用于用嵌合抗原受体治疗T细胞恶性肿瘤的CAR-T细胞的基因编辑的制作方法
本申请要求2016年8月3日提交的美国临时申请序列号62/370,485、2017年4月6日提交的美国临时申请序列号62/482,570以及2017年5月12日提交的美国临时申请序列号62/505,614的权益并且与其相关,所述申请特此以引用的方式整体并入。
本申请总体上涉及t细胞疗法。具体地,本公开涉及工程化的嵌合抗原受体(car)-t细胞及其使用方法。所公开的组合物和方法尤其可用于治疗骨髓和淋巴恶性肿瘤。
序列表的引用
序列表的纸质副本和同一序列表的计算机可读形式在以下附加并且以引用的方式并入本文。根据37c.f.r.1.821(f),以计算机可读形式记录的信息与书面序列表相同。
背景技术:
t细胞可进行基因修饰以表达嵌合抗原受体(car),所述嵌合抗原受体是包含抗原识别部分和t细胞激活结构域的融合蛋白。嵌合抗原受体t细胞表现出针对b细胞恶性肿瘤的优异临床功效。然而,部分地由于恶性t细胞与效应t细胞之间靶抗原的共有表达,针对t细胞恶性肿瘤的car-t细胞疗法的发展已证明是有问题的。car-t细胞上靶抗原的表达可诱导car-t细胞的自相残杀和功效损失,并且还减少临床益处。因此,需要不诱导自相残杀但在治疗t细胞恶性肿瘤方面有效的car-t细胞。
技术实现要素:
一方面,本公开提供一种包含嵌合抗原受体的t细胞(car-t细胞),其中所述car-t细胞缺乏所述嵌合抗原受体特异性地结合的抗原,并且其中所述嵌合抗原受体特异性地结合恶性肿瘤或癌症上的表面表达的抗原。在各个方面,所述抗原可在恶性t细胞上表达。例如,所述抗原可以是cd7、cd5、cd2、cd30或cd4。所述car-t细胞还可包含自杀基因。可替代地或另外,所述car-t细胞可包含对内源性t细胞受体α链(trac)的修饰,使得t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断。
另一方面,本公开提供一种治疗患有恶性肿瘤的哺乳动物的方法,所述方法包括向所述哺乳动物施用多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含相同的嵌合抗原受体,其中所述car-t细胞缺乏所述嵌合抗原受体特异性地结合的抗原,并且其中所述嵌合抗原受体特异性地结合在所述受试者的恶性肿瘤或癌症上表达的抗原。在各个方面,所述抗原可在恶性t细胞上表达。例如,所述抗原可以是cd7、cd5、cd2、cd30或cd4。所述多个car-t细胞还可包含自杀基因。可替代地或另外,所述多个car-t细胞可包含对内源性t细胞受体α链(trac)的修饰,使得t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断。
另一方面,本公开提供一种在需要car-t细胞疗法的受试者中预防或减少移植物抗宿主病的方法,所述方法包括向所述哺乳动物施用多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含(a)相同的嵌合抗原受体和(b)自杀基因和/或修饰,使得t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断;其中所述car-t细胞缺乏所述嵌合抗原受体特异性地结合的抗原,并且其中所述嵌合抗原受体特异性地结合在所述受试者的恶性肿瘤或癌症上表达的抗原。在各个方面,所述抗原可在恶性t细胞上表达。例如,所述抗原可以是cd7、cd5、cd2、cd30或cd4。在另外的方面,所述受试者可能需要同种异体car-t细胞疗法。
另一方面,本公开提供一种在需要同种异体car-t细胞疗法的受试者中预防或减少同种异体反应性的方法,所述方法包括向所述哺乳动物施用多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含(a)相同的嵌合抗原受体和(b)自杀基因和/或对内源性t细胞受体α链(trac)的修饰,使得t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断;其中所述car-t细胞缺乏所述嵌合抗原受体特异性地结合的抗原,并且其中所述嵌合抗原受体特异性地结合在所述受试者的恶性肿瘤或癌症上表达的抗原。在各个方面,所述抗原可在恶性t细胞上表达。例如,所述抗原可以是cd7、cd5、cd2、cd30或cd4。
以下包括本公开的另外的方面和迭代。
附图说明
申请文件包含至少一幅以彩色绘制出的绘图。在提出请求并支付必要费用后,专利局将提供具有一幅或多幅彩色绘图的本专利申请公布的副本。
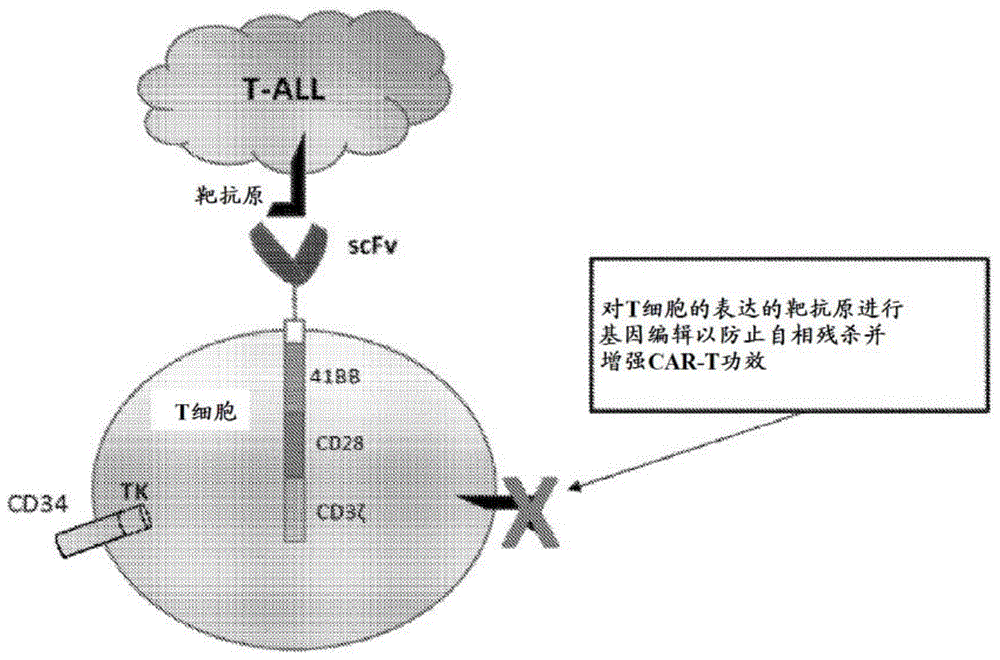
图1示出工程化的car-t细胞的示意图。
图2示出cd7基因座的基因编辑导致与野生型(wt)t细胞相比>90%的t细胞中cd7表达的损失。
图3a示出cd7基因编辑的cd7car-t(cd7δcart7)细胞在体外有效地杀死t细胞急性成淋巴细胞性白血病细胞的cd7阳性molt3细胞。
图3bcd7δcart7在体外有效地杀死t细胞急性成淋巴细胞性白血病细胞的cd7阳性hsb-2细胞。
图4a-图4d示出cd7δcart7在体内有效地杀死cd7阳性t细胞急性成淋巴细胞性白血病细胞。图4a示出治疗方案,图4b示出与对照相比cd7-car小鼠中肿瘤的减少,图4c示出与对照相比cd7car小鼠中从小鼠出去的总光子降低,并且图4d示出接受cd7car-t细胞的小鼠比用cd19car-t细胞处理的小鼠存活显著地更长。
图5a-图5j示出cart7诱导的自相残杀。图5a示出抗cd7-car构建体和抗cd19-car构建体的示意图,图5b示出在抗cd3/cd28珠的存在下在补充有50u/mlil-2和10ng/mlil15的xcyte培养基中培养的t细胞,图5c示出相比于cart19,cart7经历自相残杀并且在转导之后的数天内没有表现出稳健扩展,图5d示出cart7细胞向cd4表型倾斜,图5e示出作为相对于wt细胞的插入缺失的测序读数的百分比,靶向cd7的grna的编辑效率,图5f示出用于通过流式细胞术确定基因编辑效率的实验设计,图5g示出使用flowjov10分析的流式细胞术数据,图5h示出在用cd7g4进行基因编辑之后cd7+的细胞的百分比,图5i示出在用cd7g4进行crispr/cas9基因编辑之后cd7基因座的靶向深测序以检测非同源末端连接,并且图5j示出在用cd7g4进行crispr/cas9基因编辑之后原代t细胞的活力。
图6a-图6g示出cd7δcart7在体外和体内有效地杀死t-all细胞系。图6a示出基因编辑的car-t代的模式,图6b示出如使用具有viastaintm的nexcelom细胞计确定的细胞计数,图6c示出与cd7δcart19相比,用lenti-gfp转导的wtt细胞(gfp+t细胞)有效地通过cd7δcart7消除,图6d示出在molt-4、molt-3和hsb-2细胞中,相对于cd7δcart19,cd7δcart7有效地杀死cd7+t-all细胞系,图6e描绘用cd7δcart7或cd7δcart19处理注射有ccrf-cemcbr-gfp的nsg小鼠的实验设计,图6f示出相对于cd7δcart19处理的小鼠,cd7δcart7处理的小鼠具有显著延长的存活,并且图6g通过bli成像示出相对于cd7δcart19处理的小鼠,cd7δcart7处理的小鼠具有显著减小的肿瘤负荷。
图7a-图7g示出缺乏cd7和trac的ucart7细胞在体外有效地杀死t-all细胞系。图7a示出在抗cd3/cd28珠的存在下在补充有50u/mlil-2和10ng/mlil15的xcyte培养基中培养的t细胞的示意图(珠与细胞的比率,3:1)。在+2天激活后,将珠去除并且使用lonzanuceleofector4dtm、15μgspcas9mrna、20μgcd7grna和20μgtracgrna对4x106个t细胞进行电穿孔。根据制造商的说明书,在第+7天使用miltenyi抗cd3微珠使cd3+car-t消耗,并且额外培养两天,图7b-图7c示出多重基因编辑引起trac和cd7的高效率双缺失,如通过图7d.cd7和图7etrac基因座的facs和靶向深测序确定,图7f示出cd7δcart7和ucart7表现出稳健扩展,但是可能由于残余的非基因编辑的t细胞的自相残杀和基因编辑的细胞上持续的cd7表面表达两者而产生较少的细胞,并且图7g示出ucart7与cd7δcart7在体外杀死cd7+t-all细胞系的效率方面相等,甚至在低效应子与靶比率下也是如此。
图8a-图8b示出ucart7在体外杀死原代患者t-all胚细胞。将从具有cd7+t-all的三位个体患者获得的原代胚细胞用150nm羧基荧光素琥珀酰亚胺酯(cfse)进行标记。在进行facs分析之前,将标记的细胞与cd7δcart7、ucart7或其相应的cd19对照一式三份地在1:1比率下共孵育24小时。accucount荧光珠用于确定实际的细胞计数。使用gallios细胞仪采集数据。图8a示出代表性facs图,并且图8b示出相对于cd7δcar19和ucart19,cd7δcar7和ucart7有效地杀死t-all胚细胞。
图9a-图9f示出ucart7在体内杀死原代患者t-all胚细胞而没有诱导异种gvhd。图9a示出实验设计,图9b示出呈现来显示肿瘤和t细胞两者的血液分析的代表性流式细胞术图,图9c示出血液中总小鼠和人cd45细胞中的肿瘤细胞的百分比,图9d示出脾中总小鼠和人cd45细胞中的肿瘤细胞的百分比,图9e示出根据cooke分级的临床gvhd分数,并且图9f示出输注wtt细胞、tracδt细胞、ucart7和ucart19之后小鼠的代表性图像。
具体实施方式
在本公开的上下文中,当car-t细胞变成与靶向car-t细胞包含相同的嵌合抗原受体的另一种car-t细胞的靶标并被其杀死时,自相残杀发生,因为靶向car-t细胞表达被两种t细胞上的嵌合抗原受体特异性地识别的抗原。为了克服本领域中已知的此问题,本公开提供包含嵌合抗原受体的t细胞,其中所述t细胞缺乏所述嵌合抗原受体特异性地结合的抗原。因为本公开的car-t细胞缺乏t细胞的嵌合抗原受体特异性地结合的抗原,所以所述t细胞被称为是“抗自相残杀的”。本公开还涵盖对所述t细胞进行工程化的方法及其用途。
本发明的各个方面在以下章节中更详细地描述。
i.car-t细胞
本公开的一方面涵盖包含嵌合抗原受体的t细胞,其中所述t细胞缺乏所述嵌合抗原受体特异性地结合的抗原,即抗自相残杀的car-t细胞。
car-t细胞是表达嵌合抗原受体的t细胞。如本文所用并且通常在本领域中所用,短语“嵌合抗原受体(car)”是指重组融合蛋白,其具有与胞内结构域连接的抗原特异性胞外结构域,所述胞内结构域指导细胞在抗原结合到胞外结构域之后执行专门功能。术语“人工t细胞受体”、“嵌合t细胞受体”和“嵌合免疫受体”各自可在本文中与术语“嵌合抗原受体”互换使用。嵌合抗原受体由于其通过它们的胞内结构域结合mhc无关抗原并转导激活信号的能力而与其他抗原结合剂区分开。car的胞外部分和胞内部分在以下更详细地讨论。
嵌合抗原受体的抗原特异性胞外结构域识别并特异性地结合抗原,通常是恶性肿瘤的表面表达的抗原。例如,当抗原特异性胞外结构域以约0.1pm至约10μm、优选地约0.1pm至约1μm、更优选地约0.1pm至约100nm的亲和力常数或相互作用亲和力(kd)结合抗原时,所述抗原特异性胞外结构域特异性地结合抗原。用于确定相互作用亲和力的方法在本领域中是已知的。适用于本公开的car的抗原特异性胞外结构域可以是任何抗原结合多肽,各种各样的抗原结合多肽在本领域中是已知的。在一些情况下,所述抗原结合结构域是单链fv(scfv)。其他基于抗体的识别结构域(cabvhh(骆驼抗体可变结构域)及其人源化形式、ignarvh(鲨鱼抗体可变结构域)及其人源化形式、sdabvh(单结构域抗体可变结构域)和“骆驼化”抗体可变结构域适合使用。在一些情况下,基于t细胞受体(tcr)的识别结构域诸如单链tcr(sctv,含有v.α.v.β.的单链双结构域tcr)也适合使用。
合适的抗原可包括t细胞特异性抗原和/或对于t细胞不是特异性的抗原。在一个优选的实施方案中,由car-t细胞的嵌合抗原受体特异性地结合的抗原和car-t细胞缺乏的抗原是在恶性t细胞上表达的抗原,更优选地是与非恶性t细胞相比在恶性t细胞上过表达的抗原。“恶性t细胞”是源自t细胞恶性肿瘤的t细胞。术语“t细胞恶性肿瘤”是指广泛的高度异质分组的源自t细胞前体、成熟t细胞或自然杀伤细胞的恶性肿瘤。t细胞恶性肿瘤的非限制性实例包括t细胞急性成淋巴细胞性白血病/淋巴瘤(t-all)、t细胞大颗粒淋巴细胞(lgl)白血病、人t细胞白血病病毒1型阳性(htlv-1+)成人t细胞白血病/淋巴瘤(atl)、t细胞幼淋巴细胞白血病(t-pll)以及各种外周t细胞淋巴瘤(ptcl),包括但不限于血管免疫母细胞性t细胞淋巴瘤(aitl)、alk阳性间变性大细胞淋巴瘤和alk阴性间变性大细胞淋巴瘤。例如,通过非限制性实例的方式,cd7、cd5、cd2、cd30和cd4可以是在恶性t细胞上表达的合适抗原。在一个实施方案中,本公开的car-t细胞包含特异性地结合到cd7的嵌合抗原受体的胞外结构域。在另一个实施方案中,本公开的car-t细胞包含特异性地结合到cd5的嵌合抗原受体的胞外结构域。在又一个实施方案中,本公开的car-t细胞包含特异性地结合到cd2的嵌合抗原受体的胞外结构域。在再一个实施方案中,本公开的car-t细胞包含特异性地结合到cd30的嵌合抗原受体的胞外结构域。在再又一个实施方案中,本公开的car-t细胞包含特异性地结合到cd4的嵌合抗原受体的胞外结构域。
如上所述,在一个实施方案中,所述抗原是cd7。cd7是t细胞表面膜相关糖蛋白。cd7可在t细胞恶性肿瘤包括t细胞急性成淋巴细胞性白血病(t-all)和非霍奇金氏t细胞淋巴瘤(nhl)中过表达。本公开的car-t细胞可用于靶向过表达cd7的恶性t细胞。
本公开的嵌合抗原受体还包含胞内结构域,在抗原结合到抗原特异性胞外结构域之后,所述胞内结构域向t细胞提供胞内信号。本公开的嵌合抗原受体的胞内信号传导结构域负责激活表达嵌合受体的t细胞的效应子功能中的至少一种。术语“效应子功能”是指分化细胞的专门功能。t细胞的效应子功能例如可以是细胞溶解活性或辅助活性,包括细胞因子的分泌。幼稚t细胞、记忆t细胞或记忆型t细胞的效应子功能还可包括抗原依赖性增殖。因此,术语“胞内结构域”是指car的一部分,在抗原结合到胞外结构域之后,所述部分转导效应子功能信号并且指导t细胞执行专门功能。合适的胞内结构域的非限制性实例包括t细胞受体的ζ链或其同系物中的任一种(例如,η、δ、γ或ε)、mb1链、b29、fcriii、fcri以及信号传导分子(诸如cd3.ζ.和cd28、cd27、4-1bb、dap-10、ox40和它们的组合以及其他相似的分子和片段)的组合。可使用激活蛋白家族的其他成员的胞内信号传导部分,诸如fc.γ.riii和fc.ε.ri。虽然通常采用整个胞内结构域,但是在许多情况下,不需要使用整个胞内多肽。在胞内信号传导结构域的截短部分可使用的程度下,可使用此截短部分代替完整的链,只要所述截短部分仍然转导效应子功能信号即可。因此,术语胞内结构域旨在包括胞内结构域的足以转导效应子功能信号的任何截短部分。
通常,抗原特异性胞外结构域通过跨膜结构域连接到嵌合抗原受体的胞内结构域。跨膜结构域横贯细胞膜,将car锚固到t细胞表面,并且将胞外结构域连接到胞内信号传导结构域,从而影响car在t细胞表面上的表达。嵌合抗原受体还可进一步包含一个或多个共刺激结构域和/或一个或多个间隔子。共刺激结构域来源于共刺激蛋白的增强体内细胞因子产生、增殖、细胞毒性和/或持续性的胞内信号传导结构域。间隔子(i)将抗原特异性胞外结构域连接到跨膜结构域,(ii)将跨膜结构域连接到共刺激结构域,(iii)将共刺激结构域连接到胞内结构域,和/或(iv)将跨膜结构域连接到胞内结构域。例如,在抗原特异性胞外结构域与跨膜结构域之间包含间隔子结构域可影响抗原结合结构域的柔性并且因此影响car功能。合适的跨膜结构域、共刺激结构域和间隔子在本领域中是已知的。
本公开涵盖的car-t细胞缺乏嵌合抗原受体特异性地结合的抗原并且因此是抗自相残杀的。在一些实施方案中,t细胞的抗原被修饰,使得嵌合抗原受体不再特异性地结合修饰的抗原。例如,被嵌合抗原受体识别的抗原的表位可通过一个或多个氨基酸变化(例如,取代或缺失)来修饰,或者所述表位可从抗原缺失。在其他实施方案中,抗原的表达在t细胞中减少至少50%、至少60%、至少70%、至少80%、至少90%或更多。用于降低蛋白质的表达的方法在本领域中是已知的,并且包括但不限于修饰或替换可操作地连接到编码蛋白质的核酸序列的启动子。在再其他的实施方案中,t细胞被修饰,使得例如通过编码抗原的基因的缺失或破坏而不表达抗原。在以上实施方案中的每一个中,car-t细胞可缺乏嵌合抗原受体特异性地结合的一个或优选地全部抗原。将t细胞遗传修饰成缺乏抗原的方法在本领域中是熟知的,并且非限制性实例在以上提供。在一个示例性实施方案中,crispr/cas9基因编辑可用于将t细胞修饰成缺乏抗原,例如,如在实施例4-8的方法中所述。
本公开涵盖的car-t细胞还可缺乏内源性t细胞受体(tcr)信号传导。在各种实施方案中,可能希望减少或消除本文公开的car-t细胞中的内源性tcr信号传导。例如,当同种异体t细胞用于产生car-t细胞时,减少或消除car-t细胞中的内源性tcr信号传导可预防或减少移植物抗宿主病(gvhd)。用于减少或消除内源性tcr信号传导的方法在本领域中是已知的,并且包括但不限于修饰tcr受体的一部分(例如,tcr受体α链(trac)等)。trac修饰可阻断tcr介导的信号传导。因此,trac修饰可允许安全地使用同种异体t细胞作为car-t细胞的来源而不诱导危及生命的gvhd。
可替代地或另外,本公开涵盖的car-t细胞还可包含一种或多种自杀基因。如本文所用,“自杀基因”是指通过本领域中已知的标准方法引入到car-t细胞的核酸序列,当激活时,所述核酸序列引起car-t细胞的死亡。如果需要的话,自杀基因可促进在体内对car-t细胞的有效追踪和消除。通过激活自杀基因促进杀死可通过本领域中已知的方法发生。本领域中已知的合适的自杀基因疗法系统包括但不限于单纯疱疹病毒胸苷激酶(hsv-tk)/更昔洛韦(gcv)自杀基因疗法系统或诱导型半胱天冬酶9蛋白。在一个示例性实施方案中,自杀基因是cd34/胸苷激酶嵌合自杀基因。
在一个示例性实施方案中,本公开提供一种t细胞,其包含特异性地结合cd7的嵌合抗原受体,其中所述t细胞缺乏cd7(例如,cd7δcart7细胞)。在非限制性实例中,缺乏cd7由以下产生:(a)修饰由t细胞表达的cd7,使得嵌合抗原受体不再特异性地结合修饰的cd7,(b)修饰t细胞,使得抗原的表达在t细胞中减少至少50%、至少60%、至少70%、至少80%、至少90%或更多,或者(c)修饰t细胞,使得cd7不被表达(例如,通过编码cd7的基因的缺失或破坏)。在另外的实施方案中,t细胞包含自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在t细胞中被阻断。在非限制性实例中,在cd7δcart7细胞中表达的自杀基因编码在框内融合到人cd34cdna的胞外结构域和跨膜结构域的修饰的人-单纯疱疹病毒-1-胸苷激酶(tk)基因,并且引起阻断的tcr的修饰是对于内源性t细胞受体α链(trac)的修饰。
在另一个示例性实施方案中,本公开提供一种t细胞,其包含特异性地结合cd5的嵌合抗原受体,其中所述t细胞缺乏cd5(例如,cd5δcart5细胞)。在非限制性实例中,缺乏cd5由以下产生:(a)修饰由t细胞表达的cd5,使得嵌合抗原受体不再特异性地结合修饰的cd5,(b)修饰t细胞,使得抗原的表达在t细胞中减少至少50%、至少60%、至少70%、至少80%、至少90%或更多,或者(c)修饰t细胞,使得cd5不被表达(例如,通过编码cd5的基因的缺失或破坏)。在另外的实施方案中,t细胞包含自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在t细胞中被阻断。在非限制性实例中,在cd5δcart5细胞中表达的自杀基因编码在框内融合到人cd34cdna的胞外结构域和跨膜结构域的修饰的人-单纯疱疹病毒-1-胸苷激酶(tk)基因,并且引起阻断的tcr的修饰是对于内源性t细胞受体α链(trac)的修饰。
在另一个示例性实施方案中,本公开提供一种t细胞,其包含特异性地结合cd2的嵌合抗原受体,其中所述t细胞缺乏cd2(例如,cd2δcart2细胞)。在非限制性实例中,缺乏cd2由以下产生:(a)修饰由t细胞表达的cd2,使得嵌合抗原受体不再特异性地结合修饰的cd2,(b)修饰t细胞,使得抗原的表达在t细胞中减少至少50%、至少60%、至少70%、至少80%、至少90%或更多,或者(c)修饰t细胞,使得cd2不被表达(例如,通过编码cd2的基因的缺失或破坏)。在另外的实施方案中,t细胞包含自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在t细胞中被阻断。在非限制性实例中,在cd2δcart2细胞中表达的自杀基因编码在框内融合到人cd34cdna的胞外结构域和跨膜结构域的修饰的人-单纯疱疹病毒-1-胸苷激酶(tk)基因,并且引起阻断的tcr的修饰是对于内源性t细胞受体α链(trac)的修饰。
在另一个示例性实施方案中,本公开提供一种t细胞,其包含特异性地结合cd30的嵌合抗原受体,其中所述t细胞缺乏cd30(例如,cd30δcart30细胞)。在非限制性实例中,缺乏cd30由以下产生:(a)修饰由t细胞表达的cd30,使得嵌合抗原受体不再特异性地结合修饰的cd30,(b)修饰t细胞,使得抗原的表达在t细胞中减少至少50%、至少60%、至少70%、至少80%、至少90%或更多,或者(c)修饰t细胞,使得cd30不被表达(例如,通过编码cd30的基因的缺失或破坏)。在另外的实施方案中,t细胞包含自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在t细胞中被阻断。在非限制性实例中,在cd30δcart30细胞中表达的自杀基因编码在框内融合到人cd34cdna的胞外结构域和跨膜结构域的修饰的人-单纯疱疹病毒-1-胸苷激酶(tk)基因,并且引起阻断的tcr的修饰是对于内源性t细胞受体α链(trac)的修饰。
在另一个示例性实施方案中,本公开提供一种t细胞,其包含特异性地结合cd4的嵌合抗原受体,其中所述t细胞缺乏cd4(例如,cd4δcart4细胞)。在非限制性实例中,缺乏cd4由以下产生:(a)修饰由t细胞表达的cd4,使得嵌合抗原受体不再特异性地结合修饰的cd4,(b)修饰t细胞,使得抗原的表达在t细胞中减少至少50%、至少60%、至少70%、至少80%、至少90%或更多,或者(c)修饰t细胞,使得cd4不被表达(例如,通过编码cd4的基因的缺失或破坏)。在另外的实施方案中,t细胞包含自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在t细胞中被阻断。在非限制性实例中,在cd4δcart4细胞中表达的自杀基因编码在框内融合到人cd34cdna的胞外结构域和跨膜结构域的修饰的人-单纯疱疹病毒-1-胸苷激酶(tk)基因,并且引起阻断的tcr的修饰是对于内源性t细胞受体α链(trac)的修饰。
用于t细胞中的car设计、递送和表达的方法以及临床级car-t细胞群体的制造在本领域中是已知的。参见例如,lee等人,clin.cancerres.,2012,18(10):2780-90,其特此以引用的方式整体并入。例如,工程化的car可使用逆转录酶病毒引入到t细胞中,这将编码嵌合抗原受体的核酸序列有效并稳定地整合到靶细胞基因组中。用于病毒载体产生的一个示例性方法在实施例4-8的方法中描述。本领域中已知的其他方法包括但不限于慢病毒转导、基于转座子的系统、直接rna转染和crispr/cas系统(例如,使用合适的cas蛋白的i型、ii型或iii型系统,所述合适的cas蛋白诸如cas3、cas4、cas5、cas5e(或casd)、cas6、cas6e、cas6f、cas7、cas8a1、cas8a2、cas8b、cas8c、cas9、cas10、caslod、casf、casg、cash、csy1、csy2、csy3、cse1(或casa)、cse2(或casb)、cse3(或case)、cse4(或casc)、csc1、csc2、csa5、csn2、csm2、csm3、csm4、csm5、csm6、cmr1、cmr3、cmr4、cmr5、cmr6、csb1、csb2、csb3、csx17、csx14、csx10、csx16、csax、csx3、csz1、csx15、csf1、csf2、csf3、csf4和cu1966等)。
car-t细胞可由本领域已知的任何合适的t细胞来源生成,所述t细胞包括但不限于从受试者采集的t细胞。受试者可以是需要car-t细胞疗法的患有t细胞恶性肿瘤的患者或与需要car-t细胞疗法的患有t细胞恶性肿瘤的受试者相同种类的受试者。采集的t细胞可使用本领域中通常已知的方法进行离体扩展,之后用car转导以生成car-t细胞。
使用自体t细胞用于生成car-t细胞,虽然是可能的,但是可能呈现独特的挑战。需要car-t细胞疗法的受试者可能正在经历针对恶性肿瘤的治疗,并且此治疗可能已影响宿主的t细胞的数量和功能,从而减少可能有效地工程化为car-t细胞的t细胞的数量。另外,t细胞血液恶性肿瘤和正常t效应子可共表达许多相同表面抗原,从而使得非常难以将正常t效应子从恶性t细胞纯化以用于基因编辑和慢病毒转导。另外,如果纯化的过程不是完全的,则可能存在恶性t细胞中缺失靶抗原诸如cd7的风险,从而生成潜在地对自相残杀car-t细胞有抗性的污染t细胞癌症的群体。因此,为了避免有恶性t细胞污染正常效应t细胞的风险,使用来自患者的t细胞生成用于t细胞恶性肿瘤的car-t细胞可能不是希望的。
为了克服污染风险,来自没有t细胞恶性肿瘤的另一个受试者(供体受试者)的t细胞可用于生成同种异体疗法的car-t细胞。用于同种异体疗法的t细胞可从单个受试者或多个受试者采集。采集血细胞、分离并富集t细胞并且将它们离体扩展的方法可通过本领域中已知的方法进行。
在一个示例性实施方案中,cd7特异性cart细胞的car可通过以下生成:将商业合成的抗cd7单链可变片段(scfv)克隆到具有cd28和4-1bb内部信号传导结构域的第3代car骨架中。在添加p2a肽之后可添加胞外hcd34结构域,以便能够在病毒转导之后检测car并使用抗hcd34磁珠纯化。生成对于cd7特异性的car的一个示例性方法在实施例4-8的方法中描述。可按类似的方法制备对其他恶性t细胞抗原特异性的cars。
另一方面,可创建car-t细胞对照。对照car-t细胞可包含结合到不在恶性t细胞上表达的抗原的胞外结构域。对照car-t细胞对照结合的抗原可以是cd19。cd19是在b细胞上但不在t细胞上表达的抗原,所以具有适于结合到cd19的胞外结构域的car-t细胞将不结合到t细胞。这些car-t细胞可被称为cart19细胞,并且可用作对照以分析cart7细胞的结合效率和非特异性结合。
ii.使用car-t细胞的方法
另一方面,本公开提供一种杀死恶性t细胞的方法,所述方法包括将恶性t细胞与有效量的包含嵌合抗原受体的t细胞(car-t细胞)接触,其中所述car-t细胞缺乏所述嵌合抗原受体特异性地结合的抗原,并且其中所述嵌合抗原受体特异性地结合恶性细胞上表达的抗原。在各种实施方案中,所述恶性细胞是恶性t细胞。在另外的实施方案中,所述抗原是cd4、cd5、cd7、cd30或它们的任何组合。合适的car-t细胞在部分i中详细描述。在示例性实施方案中,car-t细胞可以是cd7δcart7细胞、cd5δcart5细胞、cd30δcart30细胞、cd4δcart4细胞或它们的任何组合。
将恶性细胞与有效量的car-t细胞接触通常涉及将car-t细胞和恶性细胞混合足以允许car-t细胞的嵌合抗原受体结合它的在恶性细胞的表面上的同源抗原一段时间。这可在体外或离体发生。如本文所用,术语“有效量”意指引起可测量的效果(例如,抗原依赖性细胞增殖、细胞因子分泌、细胞毒性杀死等)的量。有效量可通过使用本领域中已知的和/或实施例中更详细地描述的方法来确定。
另一方面,本公开提供一种用于治疗患有t细胞恶性肿瘤的受试者的方法。在一些实施方案中,所述t细胞恶性肿瘤是血液恶性肿瘤。在一些实施方案中,所述t细胞恶性肿瘤是t细胞急性成淋巴细胞性白血病(t-all)和t细胞非霍奇金淋巴瘤(t-nhl)。所述方法包括向受试者施用治疗有效量的多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含相同的嵌合抗原受体,其中所述car-t细胞缺乏由嵌合抗原受体特异性地识别的抗原,并且其中所述嵌合抗原受体特异性地结合在恶性t细胞上表达的抗原。在各种实施方案中,所述抗原可以是cd4、cd5、cd7、cd30或它们的任何组合。合适的受试者包括任何哺乳动物,优选地是人。合适的car-t细胞在部分i中详细描述。在示例性实施方案中,car-t细胞可以是cd7δcart7细胞、cd5δcart5细胞、cd30δcart30细胞、cd4δcart4细胞或它们的任何组合。所述方法可包括同种异体car-t细胞疗法或自体car-t细胞疗法,虽然同种异体car-t细胞疗法可出于在部分i中讨论的原因而是优选的。car-t细胞疗法可伴随其他疗法,包括但不限于免疫疗法、化学疗法或放射疗法。
另一方面,本公开提供一种用于治疗患有非t细胞骨髓或淋巴恶性肿瘤的受试者的方法。所述方法包括向受试者施用治疗有效量的多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含相同的嵌合抗原受体,其中所述car-t细胞缺乏由嵌合抗原受体特异性地识别的抗原,并且其中所述嵌合抗原受体特异性地结合在非t细胞骨髓或淋巴恶性肿瘤上表达的抗原。合适的受试者包括任何哺乳动物,优选地是人。合适的car-t细胞在部分i中详细描述。在一个示例性实施方案中,car-t细胞是cd7δcart7细胞。所述方法可包括同种异体car-t细胞疗法或自体car-t细胞疗法,虽然同种异体car-t细胞疗法可出于在部分i中讨论的原因而是优选的。car-t细胞疗法可伴随其他疗法,包括但不限于免疫疗法、化学疗法或放射疗法。
另一方面,本公开提供一种用于在需要car-t细胞疗法的受试者中预防或减少移植物抗宿主病的方法。在一些实施方案中,需要car-t细胞疗法的受试者是患有t细胞恶性肿瘤、非t细胞骨髓恶性肿瘤或淋巴恶性肿瘤的受试者。所述方法包括向受试者施用治疗有效量的多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含(a)相同的嵌合抗原受体和(b)自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断;其中所述car-t细胞缺乏由嵌合抗原受体特异性地识别的抗原,并且其中所述嵌合抗原受体特异性地结合在恶性肿瘤上表达的抗原。在各种实施方案中,所述恶性细胞是恶性t细胞。在另外的实施方案中,所述抗原是cd4、cd5、cd7、cd30或它们的任何组合。合适的受试者包括任何哺乳动物,优选地是人。所述方法可包括同种异体car-t细胞疗法或car-t细胞疗法,虽然同种异体car-t细胞疗法可出于在部分i中讨论的原因而是优选的。car-t细胞疗法可伴随其他疗法,包括但不限于免疫疗法、化学疗法或放射疗法。
另一方面,本公开提供一种用于在需要同种异体car-t细胞疗法的受试者中预防或减少同种异体反应性的方法。在一些实施方案中,需要同种异体car-t细胞疗法的受试者是患有t细胞恶性肿瘤、非t细胞骨髓恶性肿瘤或淋巴恶性肿瘤的受试者。所述方法包括向受试者施用治疗有效量的多个嵌合抗原受体t(car-t)细胞,每个car-t细胞包含(a)相同的嵌合抗原受体和(b)自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断;其中所述car-t细胞缺乏由嵌合抗原受体特异性地识别的抗原,并且其中所述嵌合抗原受体特异性地结合在恶性肿瘤上表达的抗原。在各种实施方案中,所述恶性细胞是恶性t细胞。在另外的实施方案中,所述抗原是cd4、cd5、cd7、cd30或它们的任何组合。合适的受试者包括任何哺乳动物,优选地是人。car-t细胞疗法可伴随其他疗法,包括但不限于免疫疗法、化学疗法或放射疗法。
在以上方面的各种实施方案中,所述多个car-t细胞可以是多个cd7δcart7细胞、多个cd5δcart5细胞、多个cd30δcart30细胞、多个cd4δcart4细胞或它们的任何组合。在另外的实施方案中,car-t细胞可包含自杀基因和/或修饰,使得内源性t细胞受体(tcr)介导的信号传导在car-t细胞中被阻断。
car-t细胞可通过静脉内途径(例如,通过静脉内输注)施用给受试者。car-t细胞可以单剂量或以多剂量施用。car-t细胞可在适用于静脉内施用的药物组合物中注射。用于iv施用的合适的药物组合物在本领域中是已知的。本公开的药物组合物还可包含另外的组分。例如,此类组分可用于维持注射的car-t细胞的活力和/或活性。在一个实施方案中,car-t细胞组合物可包含il-2以维持car-t细胞。
car-t细胞可以有效剂量施用。有效剂量可以是一个剂量或多个剂量,并且足以产生所需的治疗效果。car-t细胞的典型剂量的范围可以是约1x105-5x107个细胞/kg接受疗法的受试者的体重。有效剂量可基于恶性肿瘤的阶段、受试者的健康状况和恶性肿瘤的类型来计算。在施用多个剂量的情况下,此剂量和剂量之间的间隔可基于受试者对于疗法的反应来确定。
如本文所用的“有效剂量”或“治疗有效量”意指提供治疗性或预防性益处的量。
如本文所用的术语“治疗效果”是指可通过恶性细胞数量减少、预期寿命增加或与恶性病状相关联的各种生理症状改善等表现出来的生物效果。
实施例
包括以下实施例以证明本公开的各种实施方案。本领域技术人员应理解以下实施例中公开的技术表示由本发明人发现的在本发明实践中发挥良好作用的技术,并且因此可被认为构成本发明实践的优选模式。然而,根据本公开,本领域的技术人员应理解,在不脱离本发明的精神和范围的情况下可在已公开并仍获得类似或相似结果的特定实施方案中做出许多改变。
实施例介绍
t细胞恶性肿瘤表示在儿童和成年人中均具有高复发率和死亡率的一类毁灭性的血液癌症,对于儿童和成年人,当前没有有效疗法或靶向疗法。尽管有强化的多药剂化学疗法方案,但是患有t细胞急性成淋巴细胞性白血病(t-all)的少于50%的成年人和75%的儿童存活超过五年。对于在初始疗法之后出现复发的人,挽救化学疗法方案在20%-40%的病例中诱导缓解。同种异体干细胞移植(具有其相关联的风险和毒性)是唯一的治疗性疗法。
进行工程化以表达嵌合抗原受体(car)的t细胞是有希望的癌症免疫疗法。此类靶向疗法已显示在患有b细胞白血病和淋巴瘤的患者中诱导缓解和甚至长期无复发存活两者的巨大潜力。因此,针对t细胞恶性肿瘤的靶向疗法代表显著未满足的医疗需求。然而,若干挑战限制针对t细胞恶性肿瘤的car-t细胞的临床发展。首先,t效应细胞与t细胞恶性肿瘤之间的靶抗原的共有表达引起car-t细胞的自相残杀或自杀。第二,在没有被恶性细胞污染的情况下收获足够数量的自体t细胞最多在技术上是挑战性的且极其昂贵的。第三,当输注到免疫受损的hla匹配或失配的接受者中时,使用来自同种异体供体的基因修饰的car-t细胞可引起危及生命的移植物抗宿主病(gvhd)。
许多t细胞恶性肿瘤过表达cd7,从而为t细胞癌症的免疫疗法提供有吸引力的靶。然而,正常t细胞,包括用于对car-t进行工程化的那些t细胞,也表达cd7(>86%)。因此,cd7靶向的car-t细胞诱导t细胞自相残杀,从而限制治疗潜力。假设,使用crispr/cas9使cd7和t细胞受体α链(trac)缺失,同时还用靶向cd7的car转导这些相同的t细胞将在没有显著的效应t细胞自相残杀的情况下引起对恶性t细胞的有效靶向和杀死。trac缺失阻断tcr介导的信号传导,从而允许安全地使用同种异体t细胞作为car-t的来源而没有诱导危及生命的gvhd并且没有被cd7缺失的恶性细胞污染的风险,对cart7疗法有抗性。使用高效率crispr/cas9基因编辑,生成靶向cd7的cd7和trac缺失的car-t(ucart7)。这些ucart7细胞在体外和体内有效地杀死人t-all细胞系和来自患者的原代t-all而没有引起异种gvhd。因此,首次呈现使用car-t疗法有效地治疗t细胞恶性肿瘤的“现成”策略的临床前数据。
实施例1:car-t细胞的基因编辑
t细胞可进行遗传修饰以表达嵌合抗原受体(car),所述嵌合抗原受体是包含抗原识别部分和t细胞激活结构域的融合蛋白。car-t细胞表现出针对b细胞恶性肿瘤的优异临床功效。然而,部分地由于t细胞恶性肿瘤与效应t细胞之间共有的靶抗原表达,针对t细胞恶性肿瘤的car-t疗法的发展已证明是有问题的。car-t细胞上靶抗原的表达可诱导car-t的自相残杀和功效损失,并且还减少临床益处。通过car-t的基因编辑,证明通常在car-t(和t细胞恶性肿瘤)上表达的t细胞特异性靶抗原的有效缺失可引起car-t的有效扩展而没有显著的“自相残杀”,并且使用基因编辑的car-t细胞有效地杀死肿瘤靶。描述了t细胞产物的开发,所述t细胞产物使cd7双等位基因缺失,并且在已进行基因编辑以使cd7缺失的t细胞中过表达cd7-car(图1)。此方法可延伸以涵盖在各种t细胞癌症上并在正常t细胞上表达的其他t细胞抗原,诸如cd5、cd4和cd2。另外,将自杀基因并入在表达car的慢病毒和逆转录病毒构建体中将不仅用于防范插入突变和白血病形成两者,并且还防范长期t细胞和nk细胞血细胞减少。
实施例2:cd7基因座的基因编辑引起cd7表达的损失
粘着分子cd7在t-all(t急性成淋巴细胞性白血病;98%)和其他t细胞恶性肿瘤上高度表达,并且证明对于t细胞癌症的免疫疗法是有吸引力的靶标。然而,cd7在激活的t细胞上高度表达(>86%)。crispr/cas9用于使car-t细胞上的cd7表达缺失。设计靶向hcd7的向导rna(grna)并且通过华盛顿大学基因组工程化核心验证活性。商业合成具有最大活性的grna,从而并入修饰的碱基(2’ome和硫代磷酸酯)以增加grna稳定性。为了生成cd7car,使用商业基因合成创建抗cd7单链可变片段(scfv),并且克隆到具有cd28和4-1bb内部信号传导结构域的第3代car的骨架中(由dr.c.june,宾夕法尼亚大学提供)。通过p2a肽修饰构建体以表达人cd34(或cd34-tk75嵌合自杀基因;eissenberg等人moleculartherapy,2015)的细胞质截短突变体,以便能够在病毒转导之后检测car。使用抗cd3/cd28珠激活人原代t细胞,持续48小时,之后去除珠并且用cd7grna(20μg)和cas9mrna(15μg)进行电穿孔。然后将t细胞静置24小时。在第三天,用编码cd7-car、cd7-car-p2a-cd34、cd7-car-p2a-cd34-tk75或对照cd19-car的慢病毒颗粒转导t细胞,并且允许其扩展额外6天。通过流式细胞术确认转导效率和cd7消融。与对照t细胞相比,在>90%的t细胞中,cd7基因座的基因编辑引起cd7表达的损失(图2)。
实施例3:cd7car-t细胞在体外有效地杀死cd7阳性t-all细胞
测试基因编辑(cd7缺失)的cd7car-t细胞体外靶向cd7+t-all细胞系的能力。相比于cd19car-t细胞,cd7car-t细胞有效地杀死cd7+细胞系molt-3和hsb-2,如通过4小时铬释放测定所确定的(图3)。测试cd7缺失的cd7car-t在体内根除t-all的能力。将nsg小鼠进行亚致死辐射(200cgy),之后输注进行修饰以表达cbr(叩头虫红色)荧光素酶的5x105个molt-3t-all细胞。在第3天,静脉内注射单个剂量的car-t(3x106)。与接受cd19car-t细胞的小鼠相比,接受cd7car-t细胞的小鼠具有显著减小的肿瘤负荷,如通过生物发光成像所评估。此外,接受cd7car-t细胞的小鼠比用cd19car-t细胞处理的小鼠显著地存活更长(p=0.0018,图4)。这些数据支持t细胞过继性疗法与基因组编辑组合作为用于靶向t-all的有希望的疗法。
实施例4-8的方法
car设计
通过使用商业基因合成抗cd7单链可变片段(scfv)并克隆到具有cd28和4-1bb内部信号传导结构域的第3代car的骨架中来生成cd7-car。ef1αpelns慢病毒质粒由dr.carljune(宾夕法尼亚大学)惠赠。通过p2a肽修饰构建体以表达hcd34的胞外结构域,以便能够在病毒转导之后检测car并且使用抗hcd34磁珠纯化car-t(如果需要的话)。cart19用作非靶向对照。
病毒载体产生
为了产生慢病毒,根据制造商说明书,使用calphostm哺乳动物转染试剂盒(takara),用car慢病毒载体和包装质粒pmd.lg/prre、pmd.g、prsv.rev1,2转染lenti-x293t细胞系(takarabio,mountainview,ca)。转染后36小时收获病毒,进行过滤以去除细胞碎片,并且通过超速离心在25000rpm、4℃下离心90分钟(optimale-80k超速离心机,beckmancoulter,indianapolisi.n)。将病毒重悬浮在磷酸盐缓冲盐水中,在液氮中急速冷冻并以单一使用等分试样储存在-80℃下。
crispr/cas9基因编辑
设计向导rna,并且通过华盛顿大学基因组工程化和ipsc(seqidno:7–16)验证活性。在20μl溶液p3(程序ff-120)中,使用核转染剂4d(lonzanj)将编码grna(400ng,addgene43860)和spcas9(500ng,addgene43945)的质粒电穿孔到白血病细胞系k562中。
商业合成rna向导(trilinkbiotechnologiessandiego,ca),在grna的5’端和3’端的三个末端碱基处并入2′-o-甲基和3′硫代磷酸碱基以防止核酸酶活性。seqidno17-19是全长向导序列。酿脓链球菌cas9(spcas9)mrna(5mec,ψ)购自trilinkbiotechnologies。
基因编辑的car-t
在抗cd3/cd28珠的存在下在补充有50u/mlil-2和10ng/mlil-15的xcyte培养基中培养t细胞(珠与细胞的比率3:1)。激活之后第+2天,将珠去除,并且使用核转染剂4d、程序eo-115,用15μgspcas9(trilink,ca)和20μg的每种grna(trilink)在100μl缓冲液p3中对4x106个t细胞进行电穿孔。在第+3天,在聚凝胺(sigmaaldrich.stlouismo)(最终浓度6μg/ml)的存在下用car7或car19(对照)慢病毒颗粒转导细胞。在下游实验中使用之前,将细胞扩展额外6天。
靶向深测序
用引物,正向引物gcctgcgtgggatctacctgaggca[seqidno:1]和反向引物agctatctaggaggctgctgggggc[seqidno:2]对cd7基因座进行扩增。用正向引物tggggcaaagagggaaatga[seqidno:3]和反向引物r_gtcagatttgttgctccaggc[seqidno:4]对trac基因座进行扩增。使用illumiamiseq平台(sandiego,ca)对pcr产物进行测序。编辑效率确定为具有插入缺失的测序读数与从wt细胞获得的读数对齐的百分比。
细胞系
cd7阳性t-all细胞系、molt-3(acc84)、molt-4(acc362)、hsb-2(acc435)和ccrf-cem(acc240)直接从微生物和细胞培养物的dsmz-german采集物(leibniz,germany)获得。对细胞系进行支原体测试并通过dsmz表征。用ef1αcbr-gfp慢病毒转导ccrf-cem细胞。对gfp阳性细胞进行分选和克隆以建立ccrf-cemcbr-gfp细胞系。
铬释放测定
在补充有5%胎牛血清的rpmi中,在25:1至0.25:1范围内的效应子:靶[e:t]比率下,将car-t与molt-3、molt4、hsb2或ccrf细胞系(总计4x104个细胞/孔)一起培养。如先前所述进行铬-51释放测定。
体外原代t-all杀死测定。
来自经过同意的患者的原代t-all从siteman癌症中心(irb#201108251)获得。从所有受试者获得知情同意书。用150nm羧基荧光素琥珀酰亚胺基酯(cfse)(sigmaaldrich,mo)标记原代细胞,以便能够在t-all胚细胞与car-t之间进行区分。在1:1比率下将标记的细胞与cd7δcart7、ucart7或其相应的cd19对照共孵育24小时,之后进行facs分析。使用7-氨基放线菌素d和spheroaccucount荧光团(spherotechinc.,lakeforest,il,usa)通过流式细胞术对活的靶细胞的绝对细胞计数进行定量。使用flowjov10分析数据。
自相残杀测定
在抗cd3/cd28珠的存在下在补充有50u/mlil-2和10ng/mlil15的xcyte培养基中培养wtt细胞(珠与细胞的比率3:1)。在48小时之后将珠去除,并且用慢病毒颗粒转导t细胞以表达gfp。转导之后七十二小时,使用流式细胞术针对gfp分选t细胞,并且在补充有50u/mlil-2和10ng/mlil-15、50ng/mlscf、10ng/mlil-7和20ng/mlfl3tl的xcyte培养基中在1:1的比率下与cd7δcart7或cd7δcart19共孵育24小时。将gfp+细胞百分比计算为总活细胞的百分比,使用7-氨基放线菌素d通过流式细胞术进行定量。
t细胞表型分析
将培养的t细胞在pbs/0.1%bsa中洗涤,并且在补充有4%大鼠血清的50ulbrilliant缓冲液(bdbiosciences)中在1x106个细胞下在4c下重悬浮15分钟。然后使用以下抗体荧光团缀合物(全部来自bdbiosciences,除非另外说明),将细胞在4℃下在100μl的brilliant缓冲液中孵育30分钟:cd7bv421、cd4bv510、ccr4bv605(biolegend)、cd8bv650、cd196bv786(biolegend)、cd3af488、cd45rapercpcy5.5、cd183pe、cd197pe-cf594、cd185pe-cy7(biolegend)和ccr10apc(r&dsystems)。荧光团缀合抗体的全部细节在表1中列出。然后将细胞在pbs/0.1%bsa中洗涤两次,并且在ze5(yeti)细胞仪(biorad/propellabs)上获取数据。使用荧光减一(fmo)对照在flowjov10(treestar)上进行补偿和分析。在bonferroni事后校正的情况下,使用双向anova在graphpadprism7上进行统计分析。
表1.用于流式细胞术的缀合抗体的细节。
动物模型
动物方案遵循动物研究医学委员会的华盛顿大学学院的法规。在全部小鼠实验中使用六至十周大的nod.cg-prkdcscidii2rgtm1wjl/szj(nsg)。在全部实验中使用雄性和雌性小鼠两者,并且随机分配到处理组。
ccrf-cem异种移植物模型
使用t-all细胞系ccrf-cem(进行修饰以过表达gfp和叩头虫红色荧光素酶(cbr))在体内测试cd7δcar7的抗白血病效果。在第0天,将1x105个ccrf-cemcbr-gfp注射到nsg小鼠的尾静脉中。使用雄性和雌性小鼠两者。在第1天将car-t(2x106)注射到接受ccrf-cemcbr-gfp细胞的小鼠中。为了追踪体内ccrf-cemcbr-gfp肿瘤生长,将小鼠腹膜内注射50μg/gd-荧光素(biosynth,itasca,il,usa)并进行成像。统计考虑:使用对数秩(mantel-cox)测试来确定存活的显著差异。使用用于重复测量数据的双向anova,接着进行用于多重比较的递减bonferroni调整,确定肿瘤负荷(如通过bli成像限定)的统计分析。
来自患者的异种移植物模型
t-allpdxdfci12从异种移植物的公共储存库(proxe).www.proxe.org获得。在第0天将nsg移植有1x106个pdxdfci12细胞,接着在第+1天输注2x106个ucart7、ucart19、tracδ或wtt。在一周之后通过流式细胞术分析外周血和脾。使用红细胞裂解缓冲液(sigma-aldrich)使红细胞裂解,并且用冰冷的pbs洗涤。通过以下针对流式细胞术制备样品:将细胞重悬浮在染色缓冲液(补充有0.5%牛血清白蛋白和2mmedta的pbs)中,并且在4℃下与以下荧光染料标记的单克隆抗体的预滴定饱和稀释液一起孵育30分钟;cd34-pe、cd7-bv421、cd4-apc、cd8-pecy7、mcd45-bv510和hcd45-apc-h7。抗体的全部细节可见于表1。除非另外声明,否则抗体购自bdbiosciences。使用flowjov10分析数据。
脱靶分析
dsodn的基因组插入
通过使两种修饰的寡核苷酸(integrateddnatechnologies,ia)退火制备平双链寡脱氧核苷酸双链寡核苷酸(dsodn)。
反向_5phos/a*t*accgttattaacatatgacaactcaattaa*a*c(seqidno:5)和正向_/5phos/g*t*ttaattgagttgtcatatgttaataacggt*a*t*(seqidno:6)代表硫代磷酸酯键并且5phos代表5’磷酸化。原代t细胞的crispr/cas9基因编辑如先前所述,但是添加100pmoldsodn来进行。将细胞额外培养7天,之后收获并进行dna提取(dnaeasyqiagengmbh,germany)。
dsodn捕获
小的离散基因组基因座的杂交体捕获可证明在没有一定的诱饵设计和方案修改的情况下是困难的。利用与插入的dsodn序列互补的修饰xgen锁定探针富集含有34bpdnadsodn片段。将xgen锁定探针设计成以竞争性杂交方式参与以使杂交体下拉效率最小化。新型设计由多个探针组成,多个探针以2碱基偏移设计询问标签区域。另外,修饰的xgen锁定探针设计成增强靶序列结合到近似熔融温度的标准120ntdnaxgen锁定探针。对链霉亲合素/生物素介导的下拉机制进行修改以增大含有34bp标签的gdna的富集亚组的结果。
在靶向250bp插入物的sciclonengs仪器(perkinelmer)上利用kapahtp文库试剂盒(kapabiosystems)构建具有250ng的基因组dna的自动双重索引文库。双重索引kapa文库引物序列是seqidno:20–seqidno:54。使文库富集,持续八个pcr循环。对十六个文库进行集合预捕获,从而生成5μg文库集合体。将文库集合体与定制组的xgen锁定探针(idt)杂交,从而靶向34bpodn序列。通过qpcr(kapabiosystems)准确地确定捕获的文库集合体的浓度,以便产生适用于illuminahiseq4000平台的集群计数。2x150序列数据生成平均3.5gb的数据/样品。
guide-seq
利用针对靶向捕获实验构建的16个现有双重索引kapa文库用于guide-seq扩增。pcr反应设置有20ng的现有文库/样品、kapahifihotstartreadymix(kapabiosystems)和10μm引物。pcr条件如下:用于文库生成的pcr循环参数
用于50μl反应的混合物
25μlkapahifimastermix
1.0μl10μmp5
1.5μl10μmgsip
xμl20ng的现有双重索引文库
yμl无核酸酶的水
循环条件
95℃,持续5分钟,
15个循环的[95℃,持续30s,70℃(-1℃/循环),持续2分钟,72℃,持续30s]
10个循环的[95℃,持续30s,55℃,持续1分钟,72℃,持续30s]
72℃,持续5分钟
保持4℃
guide-seq索引引物(gsip)设计成靶向有义和反义guide-seqodn序列,同时并入p7移植序列和8bp样品指数。
通过qpcr(kapabiosystems)准确地定量三十二个扩增子文库(16个有义和16个反义),以便产生适用于illuminahiseq4000平台的集群计数。将扩增子文库归一化并集合在一起。将扩增子文库集合体和靶向捕获集合体以等摩尔浓度组合,之后生成一个通道的hiseq40002x150序列数据(illumina)。
数据分析
使用bwamemv0.7.10将序列与参考基因组(构建grch37-lite)对齐。使用guide-seq组件的修改形式来识别10-bp滑动窗口,其中靶序列以至少10个支持读数存在。为了表征脱靶对齐,检索侧接断点的两侧的35bp的参考序列并且与观察的序列对齐。需要在待保持的正向和反向方向上均具有至少一个支持读数的位点。去除也在对照样品中的一个中识别到的任何位点。密码可用性:修改的guide-seq密码在github.com/chrisamiller/guideseq处可用。
实施例4:cd7-car-t细胞诱导明显的自相残杀。
为了生成cd7-car-t(cart7),商业合成抗cd7单链可变片段(scfv),并且克隆到具有cd28和4-1bb内部信号传导结构域的第3代car骨架中。在p2a肽之后添加hcd34的胞外结构域,以便能够在病毒转导之后检测car并使用抗hcd34磁珠纯化(图5a)。靶向cd19的car-t(cart19)用作非相关car-t对照。在t细胞的转导之后,存在比cart19显著更少的cart7(图5c)。另外,当与cart19相比时,cart7偏向cd4表型(图5d)。
实施例5:通过crispr/cas9使cd7缺失
为了防止自相残杀,使用crispr/cas9基因编辑使cd7在car-t中缺失。设计十个靶向cd7的向导rna(grna),并且验证活性(seqidno:7-16)。将编码grna和cas9的质粒电穿孔到k562白血病细胞系中。cd7g4和cd7g10具有最高的基因编辑效率(如通过跨cd7基因座进行的靶向深测序所确定(图5e)),并且被选择用于进一步研究。商业合成cd7g4和cd7g10向导,在grna的5’和3’的三个末端碱基处并入2′-o-甲基和3′硫代磷酸碱基以防止核酸酶活性15。测试人原代t细胞中通过cd7g4和cd7g10两者进行的基因编辑的效率。用grna和cas9mrna对激活的t细胞进行电穿孔(图5f),并且然后在第+7天通过流式细胞术进行分析。cd7g4在使cd7表达缺失方面是最有效的,将cd7+t细胞的百分比从98.8%±0.17降低至9.1%±1.74(图5g至图5h)。通过靶向深测序确认cd7基因座的有效破坏,其中在89.14%的cd7序列读数中观察到插入缺失(图5i)。电穿孔之后24小时观察到仅最小的活力损失(图5j)。因为cd7g4在使cd7在t细胞中的表达缺失方面是最有效的,所以所有未来实验使用cd7g4执行。
实施例6:cd7δcart7在体外和体内防止自相残杀并且有效地杀死t-all。
cd7的crispr/cas9基因编辑,之后用cart7构建体转导cd7编辑的t细胞(cd7δ)如图5a所示进行,以生成cd7δcart7。在第一天用spcas9mrna(15μg)和cd7g4(20μg)对激活的t细胞进行电穿孔,之后在第3天用car7或car19对照进行病毒转导。将细胞培养额外6天(图6a)。预期在基因编辑之后由残余cd7表面表达引起的低水平的自相残杀,并且这通过相对于cd7δcart19的cd7δcart7产率的适度降低(在6天内7.5倍相对于12.6倍扩展,图6b)确认。用gfp转导的自体t细胞被cd7δcart7有效地杀死,但是不被cd7δcart19有效地杀死,从而确认当car-t靶向cd7时需要cd7缺失(图6c)。最后,相比于cd7δcart19,cd7δcart7有效地杀死cd7+t-all细胞系molt-4(70%cd7+)、molt-3(96%cd7+)和hsb-2(99%cd7+),如通过4小时cr释放测定所确定(图6d)。为了评估t-all的异种模型中cd7δcart7的活性,将1x105个叩头虫红色荧光素酶(cbr)标记的ccrf-cemt-all(通过facs,99%cd7+)细胞静脉内注射到nsg接受者中,之后在第+1天输注2x10x6个cd7δcart7或非靶向cd7δcart19对照细胞(图6e)。相比于接受cd7δcart19的小鼠或仅注射有肿瘤的小鼠,接受cd7δcart7的小鼠具有显著延长的存活(图6f)和减小的肿瘤负荷,如通过生物发光成像(bli)所确定(图6g)。为了评估cd7δcart7针对患者原代t-all细胞的功效,针对来自患者的异种移植物测试car-t。然而,t-all胚细胞仅可在仅接受肿瘤的小鼠中检测,并且在接受cd7δcart7或cd7δcart19或非操纵t细胞的小鼠中消除,从而表明cd7δcart7与非转导人t细胞和cd7δcart19两者维持在nsg小鼠中相似的体内同种异体反应性水平。
实施例7:trac和cd7在cart7中的双缺失防止自相残杀、gvhd,并且维持稳健的cd7指导的t-all杀死。
为了克服限制非自身t细胞的使用的同种异体反应性障碍,由于致死gvhd的风险,生成cd7和t细胞受体α链(trac)两者均在基因上缺失的car-t。靶向trac的grna序列从osborn等人获得。使用抗cd3/cd28珠激活t细胞两天,之后去除珠,并且用20μg的cd7g4、20μg的tracg和15μg的cas9mrna进行电穿孔(图7a)。多重crispr/cas9基因编辑在72.8%±1.92的细胞中引起cd7和trac的同时缺失,如通过facs分析所确定(图7b-e)。
为了与本领域中最近的命名保持一致,cd7δtracδcart7被称为通用cart7或ucart717。ucart7与cd7δcart7在体外杀死t-all细胞系方面一样有效。当与cd7δcart7相比时,ucart7不具有增殖缺陷,然而,如在cd7δcart7的情况下观察到的,ucart7引起相对于cd19对照cart的适度降低的car-t增殖和产率(图7f)。因为trac的不完全基因编辑留下残余的潜在同种异体反应性cd3+car-t,所以这些通过在第+8天使用抗cd3磁珠进行阴性选择来缺失。ucart7和cd7δcart7两者均以相等高的效率在体外杀死cd7+t-all细胞系molt3、ccrf-cem和hsb-2,从而证明在cd7和trac的双缺失之后没有功效损失(图7g)。有趣的是,当与cd7δcart19相比时,通过ucart19进行的非特异性杀死在高效应子与靶标(e:t)比率下削弱,从而表明在trac缺失之后同种异体反应性的损失。
接下来测试ucart7在体外杀死原代t-all胚细胞的能力。由于在原代t-all与car-t之间抗原表达的相似性,将原代t-all细胞用cfse标记以清楚地将t-all与car-t区分开。将t-all细胞在1:1的比率下与car-t一起孵育24小时。相对于相应的cd19对照car-t,cd7δcart7和ucart7两者均杀死跨全部三种原代样品的平均95%的t-all胚细胞(图8a),从而证明针对体外人原代t-all的优异的功效。
按照当wtt细胞、cd7δcart和cd7δcart19输注到携带原代t-allpdx的nsg小鼠中时的抗肿瘤活性,测试ucart7在没有同种异体反应性移植物抗白血病效应(gvl)或异种gvhd的情况下在体内杀死原代t-all的能力(图9a)。当与wtt细胞的接受者比较时,进行编辑以使trac缺失(tracδ)的t细胞的接受者在血液(图9b-c)和脾(图9d)两者中均表现出高肿瘤负荷(第+48天,脾p<0.0001,血液p=0.0001)。此外,在wtt细胞的接受者中观察到同种异体反应性t细胞的大量扩展(图9b)和严重的gvhd(平均临床gvhd分数=5.66,p<0.0001,图9e)。相比之下,在接受tracδt细胞的小鼠中,gvhd完全不存在并且t细胞不可检测(p<0.0001,图9e、f)。与接受ucart19的小鼠(其中t-all组成这些小鼠中的总cd45+细胞的>56%)相比,t-all胚细胞在接受ucart7的小鼠的外周血中不存在(p<0.0001),与在仅pdx的对照中观察到的高肿瘤负荷相似(图9c)。在脾中观察到一致的结果(图9d,ucart7<3%t-all相对于ucart19=85.87%t-all;p<0.0001),其中tracδt细胞、ucar19和仅pdx接受者小鼠表现出脾肿大。形成鲜明对比的是,ucart7接受者具有正常大小的脾。此外,与ucart19不同,ucart7可在6周注射后检测到,如通过hcd34表位所检测(图9b),从而证明体内ucart7的持续性。
实施例8:脱靶核酸酶活性
用crispr/cas9进行的高效率基因编辑可诱导不合需要的脱靶基因变化,所述变化可能对于这些t细胞的生物学具有潜在有害的效应,并且随后对于接受ucart7输注的接受者具有潜在有害的效应。使用评估人原代t细胞中的脱靶基因变化的两种不同的技术,所述两种不同的技术均依赖于在dna双链断裂处插入小双链寡脱氧核苷酸(dsodn)。第一方案利用修改形式的guide-seq,不包括条码化的索引,以便使用pcr扩增围绕插入的dsodn的靶位点;第二技术使用整合dna技术(idt)捕获探针以便富集含有靶dsodn序列的基因座。两种技术均使用下一代测序来识别插入的dsodn的基因座。为了确保识别脱靶核酸酶活性的善意位点,一式三份地进行每种条件(cd7g4、tracg和组合的cd7g+tracg),从而生成平均1.26x106个(平均值)测序读数/平行测定。分析中包括具有双向测序读数>10x覆盖的基因座。首先,评估每种技术识别中靶位点的能力。guide-seq和dsodn两者均稳健地捕获中靶活性的位点,其中每个中靶位点在每种条件中生成跨全部三个平行测定的在300x与22,000x之间的覆盖(表2和表3)。接下来,评估使用相同严格性的脱靶核酸酶活性。guide-seq揭示多重编辑的t细胞(cd7g4+tracg)中单个脱靶位点,rbm33中内含子插入存在于全部三个平行测定中。当通过dsodn捕获评估时,没有观察到脱靶位点,尽管具有高覆盖的中靶活性(表2)。在放宽guide-seq分析的严格性以包括两个或更多个平行测定中存在的潜在脱靶位点时,识别到cd7的额外四个潜在脱靶位点,trac的四个位点,以及多重cd7和trac基因编辑的潜在脱靶活性的额外一个位点(表2)。通过guide-seq识别的基因座在使用dsodn捕获获得的数据中不存在,也不是在降低的严格性的情况下这些数据的分析之后识别的脱靶核酸酶活性的额外位点。这些结果表明用cd7g4和tracgrna进行的原代t细胞的高效率crispr/cas9基因编辑(单独的或组合的)与使用这些平台的显著脱靶活性不相关。
表2:使用guide-seq识别的核酸酶活性的位点。中靶活性以深灰色突出显示。
表3:使用dsodn探针捕获识别的核酸酶活性的位点。中靶活性以深灰色突出显示。
实施例4-8的讨论
在此报告中,展示缺乏cd7和trac两者的crispr/cas9基因编辑的人t细胞的生成。用cd7-car(ucart7)进行的这些基因编辑的t细胞的慢病毒转导允许在没有随之发生的自相残杀或t细胞介导的异种gvhd的情况下在体外和体内有效地杀死cd7+原代人t-all和t-all细胞系。此工作改善诱导部分自相残杀的靶向t细胞恶性肿瘤的先前car-t。pinz等人描述cd4靶向的car-t的临床前发展,其维持cd4+靶标的cd8+car-t介导的细胞毒性,从而引起cd4+t细胞的完全丧失。同样,mamonkin等人描述仅诱导瞬时自相残杀的cd5靶向的car-t,其允许足够的cd5car-t扩展,尽管在激活的wtt细胞上具有基本上普遍的cd5扩展。
由于在大部分nhl和t-all中的高表达,cd7被选择作为靶标。除t细胞恶性肿瘤之外,cd7在~24%的aml中表达,并被认为是白血病干细胞的标记物,因此cart7可用于治疗骨髓以及淋巴恶性肿瘤。此外,需要靶向可在不影响免疫功能的情况下在t细胞中缺失的抗原。具有cd7的基因缺失的小鼠在表型上是正常的(具有正常的寿命),具有正常的淋巴细胞群,并且响应于同种异体和有丝分裂刺激维持t细胞活性。因此,cd7是用于car-t的基因编辑以靶向aml和t细胞恶性肿瘤两者的理想候选。
当在没有cd7缺失的情况下使用cart7时存在广泛的自相残杀,其中存活的cart7主要是cd4+和cd7-24。这些数据突显使用cart7的重要性,所述cart7本身缺乏cd7。此类细胞提供对于自相残杀的最佳抗性,同时允许在cd4细胞的平衡扩展的情况下细胞毒性cd8t细胞的扩展。确实,高效率crispr/cas9介导的cd7基因缺失减轻car7自相残杀,并且在cd7蛋白损失(其可在基因缺失之后)后,细胞展示完全自相残杀抗性。
使用自体t细胞用于生成cart7呈现独特的挑战。首先,患有复发的t-all和t-nhl的患者通常用t细胞毒(诸如嘌呤核苷类似物(氟达拉滨、克拉屈滨、奈拉滨)和t细胞毒性单克隆抗体(坎帕斯))进行高度预治疗。因此,t细胞的数量和功能可能明显地降低,从而限制用于治疗益处的cart7的有效生成、足够数量和功能。第二,大部分t细胞血液恶性肿瘤和正常t效应子共表达许多相同表面抗原,从而使得非常难以将正常t效应子从恶性t细胞纯化以用于基因编辑和慢病毒转导。如果纯化的过程不是完全的,则将存在恶性t细胞中缺失cd7的风险,从而生成潜在地对ucart7有抗性的污染t细胞癌症的群体。因此,有恶性t细胞潜在地污染正常效应t细胞的风险排除使用来自患者的t细胞来生成t细胞恶性肿瘤的car-t细胞。因此,通过编辑删除trac来进一步修饰cd7δcart7,从而允许在没有诱导gvhd的风险的情况下使用同种异体供体t细胞。在ucart19(从同种异体供体t细胞生成的针对cd19的trac编辑的非同种异体反应性car-t)的人试验中首次成功之后,发展了ucart7,其中通过多重crispr/cas9基因编辑以高效率并在最小脱靶效应的情况下成功地使cd7和trac两者缺失。ucart7在体外杀死t-all细胞系和原代患者t-all,与cd7δcart7一样有效。与cd7δcart7(其展示针对体内t-allpdx的同种异体反应性抗白血病活性)不同,ucart7在不诱导gvhd的情况下独立于同种异体反应性展示稳健的car7介导的杀死。这表明trac缺失不改变car介导的细胞毒性,同时完全预防gvhd。
除t细胞之外,nk细胞还表达cd7。ucart7介导的t细胞和nk细胞杀死可潜在地预防或限制同种异体ucart7排斥,并且增加ucart7持续性。尽管在免疫缺陷的pdxt模型中观察到ucart7持续性,仍然需要临床研究来完全表征在针对t细胞赘生物治疗的人中的ucart7持续性。
虽然在cd7、trac或多重基因编辑之后未观察到稳健的脱靶核酸酶活性,但是高保真度cas9(spcas9-hf1)的最近发展可进一步降低不合需要的基因事件的风险。此外,将car直接插入到trac基因座(如最近所报告)或潜在地cd7基因座中将进一步减少由于病毒载体随机整合到不合需要的基因座中而导致的致癌性转化的风险。此外,载体允许包含自杀基因诸如cd34-tk,其先前在首次人体研究中显示允许使用[18f]fhbgpet-ct成像有效地追踪基因修饰的t细胞和在体内消除t细胞两者。此策略将预防由crispr/cas9基因编辑和病毒整合引起的潜在毒性或致癌性转化。
此研究呈现针对t细胞恶性肿瘤的首次临床上可行的过继性t细胞基因疗法。具体地,显示进行人t细胞的cd7xtrac多重基因编辑,接着用第三代cd7-car进行慢病毒转导,产生ucart7,其对于自相残杀是完全有抗性的并且不表现出同种异体反应性或体内gvhd可能性。这将允许使用“现成”无肿瘤同种异体t细胞作为car-t的来源。在携带人t-all细胞系或原代人t-allpdx肿瘤的nsg小鼠中使用这些基因修饰的t细胞引起这些体内肿瘤的快速且有效消除,没有异种gvhd迹象。这些发现使得有必要进一步努力将这些观察转化为具体地用于治疗患有复发的和难治性的t细胞血液恶性肿瘤的儿童和成年人的临床学。
- 还没有人留言评论。精彩留言会获得点赞!