一种吡啶钌配合物或其晶体及制备方法与流程
本发明提供了一种过渡金属配合物,更特别地提供了一种吡啶钌配合物或其晶体及制备方法,属于席夫碱配合物技术领域。
背景技术:
席夫碱主要是指含有甲亚胺或亚胺特性基团(-RC=N-)的一类有机化合物,通常席夫碱是由胺和活性羰基缩合而成。
席夫碱类化合物及其金属配合物在医学、分析化学、催化、腐蚀以及光致变色领域的重要应用。具体而言:在医学领域:席夫碱具有抑菌、杀菌、抗肿瘤、抗病毒的生物活性。在分析领域:席夫碱作为良好的配体,可以用来鉴别,鉴定金属离子和定量分析金属离子的含量。在催化领域:席夫碱的钴和镍配合物已经作为催化剂使用。在腐蚀领域:某些芳香族的席夫碱经常作为铜的缓蚀剂。在光致变色领域:某些含有特性基团(-NO2等)的席夫碱也具有独特的应用。
也正是由于席夫碱如此重要的应用价值和研究前景,人们对新型的席夫碱及其合成方法进行了大量的深入研究,并取得了诸多成果,例如:
CN103408597A中公开了多种具有抗癌药物活性的席夫碱钌配合物,其结构式如下:
CN102304063A公开了一种席夫碱配体,其结构式如下:
该配体可与铜离子、锌离子配位从而得到两种金属配合物,该席夫碱金属配合物具有良好的大肠杆菌、金黄色葡萄球菌和欧文氏草生杆菌的抗菌作用和效果。
CN102584871A公开了一种稀土离子与水杨醛缩甘氨酸席夫碱和联吡啶的三元配合物,其中所述水杨醛缩甘氨酸席夫碱的结构式如下:
该三元配合物可用来抑制大肠杆菌,效果优异。
CN102746340A公开了一种氨基苯并噻唑席夫碱铋配合物,其结构式如下:
该配合物具有良好的金黄色葡萄球菌、枯草芽孢杆菌、大肠杆菌抑制效果。
CN103467397A公开了一种下式的结构复杂的双核铂(II)席夫碱配合物,该配合物可用来制备抗肿瘤药物,在医药领域具有良好的应用前景和生产价值,
CN104910212A公开了一种具有优异抗肿瘤性能的席夫碱铂配合物,其结构式如下:
如上所述,现有技术中公开了具有药物活性的多种席夫碱配合物,但对应新型的具有生理活性潜能的席夫碱配合物,仍存在继续研究的必要和需求,这正是目前该领域中的研究热点和重点,更是本发明得以完成的基础和动力所在。
技术实现要素:
为了研发新型的具有药物活性潜能的席夫碱配合物,本发明人进行了深入的研究,在付出了大量的创造性劳动后,从而完成了本发明。
具体而言,作为本发明的第一方面,涉及下式(5)所示的吡啶钌配合物或其晶体(即晶体配合物):
其中,R1、R2各自独立地选择H或C1-6烷基。
在本发明的所述吡啶钌配合物或其晶体中,Ru和所连苯环上的六个碳形成了一个大π键,从而形成络合结构。
在本发明的所述吡啶钌配合物或其晶体中,“+”和“-”分别表示正离子和负离子(也即该配合物是由[]内的配体阳离子与PF6-阴离子形成的)。
在本发明的所述吡啶钌配合物或其晶体中,所述C1-C6烷基的含义是指具有1-6个碳原子的直链或支链烷基,其包括了C1烷基、C2烷基、C3烷基、C4烷基、C5烷基或C6烷基,非限定性地例如可为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基或正己基等。
在本发明的所述吡啶钌配合物或其晶体中,作为一种选择,所述吡啶钌配合物为下式(5-1)所示的吡啶钌晶体配合物:
其晶胞参数为:α=90.00°,β=107.8390(10)°,γ=90.00°。
其晶体结构图见附图1。
在本发明的所述吡啶钌配合物或其晶体中,作为一种选择,所述吡啶钌配合物为下式(5-2)所示的吡啶钌晶体配合物:
其晶胞参数为:α=90.00°,β=128.261(6)°,γ=90.00°。
其晶体结构图由于高度类似于附图1的式(5-1)吡啶钌晶体配合物的晶体结构图(仅不存在形成大π键结构的苯环上的异丙基和甲基取代),故不再列出。
第二个方面,本发明还涉及式(5)所述吡啶钌配合物或其晶体的制备方法,所述制备方法反应路线如下:
其中,R1、R2的定义如上;
所述制备方法具体为:
S1:在有机溶剂中,于氮气保护下,式(1)化合物与式(2)化合物回流反应,得到式(3)化合物;
S2:待步骤S1的反应完成后,直接向所得体系中加入式(4)化合物的甲醇溶液,在氮气保护下继续回流反应;
S3:待步骤S2的反应完成后,趁热过滤,将所得滤液降至室温后,向其中加入NH4PF6,然后在氮气保护下升温回流反应,经后处理即得所述式(5)化合物。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S1中,所述有机溶剂为低级醇类化合物,例如可为甲醇、乙醇、正丙醇或异丙醇等中的任意一种。
其中,所述有机溶剂的量并没有特别的限定,本领域技术人员可根据实际情况进行合适的选择与确定,例如其用量大小以方便反应进行和后处理即可,在此不再进行详细描述。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S1中,式(1)化合物与式(2)化合物的摩尔比为1:0.5-1.5,例如可为1:0.5、1:1或1:1.5。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S1中,回流反应时间为5-8小时,例如可为5小时、6小时、7小时或8小时。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S2中,所述式(4)化合物是一种非常公知的化合物,可通过多种商业渠道或常规化学合成手段而获得,在此不再进行详细描述。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S2中,所述式(4)化合物的甲醇溶液的浓度并没有特别的限定,例如可为将式(4)化合物用适量甲醇溶解而得到的甲醇溶液,本领域技术人员可进行合适的选择与确定,在此不再进行详细描述。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S2中,所述式(4)化合物与步骤S1中所述式(1)化合物的摩尔比为1:5-10,例如可为1:5、1:7、1:9或1:10。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S2中,回流反应时间为4-6小时,例如可为4小时、5小时或6小时。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S3中,所述NH4PF6与步骤S1中所述式(1)化合物的摩尔比为1:1-3,例如可为1:1、1:2或1:3。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S3中,回流反应时间为40-80分钟,例如可为40分钟、50分钟、60分钟、70分钟或80分钟。
在本发明式(5)所述吡啶钌配合物或其晶体的制备方法中,在步骤S3中,所述后处理具体为:反应完成后,趁热过滤,将滤液静置,自然挥发,析出固体,过滤,将所得固体用甲醇充分洗涤,自然干燥完全,即得所述式(5)化合物。
第三个方面,本发明还涉及式(5-1)晶体配合物或式(5-2)晶体配合物的制备方法,所述方法包括:先根据上述制备方法而分别制备得到对应于式(5-1)或式(5-2)的产物(即R1为异丙基、R2为甲基的所述式(5)化合物,或R1和R2均为H的所述式(5)化合物)后,室温下,将产物(即得到的所述式(5)化合物)溶于乙腈中,得到乙腈溶液,再向所述乙腈溶液中缓慢加入乙醚,密封,直至晶体析出完全,即得所述式(5-1)晶体配合物或式(5-2)晶体配合物。
在本发明所述式(5-1)晶体配合物或式(5-2)晶体配合物的制备方法中,以质量克(g)计的所述产物与以体积毫升(ml)计的所述乙腈的比为1:25-35,即将每1克(g)的所述产物溶解在25-35毫升(ml)的所述乙腈中,两者的比例如可为1:25、1:30或1:35。
在本发明所述式(5-1)晶体配合物或式(5-2)晶体配合物的制备方法中,所述乙醚的用量也没有特别的限定,只要其能够促使晶体配合物充分析出即可,本领域技术人员同样可进行合适的确定与选择,在此不再进行详细描述。
如上所述,本发明提供了一种式(5)所示吡啶钌配合物及其制备方法,还提供了式(5-1)和式(5-2)的晶体配合物及其制备方法,经过研究发现,所述配合物或晶体配合物具有良好的生理活性,能够与DNA发生插入作用,从而在后续的药物研发中提供了良好的应用基础和继续研究之潜力。
附图说明
图1是式(5-1)晶体配合物的晶体结构。
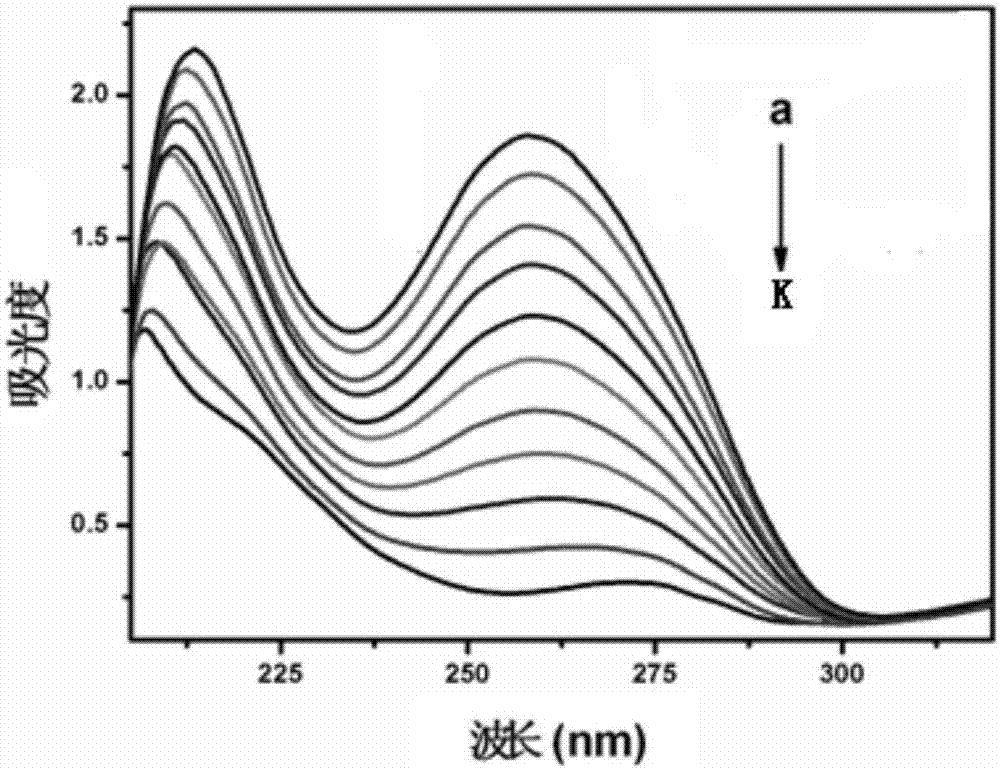
图2是表征式(5-1)晶体配合物的DNA结合常数的、在不同浓度ct-DNA溶液中的紫外吸收曲线图。
具体实施方式
下面通过具体的实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
实施例1:式(5)配合物和式(5-1)晶体配合物的制备
该实施例1实际上分为两个部分,首先是制备其中R1为异丙基、R2为甲基的式(5)配合物(见下面的反应总路线),然后再将得到的式(5)配合物进行结晶处理而得到式(5-1)的晶体配合物,但为了方便起见,在该实施例1中一并顺次描述。
A、式(5)配合物的制备,该制备方法反应总路线如下:
所述制备方法具体为:
S1:在适量有机溶剂乙醇中,于氮气保护下,100mmol式(1)化合物与50mmol式(2)化合物回流反应8小时,得到式(3)化合物(未进行分离而直接用于下一步的反应);
S2:待步骤S1的反应完成后,直接向所得体系中加入式(4)化合物的甲醇溶液(其中式(4)化合物为20mmol,甲醇为恰能将式(4)化合物溶解完全的量),然后在氮气保护下继续回流反应6小时;
S3:待步骤S2的反应完成后,趁热过滤,将所得滤液降至室温后,向其中加入35mmol NH4PF6,然后在氮气保护下升温回流反应40分钟,反应完成后,趁热过滤,将滤液静置,自然挥发,析出固体,过滤,将所得固体用适量甲醇充分洗涤,自然干燥完全,即得上式(5)化合物,产率为92.4%。
1H NMR(500MHz,d6-DMSO):δ9.07(s,1H),8.53(d,J=8.8Hz,2H),8.30(t,J=6.3Hz,2H),8.19(t,J=7.7Hz,1H),8.10(d,J=8.7Hz,2H),6.20(s,1H),5.80(d,J=5.6Hz,1H),5.70(d,J=5.7Hz,1H),5.50(s,1H),2.68(dt,J=13.5,6.8Hz,1H),2.25(s,3H),1.08(d,J=6.7Hz,3H),0.98(d,J=6.7Hz,3H)。
B、式(5-1)晶体配合物的制备
室温下,将上述A中步骤S3得到的式(5)化合物溶于乙腈中(以质量克(g)计的所述式(5)化合物与以体积毫升(ml)计的乙腈的比为1:25),得到乙腈溶液,再向所述乙腈溶液中缓慢加入适量乙醚,密封,析出红色块状晶体,直至晶体析出完全,即得所述式(5-1)晶体配合物。
其晶胞参数为:α=90.00°,β=107.8390(10)°,γ=90.00°。
其晶体结构图见附图1。
实施例2:式(5)配合物和式(5-2)晶体配合物的制备
该实施例2实际上分为两个部分,首先是制备其中R1、R2均为H的式(5)配合物(见下面的反应总路线),然后再将得到的式(5)配合物进行结晶处理而得到式(5-2)的晶体配合物,但为了方便起见,在该实施例2中一并顺次描述。
A、式(5)配合物的制备,该制备方法反应总路线如下:
所述制备方法具体为:
S1:在适量有机溶剂乙醇中,于氮气保护下,100mmol式(1)化合物与150mmol式(2)化合物回流反应5小时,得到式(3)化合物(未进行分离而直接用于下一步的反应);
S2:待步骤S1的反应完成后,直接向所得体系中加入式(4)化合物的甲醇溶液(其中式(4)化合物为10mmol,甲醇为恰能将式(4)化合物溶解完全的量),然后在氮气保护下继续回流反应4小时;
S3:待步骤S2的反应完成后,趁热过滤,将所得滤液降至室温后,向其中加入100mmol NH4PF6,然后在氮气保护下升温回流反应80分钟,反应完成后,趁热过滤,将滤液静置,自然挥发,析出固体,过滤,将所得固体用适量甲醇充分洗涤,自然干燥完全,即得上式(5)化合物,产率为75.5%。
1H NMR(500MHz,d6-DMSO):δ9.06(s,1H),8.52(d,J=8.6Hz,2H),8.30(d,J=7.9Hz,2H),8.19(t,J=7.7Hz,1H),8.10(d,J=8.6Hz,2H),6.03(s,6H)。
B、式(5-2)晶体配合物的制备
室温下,将上述A中步骤S3得到的式(5)化合物溶于乙腈中(以质量克(g)计的所述式(5)化合物与以体积毫升(ml)计的乙腈的比为1:35),得到乙腈溶液,再向所述乙腈溶液中缓慢加入适量乙醚,密封,析出红色块状晶体,直至晶体析出完全,即得所述式(5-2)晶体配合物。
其其晶胞参数为:α=90.00°,β=128.261(6)°,γ=90.00°。
其晶体结构图高度类似于附图1,故不再列出。
下面,对其生理活性进行测试。
与DNA的相互作用实验
实验方案
Tris-HCl溶液的配制:称取12.114g三羟甲基氨基甲烷(Tris)和5.844g氯化钠固体,放置到烧杯中,然后加入去离子水使其溶解,通过滴加HCl溶液来调节溶液至pH为7.2,将其定容至1L,即得到了pH为7.2的0.1mmol·L-1的目标Tris-HCl溶液。
ct-DNA溶液的配制:称取25mg小牛胸腺DNA,用去离子水溶解后,过滤,使得溶液浓度均匀,然后将其定容至50mL,即得到0.5mg/mL的ct-DNA溶液。
测试样品的制备:将所述式(5-1)晶体配合物加入到所述Tris-HCl溶液中,从而分别得到浓度为0.2mmol/L的式(5-1)晶体配合物溶液;
配制浓度分别为0.01、0.02、0.03、0.04、0.05、0.06、0.07、0.08、0.09、0.10和0.11mg/mL的ct-DNA溶液,在图2中自上而下顺次以字母a-k表示。
测试方法:分别取2ml不同浓度的上述ct-DNA溶液与1ml所述式(5-1)晶体配合物溶液混合,振荡摇匀,使用紫外分光光度法分别测定混合溶液的吸光度及吸收峰的位移变化,从而定性分析出晶体配合物与DNA的作用方式,结果见图2。
测试结果:从图2中可以看出,随着ct-DNA溶液浓度的增大,吸光度不断减小,说明了本发明的式(5-1)晶体配合物与DNA发生了插入作用;且随着ct-DNA浓度的增大,式(5-1)晶体配合物的吸收峰发生了增色和蓝移现象,可知其与ct-DNA之间存在着显著的静电作用。
根据公知的下面公式:
而计算出式(5-1)晶体配合物与ct-DNA的结合常数Kb为41.67mg/mL。
按照上述的相同方法而计算出式(5-2)晶体配合物与ct-DNA的结合常数Kb为40.89mg/mL。
由此可见,本发明的所述式(5-1)和式(5-2)晶体配合物有着显著的插入作用和静电结合作用,从而具有显著的生理活性,可用于后续药物的进一步研发之中。
综上所述,本发明提供了一种式(5)所示吡啶钌配合物及其制备方法,还提供了式(5-1)和式(5-2)的晶体配合物及其制备方法,经过研究发现,所述配合物或晶体配合物具有良好的生理活性,能够与DNA发生显著的插入作用,从而为后续的药物研发提供了良好的应用基础和继续研究之潜力。
应当理解,这些实施例的用途仅用于说明本发明而非意欲限制本发明的保护范围。此外,也应理解,在阅读了本发明的技术内容之后,本领域技术人员可以对本发明作各种改动、修改和/或变型,所有的这些等价形式同样落于本申请所附权利要求书所限定的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!