0.022mmol)与吡啶(0.0043ml,0.054mmol)溶解在二氯甲烷(0.8ml)中,在冰浴冷却下添加氯甲酸4-硝基苯酯(12mg,0.060mmol),并在相同温度下搅拌10分钟,在室温下搅拌1.5小时。在反应液中添加1-甲基哌嗪(0.010ml,0.090mmol),并在室温下搅拌3小时。在反应混合物中添加饱和碳酸氢钠水溶液,利用乙酸乙酯进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(11mg,0.017mmol)。
1h-nmr(400mhz,cdcl3)δ(ppm):0.80-0.98(m,9h),1.16-1.42(m,46h),1.52-1.77(m,4h),2.23-2.44(m,6h),2.30(s,3h),3.42-3.57(m,4h),3.90-4.04(m,4h)。
<阳离子脂质的合成(9)>
〔实例a-9〕
1-甲基哌啶-4-羧酸2-[9-(己基氧基)-9-侧氧基壬基]十二烷基酯(阳离子脂质9)
〔化54〕
按照制造例2的方法,由实例a-2-(6)中所获得的化合物(100mg,0.21mmol)、己烷-1-醇(32μl,0.26mmol)、edc(49mg,0.26mmol)、dmap(5.0mg,0.050mmol)及二氯甲烷(5.0ml)获得标题化合物(102mg,0.19mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.85-0.92(m,6h),1.19-1.39(m,37h),1.57-1.66(m,5h),1.73-1.84(m,2h),1.87-1.94(m,2h),1.96-2.07(m,2h),2.22-2.32(m,6h),2.73-2.87(m,2h),3.98(d,j=5.50hz,2h),4.06(t,j=6.79hz,2h)。
<阳离子脂质的合成(10)>
〔实例a-10〕
1-甲基哌啶-4-羧酸2-[9-(辛基氧基)-9-侧氧基壬基]十二烷基酯(阳离子脂质10)
〔化55〕
按照制造例2的方法,由实例a-2-(6)中所获得的化合物(200mg,0.42mmol)、辛烷-1-醇(67mg,0.51mmol)、edc(98mg,0.51mmol)、dmap(10mg,0.09mmol)及二氯甲烷(10ml)获得标题化合物(109mg,0.19mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.91(t,j=6.97hz,6h),1.23-1.40(m,40h),1.60-1.68(m,5h),1.76-1.85(m,2h),1.89-1.95(m,2h),1.97-2.06(m,2h),2.25-2.33(m,6h),2.78-2.86(m,2h),4.00(d,j=5.69hz,2h),4.08(t,j=6.79hz,2h)。
<阳离子脂质的合成(11)>
〔实例a-11〕
1-甲基哌啶-4-羧酸2-[9-(癸基氧基)-9-侧氧基壬基]十二烷基酯(阳离子脂质11)
〔化56〕
按照制造例2的方法,由实例a-2-(6)中所获得的化合物(200mg,0.42mmol)、癸烷-1-醇(100μl,0.51mmol)、edc(98mg,0.51mmol)、dmap(10mg,0.09mmol)及二氯甲烷(10ml)获得标题化合物(153mg,0.25mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.91(t,j=7.06hz,6h),1.17-1.42(m,44h),1.60-1.68(m,5h),1.75-1.86(m,2h),1.88-1.96(m,2h),1.97-2.07(m,2h),2.24-2.35(m,6h),2.78-2.87(m,2h),4.00(d,j=5.50hz,2h),4.08(t,j=6.79hz,2h)。
<阳离子脂质的合成(12)>
〔实例a-12〕
1-甲基哌啶-4-羧酸2-{9-侧氧基-9-[(4-戊基壬基)氧基]壬基}十二烷基酯(阳离子脂质12)
〔化57〕
按照制造例2的方法,由实例a-2-(6)中所获得的化合物(200mg,0.42mmol)、制造例7-(2)中所获得的化合物(115mg,0.60mmol)、edc(98mg,0.51mmol)、dmap(10mg,0.09mmol)及二氯甲烷(10ml)获得标题化合物(212mg,0.32mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.85-0.92(m,9h),1.16-1.34(m,49h),1.51-1.66(m,5h),1.73-1.84(m,2h),1.86-1.94(m,2h),1.95-2.05(m,2h),2.23-2.31(m,6h),2.75-2.85(m,2h),3.98(d,j=5.50hz,2h),4.04(t,j=6.79hz,2h)。
<阳离子脂质的合成(13)>
〔实例a-13〕
1-甲基哌啶-4-羧酸2-(4-侧氧基-4-(十三烷基氧基)丁基)十二烷基酯(阳离子脂质13)
〔化58〕
(1)丁烷-1,1,4-三羧酸4-苄酯1,1-二叔丁酯的合成
〔化59〕
使60%氢化钠(1.05g,26.2mmol)悬浮在thf(50ml)中,在冰浴冷却下滴加丙二酸二叔丁酯(5.86ml,26.2mmol),并在室温下搅拌20分钟。在冰浴冷却下添加碘化钠(0.37g,2.49mmol)后,滴加制造例8中所获得的化合物(6.41g,24.9mmol),在室温下搅拌18小时。利用二乙醚将反应混合物进行稀释,并利用水洗净后,利用二乙醚对水层进行萃取。合并有机相,利用饱和食盐水将其洗净,利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除,而获得标题化合物(9.88g,25.2mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.42-1.49(m,18h),1.65-1.72(m,2h),1.80-1.87(m,2h),2.39(t,j=7.52hz,2h),3.13(t,j=7.52hz,1h),5.11(s,2h),7.29-7.40(m,5h)。
(2)十四烷-1,4,4-三羧酸1-苄酯4,4-二叔丁酯的合成
〔化60〕
将实例a-13-(1)中所获得的化合物(9.88g,25.2mmol)溶解在thf(100ml)中,在水浴冷却下添加60%氢化钠(1.51g,37.8mmol),并在室温下搅拌30分钟。缓慢添加1-碘癸烷(10.7ml,50.4mmol)后,在室温下搅拌4小时。在反应混合物中添加饱和氯化铵水溶液,借由正戊烷进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(正庚烷/二乙醚)对残渣进行精制,而获得标题化合物(4.31g,8.09mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.85-0.91(m,3h),1.07-1.17(m,2h),1.21-1.33(m,16h),1.44(s,18h),1.49-1.54(m,2h),1.74-1.84(m,4h),2.36(t,j=7.34hz,2h),5.11(s,2h),7.28-7.38(m,5h)。
(3)2-[4-(苄基氧基)-4-侧氧基丁基]十二烷酸的合成
〔化61〕
将实例a-13-(2)中所获得的化合物(4.31g,8.09mmol)溶解在二氯甲烷(20ml)中,在冰浴冷却下滴加tfa(10ml),并在室温下搅拌4小时。在减压下将反应混合物浓缩后,添加甲苯并共沸2次。将所获得的粗产物溶解在二甲苯(25ml)中,加热回流8小时。将反应混合物恢复至室温,并在减压下进行浓缩。借由柱层析法(环己烷/乙酸乙酯)对所获得的残渣进行精制,而获得标题化合物(2.74g,7.28mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.82-0.89(m,3h),1.17-1.31(m,16h),1.32-1.42(m,3h),1.43-1.57(m,4h),2.16-2.22(m,1h),2.32-2.38(m,2h),5.08(s,2h),7.29-7.40(m,5h),12.06(brs,1h)。
(4)5-(羟基甲基)十五烷酸苄酯的合成
〔化62〕
将实例a-13-(3)中所获得的化合物(2.74g,7.28mmol)溶解在thf(30ml)中,在-78℃下滴加硼烷-thf络合物(1m,16.9ml,16.9mmol)。在0℃下搅拌8小时后,在15℃下搅拌22小时。添加饱和碳酸氢钠水溶液,利用二乙醚进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(环己烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(2.30g,6.34mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.85(t,j=6.88hz,3h),1.08-1.37(m,18h),1.49-1.58(m,2h),2.27-2.36(m,2h),3.21-3.29(m,2h),4.28(t,j=5.14hz,1h),5.01-5.12(m,2h),7.30-7.40(m,5h)。
(5)1-甲基哌啶-4-羧酸2-(4-(苄基氧基)-4-侧氧基丁基)十二烷基酯的合成
〔化63〕
将实例a-13-(4)中所获得的化合物(2.30g,6.34mmol)、dipea(2.2ml,12.7mmol)、1-甲基-哌啶-4-羧酸盐酸盐(2.28g,12.7mmol)及dmap(0.16g,1.27mmol)溶解在二氯甲烷(30ml)中,在冰浴冷却下添加edc(2.68g,13.9mmol),在室温下搅拌4小时。在减压下将反应混合物浓缩后,溶解在二乙醚中。依序利用饱和碳酸氢钠水溶液与水进行洗净,利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除,而获得标题化合物(2.97g,6.09mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=7.06hz,3h),1.17-1.38(m,21h),1.60-1.70(m,3h),1.77-1.87(m,2h),1.89-1.99(m,2h),2.23-2.38(m,6h),2.74-2.91(m,2h),3.94-4.02(m,2h),5.09-5.13(m,2h),7.30-7.41(m,5h)。
(6)5-{[(1-甲基哌啶-4-羰基)氧基]甲基}十五烷酸的合成
〔化64〕
将实例a-13-(5)中所获得的化合物(2.97g,6.09mmol)溶解在乙酸乙酯(35ml)中,在室温下添加10%钯-碳(0.65g,0.61mmol,50%含水),在氢气环境、常压下搅拌3小时。将反应液过滤,利用乙酸乙酯将其洗净。在减压下将滤液浓缩,而获得标题化合物(2.31g,5.81mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):0.85(t,j=6.88hz,3h),1.21-1.32(m,20h),1.44-1.66(m,5h),1.75-1.83(m,2h),1.95-1.98(m,1h),1.99-2.06(m,1h),2.13-2.23(m,5h),2.23-2.30(m,1h),2.64-2.77(m,2h),3.88-3.97(m,2h)。
(7)1-甲基哌啶-4-羧酸2-(4-侧氧基-4-(十三烷基氧基)丁基)十二烷基酯的合成
〔化65〕
将实例a-13-(6)中所获得的化合物(200mg,0.50mmol)、十三烷-1-醇(121mg,0.60mmol)及dmap(12mg,0.10mmol)溶解在二氯甲烷(5ml)中,在室温下添加edc(116mg,0.60mmol),并在室温下搅拌4小时。借由硅胶柱层析法(甲醇/乙酸乙酯)对反应混合物进行精制,而获得标题化合物(170mg,0.29mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=6.7hz,6h),1.26(brs,41h),1.57-1.69(m,6h),1.73-1.82(m,2h),1.90(brdd,j=13.5,3.03hz,2h),1.94-2.04(m,2h),2.22-2.34(m,6h),2.67-2.90(brd,j=9.8hz,2h),3.95-4.02(m,2h),4.03-4.08(t,j=6.8hz,2h)。
<阳离子脂质的合成(14)>
〔实例a-14〕
1-甲基哌啶-4-羧酸2-(4-侧氧基-4-((8-戊基十三烷基)氧基)丁基)十二烷基酯(阳离子脂质14)
〔化66〕
按照实例a-13-(7)的方法,由实例a-13-(6)中所获得的化合物(120mg,0.30mmol)、制造例4-(4)中所获得的化合物(90mg,0.33mmol)、edc(64mg,0.33mmol)、dmap(4.0mg,0.033mmol)及二氯甲烷(1.5ml)获得标题化合物(133mg,0.21mmol)。
1h-nmr(600mhz,cd3od)δ(ppm):0.87-0.97(m,9h),1.18-1.45(m,47h),1.60-1.73(m,5h),1.73-1.83(m,2h),1.88-1.99(m,2h),2.06-2.19(m,2h),2.23-2.43(m,6h),2.77-2.89(m,2h),3.99-4.15(m,4h)。
<阳离子脂质的合成(15)>
〔实例a-15〕
1-甲基哌啶-4-羧酸2-{4-[(4-壬基十三烷基)氧基]-4-侧氧基丁基}十二烷基酯(阳离子脂质15)
〔化67〕
按照实例a-13-(7)的方法,由实例a-13-(6)中所获得的化合物(110mg,0.28mmol)、制造例5-(4)中所获得的化合物(99mg,0.30mmol)、edc(58mg,0.30mmol)、dmap(4.0mg,0.033mmol)及二氯甲烷(1.3ml)获得标题化合物(106mg,0.15mmol)。
1h-nmr(600mhz,cd3od)δ(ppm):0.85-0.99(m,9h),1.15-1.44(m,55h),1.56-1.84(m,7h),1.94(m,j=13.2hz,2h),2.06-2.19(m,2h),2.23-2.44(m,6h),2.76-2.90(m,2h),3.96-4.14(m,4h)。
<阳离子脂质的合成(16)>
〔实例a-16〕
9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸二辛酯(阳离子脂质16)
〔化68〕
(1)9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸的合成
〔化69〕
将实例a-4-(5)中所获得的化合物(0.80g,1.25mmol)溶解在乙醇(5ml)中,在室温下添加10%钯-碳(0.13g,0.13mmol,50%含水),并在氢气环境、常压下搅拌18小时。利用硅藻土将反应液加以过滤,并利用乙醇将其洗净。在减压下将滤液浓缩,而获得标题化合物(0.58g,1.26mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.17-1.30(m,20h),1.43-1.52(m,4h),1.52-1.62(m,3h),1.77-1.82(m,2h),1.92-2.03(m,2h),2.12-2.22(m,7h),2.23-2.33(m,1h),2.66-2.75(m,2h),3.93(d,j=5.50hz,2h)。
(2)9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸二辛酯的合成
〔化70〕
将实例a-16-(1)中所获得的化合物(100mg,0.22mmol)、辛烷-1-醇(83μl,0.53mmol)及dmap(11mg,0.09mmol)溶解在二氯甲烷(5ml)中,在室温下添加edc(101mg,0.53mmol),并在室温下搅拌18小时。在减压下将反应混合物浓缩后,借由硅胶柱层析法(甲醇/乙酸乙酯)进行精制,而获得标题化合物(101mg,0.15mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.83-0.92(m,6h),1.21-1.39(m,40h),1.55-1.66(m,9h),1.72-1.83(m,2h),1.85-1.95(m,2h),1.95-2.06(m,2h),2.22-2.33(m,8h),2.75-2.87(m,2h),3.97(d,j=5.69hz,2h),4.05(t,j=6.69hz,4h)。
<阳离子脂质的合成(17)>
〔实例a-17〕
9-{[(1-甲基哌啶-4-羰基)氧基]甲基}十七烷二酸双(4-戊基壬基)酯(阳离子脂质17)
〔化71〕
按照实例a-16-(2)的方法,由实例a-16-(1)中所获得的化合物(0.20g,0.44mmol)、制造例7-(2)中所获得的化合物(0.23g,1.05mmol)、edc(0.20g,1.05mmol)、dmap(11mg,0.09mmol)及二氯甲烷(5ml)获得标题化合物(0.23g,0.27mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=7.24hz,12h),1.17-1.35(m,58h),1.58-1.65(m,9h),1.73-1.83(m,2h),1.87-1.93(m,2h),1.95-2.04(m,2h),2.23-2.32(m,8h),2.75-2.86(m,2h),3.97(d,j=5.50hz,2h),4.04(t,j=6.79hz,4h)。
<阳离子脂质的合成(18)>
〔实例a-18〕
5-{[(1-甲基哌啶-4-羰基)氧基]甲基}壬二酸二(十三烷基)酯(阳离子脂质18)
〔化72〕
(1)庚烷-1,4,4,7-四羧酸1,7-二苄酯4,4-二叔丁酯的合成
〔化73〕
将实例a-13-(1)中所获得的化合物(3.84g,9.78mmol)溶解在thf(30ml)中,在水浴冷却下添加60%氢化钠(0.43g,10.8mmol),并在室温下搅拌30分钟。添加4-溴丁酸苄酯(3.02g,11.7mmol)的thf(10ml)溶液后,加热回流18小时。将反应液恢复至室温,利用二乙醚进行稀释。依序利用水、饱和食盐水将有机相洗净,利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(乙酸乙酯/环己烷)对残渣进行精制,而获得标题化合物(1.88g,3.31mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.43(s,18h),1.47-1.54(m,4h),1.78-1.85(m,4h),2.36(t,j=7.34hz,4h),5.10(s,4h),7.27-7.40(m,10h)。
(2)6-(苄基氧基)-2-[4-(苄基氧基)-4-侧氧基丁基]-6-己酸的合成
〔化74〕
按照实例a-13-(3)的方法,由实例a-18-(1)中所获得的化合物(1.88g,3.31mmol)、tfa(4ml)、二氯甲烷(8ml)、及二甲苯(10ml)获得标题化合物(1.01g,2.45mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.31-1.58(m,8h),2.19-2.25(m,1h),2.33-2.37(m,4h),5.05-5.11(m,4h),7.27-7.40(m,10h),12.14(brs,1h)。
(3)5-(羟基甲基)壬二酸二苄酯的合成
〔化75〕
将实例a-18-(2)中所获得的化合物(1.01g,2.45mmol)溶解在thf(10ml)中,在-78℃下滴加硼烷-thf络合物(1m,4.9ml,4.9mmol)。在0℃下搅拌16小时后,添加饱和氯化铵水溶液,利用二乙醚进行萃取。利用饱和食盐水洗净有机相,并利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(环己烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(0.59g,1.47mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.09-1.37(m,6h),1.48-1.56(m,4h),2.32(t,j=7.34hz,4h),3.26(t,j=5.23hz,2h),4.32(t,j=5.14hz,1h),5.08(s,4h),7.28-7.41(m,10h)。
(4)5-{[(1-甲基哌啶-4-羰基)氧基]甲基}壬二酸二苄酯的合成
〔化76〕
按照实例a-13-(5)的方法,由实例a-18-(3)中所获得的化合物(0.58g,1.47mmol)、dipea(0.51ml,2.95mmol)、1-甲基-哌啶-4-羧酸盐酸盐(0.52g,2.95mmol)、dmap(36mg,0.30mmol)、edc(0.62g,3.24mmol)及二氯甲烷(7ml)获得标题化合物(0.62g,1.19mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.27-1.38(m,4h),1.59-1.69(m,5h),1.70-1.81(m,2h),1.85-1.92(m,2h),1.92-2.02(m,2h),2.26(s,4h),2.33(t,j=7.34hz,4h),2.74-2.83(m,2h),3.97(d,j=5.69hz,2h),5.11(s,4h),7.28-7.39(m,10h)。
(5)5-[{(1-甲基哌啶-4-羰基)氧基}甲基]壬二酸的合成
〔化77〕
按照实例a-16-(1)的方法,由实例a-18-(4)中所获得的化合物(0.62g,1.19mmol)、10%钯-碳(62mg,0.06mmol,50%含水)及乙醇(2ml)获得标题化合物(0.41g,1.19mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.20-1.31(m,4h),1.44-1.66(m,7h),1.72-1.81(m,4h),1.88-1.97(m,2h),2.13(s,3h),2.16-2.22(m,4h),2.22-2.30(m,1h),2.64-2.71(m,2h),3.93(d,j=5.32hz,2h)。
(6)5-{[(1-甲基哌啶-4-羰基)氧基]甲基}壬二酸二(十三烷基)酯的合成
〔化78〕
按照实例a-13-(7)的方法,由实例a-18-(5)中所获得的化合物(135mg,0.39mmol)、十三烷-1-醇(189mg,0.94mmol)、dmap(9mg,0.08mmol)、edc(166mg,0.87mmol)及二氯甲烷(5ml)获得标题化合物(107mg,0.15mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=7.06hz,6h),1.20-1.41(m,45h),1.58-1.71(m,9h),1.71-1.82(m,2h),1.86-1.94(m,2h),1.95-2.04(m,2h),2.23-2.30(m,8h),2.77-2.83(m,2h),4.00(d,j=5.5hz,2h),4.05(t,j=6.88hz,4h)。
<阳离子脂质的合成(19)>
〔实例a-19〕
5-{[(1-甲基哌啶-4-羰基)氧基]甲基}壬二酸双(8-戊基十三烷基)酯(阳离子脂质19)
〔化79〕
按照实例a-13-(7)的方法,由实例a-18-(5)中所获得的化合物(80mg,0.23mmol)、制造例4-(4)中所获得的化合物(151mg,0.56mmol)、dmap(6mg,0.05mmol)、edc(98mg,0.51mmol)及二氯甲烷(5ml)获得标题化合物(79mg,0.09mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=7.24hz,12h),1.15-1.40(m,60h),1.59-1.71(m,10h),1.72-1.82(m,2h),1.86-1.94(m,2h),1.94-2.04(m,2h),2.23-2.32(m,8h),2.77-2.85(m,2h),4.00(d,j=5.5hz,2h),4.05(t,j=6.88hz,4h)。
<阳离子脂质的合成(20)>
〔实例a-20〕
5-{[(1-甲基哌啶-4-羰基)氧基]甲基}壬二酸双(4-壬基十三烷基)酯(阳离子脂质20)
〔化80〕
按照实例a-13-(7)的方法,由实例a-18-(5)中所获得的化合物(80mg,0.23mmol)、制造例5-(4)中所获得的化合物(198mg,0.61mmol)、dmap(6mg,0.05mmol)、edc(98mg,0.51mmol)及二氯甲烷(5ml)获得标题化合物(46mg,0.05mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=6.97hz,12h),1.17-1.41(m,77h),1.57-1.72(m,9h),1.72-1.81(m,2h),1.85-1.94(m,2h),1.94-2.03(m,2h),2.23-2.31(m,8h),2.80(m,j=10.50hz,2h),4.00(d,j=5.50hz,2h),4.03(t,j=6.88hz,4h)。
<阳离子脂质的合成(21)>
〔实例a-21〕
7-{[(1-甲基哌啶-4-羰基)氧基]甲基}十三烷二酸二(十一烷基)酯(阳离子脂质21)
〔化81〕
(1)己烷-1,1,6-三羧酸6-苄酯1,1-二叔丁酯的合成
〔化82〕
使60%氢化钠(0.80g,20.0mmol)悬浮在thf(35ml)中,在冰浴冷却下滴加丙二酸二叔丁酯(4.48ml,20.0mmol),并在室温下搅拌15分钟。在冰浴冷却下添加碘化钠(0.29g,1.90mmol)后,滴加制造例9中所获得的化合物(5.43g,19.0mmol)的thf(10ml)溶液,在室温下搅拌18小时。利用二乙醚将反应混合物进行稀释,并利用水洗净后,利用二乙醚对水层进行萃取。合并有机相,利用饱和食盐水将其洗净,利用无水硫酸镁加以干燥。过滤后,在减压下将溶剂蒸馏去除。借由硅胶柱层析法(环己烷/乙酸乙酯)对残渣进行精制,而获得标题化合物(6.01g,14.3mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.27-1.38(m,4h),1.45(s,18h),1.62-1.68(m,2h),1.73-1.82(m,2h),2.35(t,j=7.52hz,2h),3.09(t,j=7.61hz,1h),5.11(s,2h),7.29-7.40(m,5h)。
(2)十一烷-1,6,6,11-四羧酸1,11-二苄酯6,6-二叔丁酯的合成
〔化83〕
按照实例a-18-(1)的方法,由实例a-21-(1)中所获得的化合物(4.0g,9.51mmol)、60%氢化钠(0.42g,10.5mmol)、制造例9中所获得的化合物(3.25g,11.4mmol)及thf(30ml)获得标题化合物(5.18g,8.29mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.09-1.19(m,4h),1.28-1.36(m,5h),1.43(s,18h),1.60-1.69(m,4h),1.72-1.80(m,4h),2.34(t,j=7.52hz,4h),5.05-5.15(m,4h),7.28-7.41(m,9h)。
(3)8-(苄基氧基)-2-[6-(苄基氧基)-6-侧氧基己基]-8-侧氧基辛酸的合成
〔化84〕
按照实例a-13-(3)的方法,由实例a-21-(2)中所获得的化合物(5.18g,8.29mmol)、tfa(10ml)、二氯甲烷(20ml)及二甲苯(25ml)获得标题化合物(2.49g,5.31mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.15-1.57(m,16h),2.11-2.20(m,1h),2.33(t,j=7.34hz,4h),5.08(s,4h),7.23-7.43(m,10h),12.01(brs,1h)。
(4)7-(羟基甲基)十三烷二酸二苄酯的合成
〔化85〕
按照实例a-18-(3)的方法,由实例a-21-(3)中所获得的化合物(2.49g,5.31mmol)、硼烷-thf络合物(1m,13.2ml,13.2mmol)及thf(20ml)获得标题化合物(1.95g,4.29mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.11-1.36(m,13h),1.39-1.46(m,1h),1.60-1.70(m,4h),2.35(t,j=7.52hz,4h),3.51(d,j=5.32hz,2h),5.11(s,4h),7.29-7.40(m,10h)。
(5)7-{[(1-甲基哌啶-4-羰基)氧基]甲基}十三烷二酸二苄酯的合成
〔化86〕
按照实例a-13-(5)的方法,由实例a-21-(4)中所获得的化合物(1.95g,4.29mmol)、dipea(1.5ml,8.58mmol)、1-甲基-哌啶-4-羧酸盐酸盐(1.54g,8.58mmol)、dmap(0.11g,0.86mmol)、edc(1.81g,9.44mmol)及二氯甲烷(20ml)获得标题化合物(2.97g,6.09mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):1.22-1.35(m,12h),1.61-1.68(m,4h),1.70-1.84(m,3h),1.85-1.93(m,2h),1.95-2.06(m,2h),2.21-2.30(m,4h),2.35(t,j=7.52hz,4h),2.74-2.85(m,2h),3.96(d,j=5.50hz,2h),5.11(s,4h),7.29-7.39(m,10h)。
(6)7-{[(1-甲基哌啶-4-羰基)氧基]甲基}十三烷二酸的合成
〔化87〕
按照实例a-16-(1)的方法,由实例a-21-(5)中所获得的化合物(2.28g,3.93mmol)、10%钯-碳(0.42g,0.39mmol,50%含水)及乙醇(20ml)获得标题化合物(1.34g,3.35mmol)。
1h-nmr(600mhz,dmso-d6)δ(ppm):1.19-1.30(m,12h),1.44-1.53(m,4h),1.53-1.63(m,3h),1.72-1.81(m,2h),1.90-1.98(m,2h),2.14(s,3h),2.18(t,j=7.43hz,4h),2.23-2.31(m,1h),2.64-2.73(m,2h),3.93(d,j=5.50hz,2h)。
(7)7-{[(1-甲基哌啶-4-羰基)氧基]甲基}十三烷二酸二(十一烷基)酯的合成
〔化88〕
将实例a-21-(6)中所获得的化合物(200mg,0.50mmol)、十一烷-1-醇(83μl,1.20mmol)及dmap(24mg,0.20mmol)溶解在二氯甲烷(10ml)中,在室温下添加edc(230mg,1.20mmol),并在室温下搅拌18小时。添加饱和碳酸氢钠水溶液,利用二氯甲烷加以萃取后,在减压下将有机相浓缩。借由硅胶柱层析法(乙酸乙酯/环己烷/甲醇)对残渣进行精制,而获得标题化合物(220mg,0.32mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=7.06hz,6h),1.19-1.39(m,44h),1.57-1.68(m,9h),1.71-1.86(m,2h),1.84-1.95(m,2h),1.95-2.06(m,2h),2.21-2.34(m,8h),2.75-2.87(m,2h),3.97(d,j=5.69hz,2h),4.05(t,j=6.79hz,4h)。
<阳离子脂质的合成(22)>
〔实例a-22〕
7-{[(1-甲基哌啶-4-羰基)氧基]甲基}十三烷二酸双(4-庚基十一烷基)酯(阳离子脂质22)
〔化89〕
按照实例a-16-(2)的方法,由实例a-21-(6)中所获得的化合物(200mg,0.25mmol)、制造例6-(2)中所获得的化合物(160mg,0.60mmol)、dmap(12mg,0.10mmol)、edc(120mg,0.60mmol)及二氯甲烷(5ml)获得标题化合物(88mg,0.10mmol)。
1h-nmr(600mhz,cdcl3)δ(ppm):0.88(t,j=6.97hz,12h),1.11-1.36(m,66h),1.49-1.67(m,9h),1.71-1.84(m,2h),1.84-1.95(m,2h),1.95-2.05(m,2h),2.21-2.33(m,8h),2.74-2.88(m,2h),3.97(d,j=5.50hz,2h),4.04(t,j=6.88hz,4h)。
将所合成的阳离子脂质1-22示在下述表a。
〔表a〕
表a:所合成的阳离子脂质1~22
b.组合物的制备及解析
<组合物的制备(1)>
〔实例b-1〕
使用实例a-1的阳离子脂质1而制备组合物。作为核酸,使用如下所述的已退火(annealing)的sirna(genedesign股份有限公司,以下有时称为“因子viisirna”),其是抑制因子vii(血液凝固第vii因子)基因的表达者,且包含正义链5′-ggafufcafufcfufcaagfufcfufuafct*t-3′(t:dna,fu、fc=2′-氟rna(2′-fluororna),*=硫代磷酸酯键(phosphorothioatelinkage))(序列编号1)、反义链5′-gfuaagafcfufugagafugafufcfct*t-3′(t:dna,fu、fc=2′-氟rna,*=硫代磷酸酯键)(序列编号2)的碱基序列。
利用25mm乙酸钠(ph值为4.0)将因子viisirna溶解为80μg/ml,制成sirna稀释液。此外,以总脂质浓度成为7.2mm的方式将阳离子脂质1、dspc(日本精化股份有限公司)、胆固醇(cholesterol)(日本精化股份有限公司)、mpeg2000-dmg(日油股份有限公司)以60/8.5/30/1.5(摩尔比)的比例溶解在乙醇中,制成脂质溶液。将sirna稀释液与脂质溶液分别以3ml/min、1ml/min的流速投入并加以混合,借此获得脂质复合体水溶液。将所获得的脂质复合体水溶液供在使用透析膜(商品名“float-a-lyzerg2”,spectrum公司,50kmwco)的透析,将外液置换为磷酸缓冲液(pbs,ph值为7.4)。透析后,进行浓缩及过滤杀菌,而获得实例b-1的液体组合物。
〔实例b-2〕
作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例a-7的组合物。
〔实例b-3〕
作为阳离子脂质,使用实例a-3的阳离子脂质3代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例b-3的组合物。
〔实例b-4〕
作为阳离子脂质,使用实例a-4的阳离子脂质4代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例b-4的组合物。
〔实例b-5〕
作为阳离子脂质,使用实例a-5的阳离子脂质5代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例b-5的组合物。
〔实例b-6〕
作为阳离子脂质,使用实例a-6的阳离子脂质6代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例b-6的组合物。
〔实例b-7〕
作为阳离子脂质,使用实例a-7的阳离子脂质7代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例b-7的组合物。
〔实例b-8〕
作为阳离子脂质,使用实例a-8的阳离子脂质8代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得实例b-8的组合物。
〔比较例b-1〕
作为阳离子脂质,依照专利文献2所记载的方法合成并使用专利文献2所记载的下述式(12)所表示的9-((4-(二甲胺基)丁酰基)氧基)十七烷二酸二((z)-壬烷-2-烯-1-基)酯(以下有时称为“aln-319”)代替阳离子脂质1,除此以外,以与实例b-1相同的方式获得比较例b-1的组合物。
〔化90〕
<组合物的解析(1)>
对在实例b-1~实例b-8及比较例b-1的组合物,测量在脂质复合体中的sirna的封入率。
具体而言,利用不含rnase的水(rnasefreewater)稀释组合物,以使用quant-itribogreenrna试剂(invitrogen公司)所测得的sirna浓度(a)作为存在在脂质复合体外液中的sirna。此外,以利用1%tritonx-100稀释组合物所测得的sirna浓度(b)作为组合物中的总sirna浓度。继而,根据下式(f1)算出核酸的封入率。
封入率(%)=100-(a/b)×100(f1)
此外,利用粒径测量装置(商品名“zetasizernanozs”,malvern公司制造)测量脂质复合体的平均粒径。
将sirna的封入率以及脂质复合体的平均粒径(z-平均)及多分散指数示在表1。
〔表1〕
表1
确认到实例b-1~实例b-8的组合物显示出与比较例b-1的组合物同等高的sirna的封入率。
<组合物的制备(2)>
〔实例b-9〕
使用实例a-1的阳离子脂质1制备组合物。作为核酸,使用萤火虫荧光素酶(fireflyluciferase)(fluc)的mrna(trilinkbiotechnologies,以下有时称为“flucmrna」)。
利用50mm乙酸钠(ph值为4.0)将flucmrna溶解为27μg/ml,制成mrna稀释液。此外,以总脂质浓度成为2.4mm的方式将阳离子脂质1、dspc(日本精化股份有限公司)、胆固醇(日本精化股份有限公司)、mpeg2000-dmg(日油股份有限公司)以60/8.5/30/1.5(摩尔比)的比例溶解在乙醇中,制成脂质溶液。将mrna稀释液与脂质溶液分别以3ml/min、1ml/min的流速投入并加以混合,借此获得脂质复合体水溶液。将所获得的脂质复合体水溶液供在使用透析膜(商品名“float-a-lyzerg2”,spectrum公司,50kmwco)的透析,将外液置换为磷酸缓冲液(pbs,ph值为7.4)。透析后,进行浓缩及过滤杀菌,而获得实例b-9的组合物。
〔实例b-10〕
作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质1,除此以外,以与实例b-9相同的方式获得实例b-10的组合物。
〔比较例b-2〕
作为阳离子脂质,使用上述的aln-319代替阳离子脂质1,除此以外,以与实例b-9相同的方式获得比较例b-2的组合物。
<组合物的解析(2)>
以与组合物的解析(1)相同的方式,对在实例b-9、实例b-10及比较例b-2的组合物,测量在脂质复合体中的mrna的封入率、及脂质复合体的平均粒径。将mrna的封入率及脂质复合体的平均粒径(z-平均)示在表2。
〔表2〕
表2
确认到实例b-9~实例b-10的组合物显示出与比较例b-2的组合物同等的高mrna封入率。
<组合物的制备(3)>
〔实例b-11〕
使用实例a-2的阳离子脂质2制备组合物。作为阳离子脂质,使用阳离子脂质2代替阳离子脂质1,作为核酸,使用人红血球生成素(humanerythropoietin)(hepo)的mrna(trilinkbiotechnologies,以下有时称为“epomrna”)代替flucmrna,除此以外,以与实例b-9相同的方式获得实例b-11的组合物。
〔比较例b-3〕
作为阳离子脂质,使用上述的aln-319代替阳离子脂质2,除此以外,以与实例b-11相同的方式获得比较例b-3的组合物。
<组合物的解析(3)>
以与组合物的解析(1)相同的方式,对在实例b-11及比较例b-3的组合物,测量在脂质复合体中的mrna的封入率、及脂质复合体的平均粒径。将mrna的封入率及脂质复合体的平均粒径(z-平均)示在表3。
〔表3〕
表3
确认到实例b-11的组合物显示出与比较例b-3的组合物同等的高mrna封入率。
<组合物的制备(4)>
〔实例b-12〕
以与组合物的制备(1)相同的方式,使用实例a-1的阳离子脂质1,而获得含有因子viisirna的实例b-12的组合物。
〔实例b-13〕
作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质1,除此以外,以与实例b-12相同的方式获得实例b-13的组合物。
〔比较例b-4〕
作为阳离子脂质,使用上述的aln-319代替阳离子脂质1,除此以外,以与实例b-12相同的方式获得比较例b-4的组合物。
<组合物的解析(4)>
以与组合物的解析(1)相同的方式,对实例b-12、实例b-13及比较例b-4的组合物测量脂质复合体的平均粒径(保管前平均粒径)。进而,在4℃下将各组合物在密闭的小瓶中保管3个月,并测量脂质复合体的平均粒径(保管后平均粒径)。将脂质复合体的平均粒径(z-平均)的变化示在表4。表中,平均粒径的变化(%)是借由保管后平均粒径/保管前平均粒径×100而算出。
〔表4〕
表4
实例b-12及实例b-13的组合物即使在保管3个月后,平均粒径亦几乎无变化,显示出与比较例b-4的组合物相比物理上更稳定。
<组合物的制备(5)>
〔实例b-14〕
以与组合物的制备(1)相同的方式,使用实例a-9的阳离子脂质9,而获得含有因子viisirna的实例b-14的组合物。
〔实例b-15〕
作为阳离子脂质,使用实例a-10的阳离子脂质10代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-15的组合物。
〔实例b-16〕
作为阳离子脂质,使用实例a-11的阳离子脂质11代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-16的组合物。
〔实例b-17〕
作为阳离子脂质,使用实例a-12的阳离子脂质12代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-17的组合物。
〔实例b-18〕
作为阳离子脂质,使用实例a-13的阳离子脂质13代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-18的组合物。
〔实例b-19〕
作为阳离子脂质,使用实例a-14的阳离子脂质14代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-19的组合物。
〔实例b-20〕
作为阳离子脂质,使用实例a-15的阳离子脂质15代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-20的组合物。
〔实例b-21〕
作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质9,除此以外,以与实例b-14相同的方式获得实例b-21的组合物。
<组合物的解析(5)>
以与组合物的解析(1)相同的方式,对实例b-14~实例b-21的组合物测量在脂质复合体中的sirna的封入率、及脂质复合体的平均粒径。将sirna的封入率以及脂质复合体的平均粒径(z-平均)及多分散指数示在表5。
〔表5〕
表5
<组合物的制备(6)>
〔实例b-22〕
以与组合物的制备(1)相同的方式,使用实例a-16的阳离子脂质16,而获得含有因子viisirna的实例b-22的组合物。
〔实例b-23〕
作为阳离子脂质,使用实例a-17的阳离子脂质17代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-23的组合物。
〔实例b-24〕
作为阳离子脂质,使用实例a-18的阳离子脂质18代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-24的组合物。
〔实例b-25〕
作为阳离子脂质,使用实例a-19的阳离子脂质19代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-25的组合物。
〔实例b-26〕
作为阳离子脂质,使用实例a-20的阳离子脂质20代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-26的组合物。
〔实例b-27〕
作为阳离子脂质,使用实例a-21的阳离子脂质21代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-27的组合物。
〔实例b-28〕
作为阳离子脂质,使用实例a-22的阳离子脂质22代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-28的组合物。
〔实例b-29〕
作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质16,除此以外,以与实例b-22相同的方式获得实例b-29的组合物。
<组合物的解析(6)>
以与组合物的解析(1)相同的方式,对实例b-22~实例b-29的组合物测量在脂质复合体中的sirna的封入率、及脂质复合体的平均粒径。将sirna的封入率以及脂质复合体的平均粒径(z-平均)及多分散指数示在表6。
〔表6〕
表6
<组合物的制备(7)>
〔实例b-30〕
以与组合物的制备(1)相同的方式,使用实例a-1的阳离子脂质1,而获得含有因子viisirna的实例b-30的组合物。
〔实例b-31〕
作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质1,除此以外,以与实例b-30相同的方式获得实例b-30的组合物。
〔实例b-32〕
作为阳离子脂质,使用实例a-3的阳离子脂质3代替阳离子脂质1,除此以外,以与实例b-30相同的方式获得实例b-32的组合物。
〔实例b-33〕
作为阳离子脂质,使用实例a-4的阳离子脂质4代替阳离子脂质1,除此以外,以与实例b-30相同的方式获得实例b-33的组合物。
〔比较例b-5〕
作为阳离子脂质,使用上述的aln-319代替阳离子脂质1,除此以外,以与实例b-30相同的方式获得比较例b-5的组合物。
<组合物的解析(7)>
以与组合物的解析(1)相同的方式,对实例b-30~实例b-33及比较例b-5的组合物测量脂质复合体的平均粒径(保管前平均粒径)。进而,在4℃下将各组合物保管在密闭的小瓶中,并测量3个月后及6个月后的脂质复合体的平均粒径(保管后粒径)。将脂质复合体的平均粒径(z-平均)的变化示在表7。表中,平均粒径的变化(%)是借由保管后平均粒径(6个月后)/保管前平均粒径×100而算出。
〔表7〕
表7
实例b-30~实例b-33的组合物即使在保管6个月后,平均粒径亦几乎无变化,显示出与比较例b-5的组合物相比物理上更稳定。
<组合物的制备(8)>
〔实例b-34〕
使用实例a-2的阳离子脂质2制备组合物。作为核酸,使用epomrna。
利用10mm柠檬酸钠(ph值为4.0)将epomrna溶解为80μg/ml,制成mrna稀释液。此外,以总脂质浓度成为2.25mm的方式将阳离子脂质2、dope(日油股份有限公司)、胆固醇(日本精化股份有限公司)、mpeg2000-dmg(日油股份有限公司)以60/5.0/33.5/1.5(摩尔比)的比例溶解在乙醇中,制成脂质溶液。将mrna稀释液与脂质溶液分别以3ml/min、1ml/min的流速投入并加以混合,借此获得脂质复合体水溶液。将所获得的脂质复合体水溶液供在使用透析膜(商品名“float-a-lyzerg2”,spectrum公司,50kmwco)的透析,将外液置换为磷酸缓冲液(pbs,ph值为7.4)。透析后,进行浓缩及过滤杀菌,而获得实例b-34的组合物。
〔实例b-35〕
作为核酸,使用荧光素酶sirna(luciferasesirna)代替因子viisirna,作为阳离子脂质,使用实例a-2的阳离子脂质2代替阳离子脂质1,除此以外,以与组合物的制备(1)相同的方式获得实例b-35的组合物。再者,荧光素酶sirna是使用包含正义链5′-cuuacgcugaguacuucgat*t-3′(t:dna,*=硫代磷酸酯键)(序列编号3)、反义链5′-ucgaaguacucagcguaagt*t-3′(t:dna,*=硫代磷酸酯键)(序列编号4)的碱基序列且经退火(annealing)者(genedesign股份有限公司)。
<组合物的解析(8)>
以与组合物的解析(1)相同的方式,对实例b-34及实例b-35的组合物测量在脂质复合体中的mrna或sirna的封入率、及脂质复合体的平均粒径。将mrna或sirna的封入率及脂质复合体的平均粒径(z-平均)示在表8。
〔表8〕
表8
确认到实例b-34及实例b-35的组合物显示出较高的核酸封入率。该结果表明,无论核酸的种类为何,实例的组合物均可用于核酸传递。
c.试验例
〔试验例1〕
以封入至脂质复合体中的因子viisirna浓度成为1μg/ml或5μg/ml的方式,利用pbs稀释实例b-1~实例b-3及比较例b-1的组合物。以10ml/kg的给予体积将各组合物尾静脉内给予至icr小鼠(5周龄,雌性,平均体重25g,n=3),从给予起24小时后,在麻醉下采血及采集肝脏。借由离心将血浆从血液分离,利用市售的试剂盒(商品名“biophenfvii”,hyphenbiomed公司)对血浆中的因子vii蛋白质浓度进行定量。作为阴性对照,对给予pbs的组进行相同的处理。
将pbs给予组的因子vii蛋白质浓度设为100%,以相对值的形式算出组合物给予组的因子vii蛋白质浓度。将结果示在图1及表9。
〔表9〕
表9
确认到实例b-1~实例b-3的组合物与比较例b-1的组合物相比,因子vii蛋白质的表达抑制效果更高。该结果表明,实例的组合物将核酸高效率地释放至细胞质中。
〔试验例2〕
以与试验例1相同的方式将实例b-4及比较例b-1的组合物给予至icr小鼠(5周龄,雌性,平均体重25g,n=3),并算出从给予起24小时后的血浆中的因子vii蛋白质浓度的相对值。将结果示在图2及表10。
〔表10〕
表10
确认到实例b-4的组合物与比较例b-1的组合物相比,因子vii蛋白质的表达抑制效果较高。该结果表明,实例的组合物将核酸高效率地释放至细胞质中。
〔试验例3〕
以与试验例1相同的方式将实例b-5的组合物给予至icr小鼠(5周龄,雌性,平均体重25g,n=3),并算出从给予起24小时后的血浆中的因子vii蛋白质浓度的相对值。将结果示在表11。
〔表11〕
表11
确认到实例b-5的组合物的因子vii蛋白质的表达抑制效果较高。该结果表明,实例的组合物将核酸高效率地释放至细胞质中。
〔试验例4〕
以封入至脂质复合体中的hepomrna浓度成为1μg/ml或3μg/ml的方式,利用pbs稀释实例b-11与比较例b-3的组合物。以10ml/kg的给予体积将各组合物尾静脉内给予至icr小鼠(5周龄,雌性,平均体重25g,n=3),从给予起24小时后在麻醉下实施采血。借由离心将血浆从血液分离,利用市售的试剂盒(商品名“humanerythropoietinquantikineivdelisakit”,r&dsystems)对血浆中的hepo蛋白质浓度进行定量。作为阴性对照,对未给予任何物质的(未处理)组与给予pbs的组进行相同的处理。将结果示在表12。
〔表12〕
表12
确认到实例b-11的组合物与比较例b-3的组合物相比,hepo蛋白质的表达效果较高。该结果表明,实例的组合物将核酸高效率地释放至细胞质中。
〔试验例5〕
以封入至脂质复合体中的因子viisirna浓度成为3μg/ml或30μg/ml的方式,利用pbs稀释实例b-14~实例b-21的组合物。以10ml/kg的给予体积将各组合物尾静脉内给予至icr小鼠(5周龄,雌性,平均体重25g,n=3),从给予起24小时后,在麻醉下采血及采集肝脏。借由离心将血浆从血液分离,利用市售的试剂盒(商品名“biophenfvii”,hyphenbiomed公司)对血浆中的因子vii蛋白质浓度进行定量。作为阴性对照,对给予pbs的组进行相同的处理。
将pbs给予组的因子vii蛋白质浓度设为100%,以相对值的形式算出组合物给予组的因子vii蛋白质浓度。将结果示在表13。
〔表13〕
表13
〔试验例6〕
以与试验例5相同的方式将实例b-22~实例b-29的组合物给予至icr小鼠(5周龄,雌性,平均体重25g,n=3),并算出从给予起24小时后的血浆中的因子vii蛋白质浓度的相对值。将结果示在表14。
〔表14〕
表14
〔试验例7〕
以封入至脂质复合体中的rna浓度成为30μg/ml的方式,利用pbs稀释封入有epomrna的实例b-34的组合物。以10ml/kg的给予体积将各组合物尾静脉内给予至balb/c小鼠(雌性,n=4),从给予起1天及4天后在麻醉下实施采血。借由离心将血浆从血液分离,利用市售的试剂盒(商品名“humanerythropoietinquantikineivdelisakit”,r&dsystems)对血浆中的hepo蛋白质浓度进行定量。此外,测量网状红血球数。作为阴性对照,对未给予任何物质的(未处理)组及给予封入有荧光素酶sirna的实例b-35的组合物的组进行相同的处理。将结果示在表15
〔表15〕
表15
在未处理组及lucsirna给予组中未检测到hepo,在epomrna给予组中,在给予1天后检测到hepo。此外,借由所产生的epo的作用,在给予4天后确认到网状红血球数增加。该结果表明,实例的组合物将核酸高效率地释放至细胞质中。
根据以上的结果,利用本发明的阳离子脂质,可将核酸高效率地释放至细胞质中。此外,利用本发明的阳离子脂质,在保管一定时间时,能够抑制脂质复合体的粒径增大。
产业上的可利用性
根据本发明,可提供一种能够将核酸高效率地释放至细胞质中的阳离子脂质。
- 还没有人留言评论。精彩留言会获得点赞!