一种聚酰胺薄膜复合膜的改性方法、聚酰胺薄膜复合膜及其应用与流程
本发明属于膜分离领域,更具体地,涉及一种聚酰胺薄膜复合膜的改性方法、一种聚酰胺薄膜复合膜及其应用。
背景技术:
薄膜复合膜(tfc膜)是膜分离过程中常用的一种聚合物膜,聚酰胺薄膜复合膜是通过界面聚合的方法由水相多元胺或醇(酚)和油相多元酰氯或异氰酸酯反应制备得到,在多孔支撑层表面形成一层致密的几百纳米厚的具有峰-谷粗糙形貌的超薄活性层。
传统的聚酰胺薄膜复合膜是由水相间苯二胺和油相均苯三甲酰氯反应得到的一种高交联的芳香聚酰胺结构,这种具有高交联度的聚酰胺通常相对疏水,并且这种聚酰胺活性层通常具有典型的峰-谷形貌,使得膜表面粗糙度大。亲水性差和表面粗糙度大导致了这种膜水通量低且具有一定的膜污染倾向。因此,如何以一种简单的方法制备得到一种表面亲水性好聚酰胺薄膜复合膜来实现高的水通量和更佳的抗污染性能,是目前面临的一个问题。
技术实现要素:
针对现有技术的以上缺陷或改进需求,本发明提供了聚酰胺薄膜复合膜的改性方法、一种用于水处理的改性的高性能聚酰胺薄膜复合膜及其应用,其目的在于通过对聚酰胺薄膜复合膜的聚酰胺层进行表面改性,制备得到一种亲水性好、水通量大、反向盐通量低、膜污染倾向低且抗菌的薄膜复合膜,由此解决现有技术制备得到的聚酰胺薄膜复合膜水通量低以及膜污染倾向高的技术问题。
为实现上述目的,按照本发明的一个方面,提供了一种聚酰胺薄膜复合膜的改性方法,所述聚酰胺薄膜复合膜包括聚合物支撑层以及初始聚酰胺层,所述初始聚酰胺层通过多元胺和多元酰氯通过界面聚合反应得到,对所述初始聚酰胺层进行表面改性得到改性的聚酰胺层,所述表面改性包括对所述初始聚酰胺层进行表面接枝多胺化合物处理。
优选地,所述多胺化合物包括聚乙烯亚胺、乙二胺、丙二胺、丁二胺、己二胺、n,n′-二(2-氨乙基)-1,3-丙二胺、n,n′-双(2-羟乙基)乙二胺、三亚乙基四胺和二乙烯三胺中的一种或多种。
优选地,所述表面接枝多胺化合物处理具体步骤包括:将所述初始聚酰胺层置于20~60℃含1~5wt%的ph为9~11的多胺化合物中浸泡20~60min,取出后水洗,得到表面接枝多胺化合物的聚酰胺层。
优选地,对所述初始聚酰胺层进行表面接枝多胺化合物处理后,还对其进行表面接枝有机酸处理。
优选地,所述有机酸包括二乙烯三胺五乙酸、三乙烯四胺六乙酸、乙二胺四乙酸、n-羟乙基乙二胺-n,n′,n′-三乙酸、二乙烯三胺五甲叉膦酸、己二胺四甲叉膦酸、乙二胺四亚甲基膦酸和氨基三亚甲基膦酸中的一种或多种。
优选地,所述表面接枝有机酸处理具体步骤包括:将表面接枝多胺化合物的聚酰胺层置于ph为2~5的1wt%~5wt%的有机酸的水溶液中浸泡1~6h,取出后水洗,得到表面接枝有机酸的聚酰胺层。
优选地,对所述聚酰胺层依次进行表面接枝多胺化合物处理和接枝有机酸处理后,还对其进行表面矿化处理。
优选地,所述表面矿化处理采用无机盐进行表面矿化,所述表面矿化处理采用无机盐进行表面矿化,使无机盐金属离子通过与表面接枝的有机酸通过鳌合作用结合在所述聚酰胺层的表面;用于表面矿化的无机盐包括可溶性铜盐、可溶性铁盐、可溶性银盐、可溶性钙盐和可溶性钡盐中的一种或多种。
优选地,所述表面矿化处理具体步骤包括:将表面接枝有机酸的聚酰胺层置于ph为9~11的碱性水溶液中浸泡10~60min,取出水洗后,再置于5wt%~10wt%的无机金属盐水溶液中室温浸泡处理6~12h,得到表面矿化处理后的聚酰胺层。
优选地,对所述聚酰胺层依次进行表面接枝多胺化合物处理、表面接枝有机酸处理和表面矿化处理后,还对其进行表面原子化处理。
优选地,所述表面原子化处理具体为:在还原剂的作用下,将可溶性金属盐还原为金属原子;所述可溶性金属盐包括可溶性铜盐或可溶性银盐。
优选地,所述表面原子化处理的具体步骤包括:将表面矿化处理后的聚酰胺层浸泡在浓度为0.05~0.3mol/l铜盐或银盐溶液中6~12h,然后再浸泡在0.005~0.03mol/l的硼氢化钠水溶液中10~30min,取出水洗后得到改性后的聚酰胺层。
按照本发明的另一个方面,提供了一种聚酰胺薄膜复合膜,所述聚酰胺薄膜复合膜包括聚合物支撑层和改性的聚酰胺层,所述改性的聚酰胺层按照如上所述的改性方法得到。
优选地,所述初始聚酰胺层的制备方法包括如下步骤:将在多元胺溶液中浸泡处理后的聚合物支撑层与多元酰氯溶液接触,发生界面聚合反应得到初始聚酰胺层。
优选地,所述多元胺为对苯二胺、间苯二胺、邻苯二胺、对环己二胺、己二胺、聚乙烯亚胺和对二氮己环中的一种或多种。
优选地,所述聚合物支撑层为微滤膜或超滤膜,所述聚合物支撑层的材料选自聚丙烯腈、聚醚砜、聚砜、聚酰亚胺、聚酰胺、聚醚酰亚胺、聚酰胺酰亚胺或聚偏氟乙烯。
优选地,所述浸泡处理时间为1~10min,优选为1~2min。
优选地,所述多元酰氯为均苯四甲酰氯、均苯三甲酰氯、对苯二甲酰氯、邻苯二甲酰氯和己二酰氯中的一种或多种,所述多元酰氯的质量体积浓度为0.05%~0.5%,所述多元酰氯溶液的溶剂为正己烷、正庚烷、环己烷和甲苯中的一种或多种。
优选地,所述界面聚合反应时间为1~10分钟。
优选地,所述改性的聚酰胺层水接触角为27.48°±1.58°~17.5°±0.8°。
按照本发明的另一个方面,提供了一种所述的聚酰胺薄膜复合膜的应用,应用于膜分离。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果。
1、本发明提供了一种用于水处理的高性能聚酰胺薄膜复合膜,该复合膜包括聚合物支撑层以及初始聚酰胺层,初始聚酰胺层由多元胺和多元酰氯通过界面聚合反应制备得到,再通过对该初始聚酰胺层进行一系列表面改性制备获得改性的聚酰胺层,该表面改性包括按照一定的次序进行表面接枝多胺化合物、接枝有机酸、表面矿化和表面原子化中一种或多种改性方式。通过选择表面接枝多胺化合物和有机酸具体种类及添加比例,以及表面矿化和原子化的金属种类,反应条件的控制等,制备得到一种水通量大、反向盐通量低、膜污染倾向低且抗菌的薄膜复合膜。
2、本发明经过改性处理的聚酰胺薄膜复合膜由于在聚酰胺层表面进行亲水性多胺化合物和有机酸的接枝,加上表面矿化,使得制备得到的聚酰胺薄膜复合膜亲水性增加,用于膜分离测试水通量和盐通量时得到更高的水通量,相对于传统的未经改性的复合膜的水通量盐通量提高了23~127%;同时,由于膜表面电荷的改变,经有机酸改性后的膜的反向盐通量相比较于未改性膜下降了约10%。
3、本发明经过改性处理的聚酰胺薄膜复合膜由于膜表面亲水性的提升,接枝化合物的立体位阻效应、更少活性络合位点(羧基)和具有抗菌效应的矿化层和和原子层的引入,经验证,对于常见的污染物海藻酸钠和本地湖水具有良好的抗污染性能,污染后的水通量降低不超过33%,并且污染后较易清洗,水通量恢复率不低于90%。
4、本发明聚酰胺薄膜复合膜的改性方法中涉及包括接枝多胺化合物、接枝有机酸、表面矿化和表面原子化处理的多个方案,对于包括两个以上步骤的方案,其改性处理步骤的先后次序不能颠倒或改变。初始形成的聚酰胺层表面有残留的酰氯基团,多胺化合物可以与之反应,提高膜表面亲水性,并为后续接枝有机酸提供氨基反应位点,酸功能基团的引入一方面可以提高膜表面亲水性,另一方面也可以为后续金属离子在膜表面矿化提供螯合配位反应位点,矿化层的引入可以进一步提升膜表面亲水性,并且银盐和铜盐的引入还可以提高膜的抗菌性能,最后再将引入的银盐或铜盐还原得到更稳定的原子态,可以得到抗菌效果更佳的复合膜。
附图说明
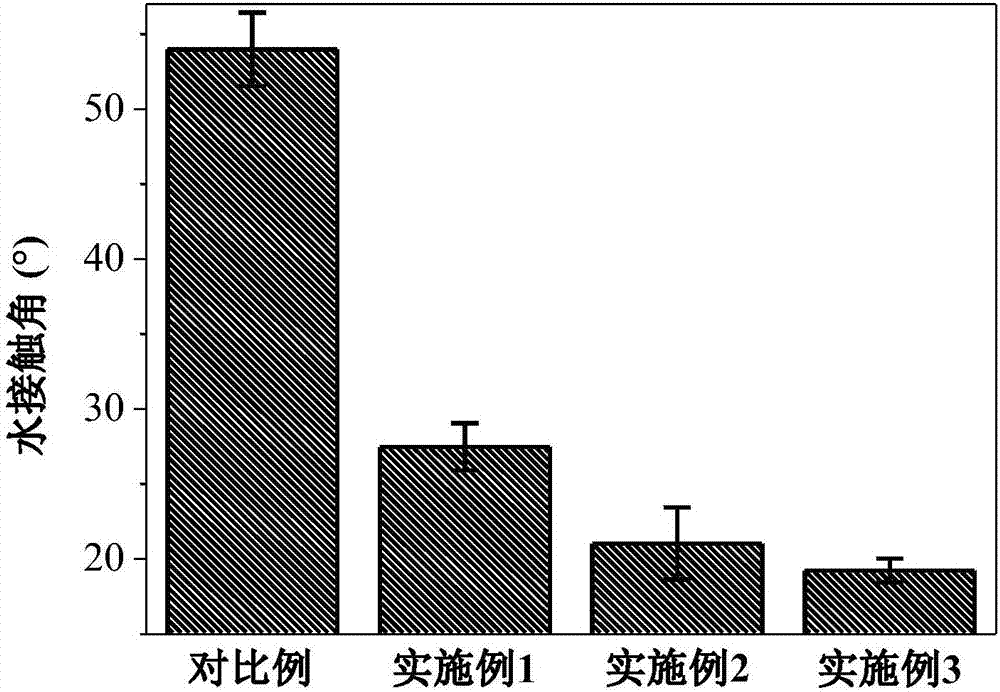
图1是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的水接触角图;
图2(a)是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的水通量测试比较图;图2(b)是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的反向盐通量测试比较图;
图3(a)是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的含海藻酸钠人工污水抗污染后均一化水通量测试结果图;图3(b)是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的含海藻酸钠人工污水抗污染反洗后均一化水通量测试结果图;
图4(a)是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的本地湖水水抗污染后均一化水通量测试结果图;图4(b)是对比例和实施例1~3制备得到的聚酰胺薄膜复合膜的本地湖水抗污染反洗后均一化水通量测试结果图;
图5是对比例和实施例2、3、6和8制备得到的聚酰胺薄膜复合膜的水接触角图;
图6(a)是对比例和实施例2、3、6和8制备得到的聚酰胺薄膜复合膜的水通量测试比较图;图6(b)是对比例和实施例2、3、6和8制备得到的聚酰胺薄膜复合膜的反向盐通量测试比较图;
图7是对比例和实施例2、10~13制备得到的聚酰胺薄膜复合膜的水接触角图;
图8(a)是对比例和实施例10~13制备得到的聚酰胺薄膜复合膜的水通量测试比较图;图8(b)是对比例和实施例10~13制备得到的聚酰胺薄膜复合膜的反向盐通量测试比较图;
图9(a)是对比例和实施例10~13制备得到的聚酰胺薄膜复合膜的本地湖水抗污染后均一化水通量测试结果图;图9(b)是对比例和实施例10~13制备得到的聚酰胺薄膜复合膜的本地湖水抗污染反洗后均一化水通量测试结果图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
本发明提供了一种聚酰胺薄膜复合膜的改性方法,该聚酰胺薄膜复合膜包括聚合物支撑层以及初始聚酰胺层,初始聚酰胺层为多元胺和多元酰氯通过界面聚合反应得到,具体地,初始聚酰胺层的获得方法为:将在多元胺溶液中浸泡处理后的聚合物支撑层与多元酰氯溶液接触,使得多元胺与多元酰氯发生界面聚合反应得到初始聚酰胺层。多元胺为对苯二胺、间苯二胺、邻苯二胺、对环己二胺、己二胺、聚乙烯亚胺和对二氮己环中的一种或多种。水相多元胺溶液质量分数为1%-8%,优选为1%-5%,多元胺浓度太低会导致界面聚合形成的聚酰胺活性层不够致密,复合膜选择性低,浓度太高会使形成的聚酰胺活性层太过致密,导致水通量降低。充分浸泡所需的时间与水相溶液的浓度相关,通常1min~30min以上可使得多元胺的吸附达到饱和,多元胺溶液中浸泡处理时间可选择为1~10min,优选为1~2min。多元酰氯为均苯四甲酰氯、均苯三甲酰氯、对苯二甲酰氯、邻苯二甲酰氯和己二酰氯中的一种或多种,所述多元酰氯的质量体积浓度为0.05%~0.5%,所述多元酰氯溶液的溶剂为正己烷、正庚烷、环己烷和甲苯中的一种或多种。与多元酰氯溶液接触发生的界面聚合反应的时间为1min~30min,优选1~10分钟;接触的时间太短,多元酰氯与混合胺的聚合反应不完全,接触的时间太长,则反应形成的聚酰胺活性层太厚,从而降低复合膜的水通量。
本发明对上述初始聚酰胺层进行表面改性得到改性的聚酰胺层,作为其中的一种方案,该表面改性包括对所述聚酰胺层进行表面接枝多胺化合物处理。所述多胺化合物包括聚乙烯亚胺、乙二胺、丙二胺、丁二胺、己二胺、n,n′-二(2-氨乙基)-1,3-丙二胺、n,n′-双(2-羟乙基)乙二胺、三亚乙基四胺和二乙烯三胺中的一种或多种。表面接枝多胺化合物处理具体步骤包括:将所述聚酰胺层置于20~60℃含1~5wt%的ph为9~11的多胺化合物中浸泡20~60min,取出后水洗,得到表面接枝多胺化合物的聚酰胺层,浸泡溶液温度不能太高,多胺溶液ph不能超过11,否则会破坏聚酰胺层,导致反向盐通量增大。
作为优选的方案,对所述聚酰胺层进行表面接枝多胺化合物处理后,还对其进行表面接枝有机酸处理;优选地,所述有机酸包括二乙烯三胺五乙酸、三乙烯四胺六乙酸、乙二胺四乙酸、n-羟乙基乙二胺-n,n′,n′-三乙酸、二乙烯三胺五甲叉膦酸、己二胺四甲叉膦酸、乙二胺四亚甲基膦酸和氨基三亚甲基膦酸中的一种或多种;优选地,所述表面接枝有机酸处理具体步骤包括:将表面接枝多胺化合物的聚酰胺层置于ph为2~5的1wt%~5wt%的有机酸的水溶液中浸泡1h,取出后水洗,得到表面接枝有机酸的聚酰胺层。有机酸溶液的ph值不能低于2,否则会使聚酰胺层水解,导致膜反向盐通量增大,但ph也不能太高,否则会导致大部分酸功能基团去质子化,失去与氨基反应的活性,使改性效果不明显。
作为进一步优选的方案,对所述聚酰胺层依次进行表面接枝多胺化合物处理和接枝有机酸处理后,还对其进行表面矿化处理;优选地,所述表面矿化处理采用无机盐进行表面矿化,用于表面矿化的无机盐包括可溶性铜盐、可溶性铁盐、可溶性银盐、可溶性钙盐和可溶性钡盐中的一种或多种;优选地,所述表面矿化处理具体步骤包括:将表面接枝有机酸的聚酰胺层置于ph为9~11的碱性水溶液中浸泡10~60min,取出水洗后,再置于5wt%~10wt%的无机金属盐水溶液中常温浸泡处理6~12h,得到表面矿化处理后的聚酰胺层。这里的常温即室温,为20~30℃,一般控制为25℃。碱性水溶液的ph不能超过11,否则会使聚酰胺层水解,导致膜反向盐通量增大,ph也不能太低,否则酸功能基团的去质子化的程度不够,不能提供足够多的螯合位点,导致矿化程度不高。
进一步地,对所述聚酰胺层依次进行表面接枝多胺化合物处理、接枝有机酸处理和表面矿化处理后,还对其进行表面原子化处理;优选地,所述表面原子化处理具体为:在还原剂的作用下,将可溶性金属盐还原为金属原子;所述可溶性金属盐包括可溶性铜盐或可溶性银盐;优选地,所述表面原子化处理的具体步骤包括:将表面矿化处理后的聚酰胺层浸泡在浓度为0.05~0.3m的铜盐或银盐溶液中,然后再浸泡在0.005~0.03mol/l的硼氢化钠水溶液中10~30min,取出水洗后得到改性后的聚酰胺层。
本发明提供了一种改性的聚酰胺薄膜复合膜,其包括聚合物支撑层以及改性的聚酰胺层,改性的聚酰胺层为按照上述任一改性方法对初始聚酰胺层进行表面改性得到。该改性的聚酰胺层的水接触角相对于现有技术的聚酰胺薄膜复合物大大减小,为27.48°±1.58°~17.5°±0.8°。该聚酰胺薄膜复合膜可应用于膜分离中的水处理,比如海水淡化处理。
通过改性本发明提供了一种高性能聚酰胺薄膜复合膜,改性主要是针对初始聚酰胺层进行的表面改性,初始的聚酰胺层是由多元胺和多元酰氯通过界面聚合反应制备得到的,本发明提到的表面改性包括四种方式:
第一种:对初始聚酰胺层进行表面接枝多胺化合物处理。由于多元胺水溶液与多元酰氯发生界面聚合反应,形成的初始聚酰胺层表面含有残留的酰氯基团,因此通过控制接枝工艺,接枝多胺化合物之后可以改善膜表面的亲水性,从而提高膜的抗污染性能。
第二种:对初始聚酰胺层依次进行表面接枝多胺化合物处理和表面接枝有机酸处理。在对初始聚酰胺层进行表面接枝多胺化合物以后,由于表面含有氨基基团,因此可以保证有机酸的羧基或者膦酸基团与之反应而接枝上有机酸,从而提高膜表面亲水性,以及为后续矿化改性提供螯合位点,并且提高膜的抗污染性能。
第三种:对初始聚酰胺层依次进行表面接枝多胺化合物处理、表面接枝有机酸处理和表面矿化处理。依次进行接枝多胺化合物和有机酸处理的聚酰胺层表面含有酸功能基团,在此基础上进行表面矿化,即将酸化改性后的膜浸泡在无机盐溶液中,使得酸功能基团与无机盐的的金属离子通过螯合作用结合,改善膜表面的亲水性以及抗污染性能,矿化银盐和铜盐的膜同时具有一定的抗菌性。但是表面矿化钙盐的方案不适用于处理含有机海藻类污染物的废水,这是因为表面矿化钙盐后的聚酰胺薄膜复合膜会与废水中有机污染物的羧基发生配位反应,在表面形成一层污染层,影响该复合膜的水通量。但是对于含微生物污染物的废水比如本发明的本地湖水,该改性的复合膜水通量以及抗污染性能很好。
第四种:对初始聚酰胺层依次进行表面接枝多胺化合物处理、表面接枝有机羧酸处理、表面矿化处理以及表面原子化处理,制备得到一种水通量大、膜污染倾向低且抗菌的薄膜复合膜。表面原子化即将矿化银盐和铜盐还原成相应的原子态,使得具有更好的稳定性,从而具有更好更稳定的抗菌性。
本发明的改性方法中涉及四个步骤,即表面接枝多胺化合物处理、表面接枝有机酸处理、表面矿化处理以及表面原子化处理,这四个步骤必须依次进行,不可改变次序,因为后一步都必须在前一步的基础上进行改性。初始形成的聚酰胺层会有残留的酰氯基团,多胺化合物可以与之反应,提高膜表面亲水性,并为后续接枝有机酸提供氨基反应位点,酸功能基团的引入一方面可以提高膜表面亲水性,另一方面也可以为后续金属离子再膜表面矿化提供螯合反应位点,矿化层的引入可以进一步提升膜表面亲水性,并且银盐和铜盐的引入还可以提高膜的抗菌性能,最后再将引入的银盐或铜盐还原得到更稳定的原子态,可以得到抗菌效果更佳的复合膜。
本发明通过对初始聚酰胺层进行一系列表面改性得到改性后的聚酰胺层,其表面亲水性大幅度提升,并具有抗菌层,比传统不改性复合膜具有更高的水通量和更佳的抗污染和抗菌性能。
以下为实施例:
对比例
该对比例制备的聚酰胺薄膜复合膜的聚合物支撑层为聚砜微滤膜。该复合膜的制备过程包括以下步
(1)配制水相溶液,所述水相溶液含有质量分数为3.4%的间苯二胺多元胺。配制多元酰氯有机相溶液,所述溶液以正己烷作为溶剂,以质量/体积分数0.15%的均苯三甲酰氯作为溶质。将聚砜基膜完全浸泡在水相溶液中2min后取出。
(2)赶除表面多余水相溶液后,将有机相溶液倒在润湿的聚砜膜上表面,1分钟接触时间后,将多余的有机相溶液倒掉。胺单体与酰氯单体在两相界面处发生界面聚合反应,形成聚酰胺薄膜复合膜。
实施例1
实施例1的复合膜的聚合物支撑层为聚砜微滤膜。该复合膜的制备过程包括以下步
(1)配制水相溶液,所述水相溶液含有质量分数为3.4%的间苯二胺多元胺。配制多元酰氯有机相溶液,所述溶液以正己烷作为溶剂,以质量/体积分数0.15%的均苯三甲酰氯作为溶质。将聚砜基膜完全浸泡在水相溶液中2min后取出。
(2)赶除表面多余水相溶液后,将有机相溶液倒在润湿的聚砜膜上表面,1分钟接触时间后,将多余的有机相溶液倒掉。胺单体与酰氯单体在两相界面处发生界面聚合反应,形成聚酰胺薄膜复合膜。
(3)将上述刚形成的聚酰胺薄膜复合膜转移至40℃含2wt%的聚乙烯亚胺溶液中反应20min,然后用水冲洗并置于纯水中保存使用。
实施例2
实施例2的复合膜的聚合物支撑层为聚砜微滤膜。该复合膜的制备过程包括以下步
(1)配制水相溶液,所述水相溶液含有质量分数为3.4%的间苯二胺多元胺。配制多元酰氯有机相溶液,所述溶液以正己烷作为溶剂,以质量/体积分数0.15%的均苯三甲酰氯作为溶质。将聚砜基膜完全浸泡在水相溶液中2min后取出。
(2)赶除表面多余水相溶液后,将有机相溶液倒在润湿的聚砜膜上表面,1分钟接触时间后,将多余的有机相溶液倒掉。胺单体与酰氯单体在两相界面处发生界面聚合反应,形成聚酰胺薄膜复合膜。
(3)将上述刚形成的聚酰胺薄膜复合膜转移至40℃含2wt%的聚乙烯亚胺溶液中反应20min,然后用水冲洗。
(4)将步骤(3)得到的所述复合膜转移到常温下的ph为3的1wt%的二乙烯三胺五甲叉膦酸水溶液中1h,然后用水冲洗并置于纯水中保存使用。
实施例3
实施例3的复合膜的聚合物支撑层为聚砜微滤膜。该复合膜的制备过程包括以下步
(1)配制水相溶液,所述水相溶液含有质量分数为3.4%的间苯二胺多元胺。配制多元酰氯有机相溶液,所述溶液以正己烷作为溶剂,以质量/体积分数0.15%的均苯三甲酰氯作为溶质。将聚砜基膜完全浸泡在水相溶液中2min后取出。
(2)赶除表面多余水相溶液后,将有机相溶液倒在润湿的聚砜膜上表面,1分钟接触时间后,将多余的有机相溶液倒掉。胺单体与酰氯单体在两相界面处发生界面聚合反应,形成聚酰胺薄膜复合膜。
(3)将上述刚形成的聚酰胺薄膜复合膜转移至40℃含2wt%的聚乙烯亚胺溶液中反应20min,然后用水冲洗。
(4)将步骤(3)得到的所述复合膜转移到常温下的ph为3的1wt%的二乙烯三胺五甲叉膦酸水溶液中1h,然后用水冲洗。
(5)将步骤(4)得到的所述复合膜转移到常温下的ph为11的氢氧化钠水溶液中10min,然后用水冲洗,再置于5wt%的氯化钙水溶液中常温处理6h,取出清洗并置于纯水中保存。
实施例4
以所述的相同步骤重复实施例2,区别在于,在所述步骤(4)中,有机酸为氨基三亚甲基膦酸。
实施例5
以所述的相同步骤重复实施例2,区别在于,在所述步骤(4)中,有机酸为乙二胺四亚甲基膦酸。
实施例6
以所述的相同步骤重复实施例2,区别在于,在所述步骤(4)中,有机酸为二乙烯三胺五乙酸。
实施例6
以所述的相同步骤重复实施例3,区别在于,在所述步骤(4)中,有机酸为氨基三亚甲基膦酸。
实施例7
以所述的相同步骤重复实施例3,区别在于,在所述步骤(4)中,有机酸为乙二胺四亚甲基膦酸。
实施例8
以所述的相同步骤重复实施例3,区别在于,在所述步骤(4)中,有机酸为二乙烯三胺五乙酸。
实施例9
以所述的相同步骤重复实施例3,区别在于,在所述步,无机盐为氯化铁。
实施例10
以所述的相同步骤重复实施例3,区别在于,在所述步骤(5)中,无机盐为硫酸铜。
实施例11
以所述的相同步骤重复实施例3,区别在于,在所述步骤(5)中,无机盐为硝酸银。
实施例12
以所述的相同步骤重复实施例9,区别在于,增加步骤(6):将步骤(5)用硫酸铜浸泡处理得到的所述复合膜浸泡在0.01mol/l的硼氢化钠水溶液中常温下进行还原20min,然后用水冲洗。
实施例13
以所述的相同步骤重复实施例11,区别在于,增加步骤(6):将步骤(5)用硝酸银浸泡处理得到的所述复合膜浸泡在0.01mol/l的硼氢化钠水溶液中常温下进行还原20min,然后用水冲洗。
实施例14~实施例20
为了简化描述,故将实施例5~实施例10的制备参数列入表1,表中未列的参数与实施例3相同。
表1实施例14~实施例20聚酰胺薄膜复合膜的制备参数
实验结果分析:
图1显示了对比例和实施例1~3的水接触角图,相比较于对比例,实施例1~3均具有更低接触角,表明亲水性的提高,这是由于氨基(聚乙烯亚胺)、磷酸基团(二乙烯三胺五甲叉膦酸)和矿化层的引入。
用2m氯化钠水溶液作为汲取液,去离子水作为料液,用正向渗透测试装置测试实施例1~4制备的复合膜的水通量和反向盐通量,测试时间为1h,每种膜测三个样。图2(a)和图2(b)显示了对比例和实施例1~3的聚酰胺薄膜复合膜的正渗透分离性能,其中图2(a)为对比例和实施例1~3的聚酰胺薄膜复合膜的水通量测试数据;图2(b)为对比例和实施例1~3的聚酰胺薄膜复合膜的反向盐通量测试数据。如图2(a)所示,对比例的复合膜在正渗透模式和压力延缓渗透模式下的水通量为16.5±1.3lmh和31.0±2.2lmh,而对于实施例1~3改性复合膜其水通量明显提升,最大可提升至37.2±2.2lmh和68.2±2.2lmh。经一系列表面改性后,复合膜的水通量得到提升,反向盐通量略微增加,对于实施例2,其反向盐通量甚至下降。如图2(b)所示,实施例1制备的空白复合膜在正渗透模式和压力延缓渗透模式下的反向盐通量为14.8±2.3gmh和25.8±2.1gmh。而实施例3制备的改性复合膜的反向盐通量可降低至12.3±1.0gmh和22.9±0.5gmh。并且相对于已报导的大多数聚酰胺薄膜复合膜在相同的测试条件下,其水通量有明显提升。
用正渗透设备对对比例和实施例1~3制备的复合膜进行了对海藻酸钠合成废水的抗污染性能测试,其中海藻酸钠合成废水中的成分及浓度为:0.45mmkh2po4,9.20mmnacl,0.61mmmgso4,0.5mmnahco3,0.5mmcacl2,and0.93mmnh4cl。结果如图3(a)和图3(b)所示。用含有250mg/l海藻酸铵的合成废水作为污染物料液,以2m的氯化钠水溶液作为汲取液,进行18小时的连续性污染测试,实时记录复合膜在测试过程中的水通量变化(污染时通量)。污染测试过程中,料液和汲取液的流速均为0.3l/min。测试结束后,用去离子水作为料液在加大一倍流速的条件下对膜进行反洗20min,然后用纯水作为料液实时测量水通量恢复情况。结果如图3(a)和图3(b)所示,对比例空白复合膜污染后水通量大幅度下降,并且反洗后水通量恢复率也才约70%。然而对于改性后的复合膜(实施例1~2),表现出更佳的抗污染性能,污染18小时后,水通量下降率都低于33%,且反洗后水通量恢复率都高达89%以上。实施例3表现出较差的抗污染性能是由于膜表面的钙离子加剧了膜污染。由于实施例3改性的复合膜表面有钙离子,而配制人工污水中的海藻酸钠含有羧基,复合膜表面本身的钙离子与海藻酸钠中的羧基发生配位反应使得复合膜表面形成的比较厚且较致密的凝胶层,大大降低了水通量。因此,表面钙盐矿化处理的改性膜不适用于有机污染废水,选用其他的污染物体系,比如无机污染物或微生物污染物体系,可以表现出较好的抗污染性能。
图4(a)和图4(b)显示了对比例和实施例1-3制备复合膜对本地湖水的抗污染测试结果。本地湖水主要为微生物污染和一定的无机污染。从图4(a)和图4(b)中可以看出,对比例空白复合膜污染后水通量大幅度下降,并且反洗后水通量恢复率也才约76%。然而对于改性后的复合膜,表现出更佳的抗污染性能,污染18小时后,水通量下降率都低于22%,且反洗后水通量恢复率都高达93%以上。
图5显示了对比例和实施例2、3、6、8的水接触角图,相比较于对比例,实施例2、3、6、8均具有更低的接触角,表明亲水性的提高,这是由于氨基(聚乙烯亚胺)、磷酸基团(二乙烯三胺五甲叉膦酸)和矿化层的引入。并且有机膦酸改性的实施例2比有机羧酸改性的实施例6具有更低的水接触角,这是由于膦酸具有更低的pka值。相应的,实施例3比实施例8的接触角更低,这是由于膦酸基团对钙离子的络合能力比羧酸基团强。
图6(a)和图6(b)显示了对比例和实施例2、3、6、8的正渗透分离性能。其中图6(a)为对比例和实施例2、3、6、8的聚酰胺薄膜复合膜的水通量测试数据;图6(b)为对比例和实施例2、3、6、8的聚酰胺薄膜复合膜的反向盐通量测试数据。相比较于对比例,对于实施例改性复合膜其水通量明显提升,对于实施例2和6,其反向盐通量甚至下降。
图7显示了对比例和实施例2、10~13的水接触角图,相比较于对比例,实施例均具有更低的接触角,表明亲水性的提高,这是由于磷酸基团(二乙烯三胺五甲叉膦酸)、矿化层和原子层的引入。并且引入银矿化层和银原子的实施例10和12比相应的引入铜矿化层和铜原子的实施例11和13具有更低的水接触角,这是由于膦酸基团对银离子的络合能力比铜离子强。
图8(a)和图8(b)显示了对比例和实施例10~13的正渗透分离性能,图8(a)为对比例和实施例10~13的聚酰胺薄膜复合膜的水通量测试数据;图8(b)为对比例和实施例10~13的聚酰胺薄膜复合膜的反向盐通量测试数据。相比较于对比例,对于实施例改性复合膜其水通量明显提升,其反向盐通量增加不多。与图7接触角结果一致,实施例10和12的水通量比相应的实施例11和13的大。
图9(a)和图9(b)显示了对比例和实施例10-13制备复合膜对本地湖水的抗污染测试结果。从图中可以看出,对比例空白复合膜污染后水通量大幅度下降,并且反洗后水通量恢复率也才约76%。然而对于改性后的复合膜,表现出更佳的抗污染性能,污染18小时后,水通量下降率都低于17%,且反洗后水通量恢复率都高达97%以上。并且原子化后的复合膜的抗污染性能更优于矿化后的复合膜,且银原子化的复合膜的抗污染性能更优于铜原子化后的复合膜,这是由于相比较于铜原子,银原子负载的量更多,且抗菌效果更好。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!