一种评价三阴性乳腺癌风险评估方法与流程
本发明涉及癌症诊断治疗领域,特别是涉及一种评价三阴性乳腺癌风险评估方法。
背景技术:
目前癌症是现今危害人们健康的主要病症。乳腺癌则是全球范围内女性最常见的恶性肿瘤之一,也是引起女性死亡的重要原因。在我国,与其他大多数国家一样,乳腺癌也成为了中国女性最常见的癌症;每年中国乳腺癌新发数量和死亡数量分别占世界人口的12.2%与9.6%,每年新发病例约21万,我国乳腺癌发病率增长速度,高出发达国家1-2个百分点。随着新技术新药物等的应用,早期乳腺癌的治愈率可以达到90%,然而仍有30%-40%的患者会死于乳腺癌复发。
肿瘤标识物是可以在血清、血浆、其他体液、组织提取物或石蜡固定的组织中检测到的自然生产的分子,可以为乳腺癌的诊断、治疗方案的选择、疾病进程和复发的预测及治疗效果的监测等提供有价值的信息。乳腺癌作为女性高发恶性肿瘤,非常缺乏高效的肿瘤标识物。目前我国在临床上应用的乳腺癌标识物有雌激素受体(ER)、孕激素受体(PR)和HER2基因,常见的乳腺癌分类也是根据这三个标识物分类的。三阴性乳腺癌是这三种标识物均不表达的一类乳腺癌,也是所有乳腺癌中最难治疗复发率最高的一种。如何对三阴性乳腺癌进行准确的分型、尽早判断肿瘤是否会复发、转移进而选择合理的治疗手段,提高三阴性乳腺癌患者的生存率和生存质量是三阴性乳腺癌治疗的重要的发展方向。
预后是指发病后,疾病未来过程的一种预先估计。在医学上,“预后”是指根据经验预测的疾病发展情况。预后主要涉及到三个方面,将发生什么结果、发生不良结果的可能性有多大、什么时候发生。预后分析是对疾病发病后发展为各种不同结局的预测;是临床非常实用、对临床很有指导作用的临床研究。研究和评级预后的目的,为了便于了解各种疾病对人类危害性的大小、探索影响预后的因素、研究改善预后的具体措施。
可变聚腺苷酸化(Alternative polyadenylation,APA)是一个基础的有关基因调控的分子机制,它通过信使RNA(mRNA)3’端的非编码区来进行生理和病理上的调控。有70%的人类基因有多种polyA位点,可以产生不同长度的3’UTR从而导致转录本的多样化。前期一个关于3’UTR APA dynamics与前列腺癌的研究证明了3’UTR信号可以应用于对肿瘤风险程度的分类。但是在不同3’UTR对肿瘤发病及发展和转移上的研究还是很少。在乳腺癌的研究中,科研人员更多地将注意力集中在短的3’UTR在预后中的显著表现上,而忽略了不同的分子分型间的异质性。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种评价三阴性乳腺癌风险评估方法,用于解决现有技术中的问题。
为实现上述目的及其他相关目的,本发明第一方面提供3’UTR信号作为生物标志物在制备用于评估乳腺癌风险的试剂盒中的用途,
所述3’UTR信号选自如下基因中的一种或多种的3’UTR信号:
SMAD6基因、CXCL8基因、CLIC2基因、PRCKB基因、RTN1基因、ZCCHC14基因、PPIC基因、SIK3基因、UBE2G2基因、SCL2A3基因、SYNGR1基因、NPL基因、PRSS12基因、ADGRL2基因、ZER1基因、WDHD1基因、N4BP2L2基因。
在本发明一些实施方式中,所述乳腺癌选自三阴性乳腺癌。
本发明第二方面提供用于检测3’UTR信号的试剂在制备用于评估乳腺癌风险的试剂盒中的用途,
所述3’UTR信号选自如下基因中的一种或多种的3’UTR信号:
SMAD6基因、CXCL8基因、CLIC2基因、PRCKB基因、RTN1基因、ZCCHC14基因、PPIC基因、SIK3基因、UBE2G2基因、SCL2A3基因、SYNGR1基因、NPL基因、PRSS12基因、ADGRL2基因、ZER1基因、WDHD1基因、N4BP2L2基因。
在本发明一些实施方式中,所述用于检测3’UTR信号的试剂用于获得各基因的各3’UTR的表达量。
在本发明一些实施方式中,所述用于检测3’UTR信号的试剂用于获得各基因的各3’UTR的表达量选自:各基因的各3’UTR的PCR扩增所需试剂。
在本发明一些实施方式中,所述用于检测3’UTR信号的试剂包括各基因的各3’UTR的所对应的引物、探针。
在本发明一些实施方式中,所述乳腺癌选自三阴性乳腺癌。
本发明第三方面提供一种用于评估乳腺癌风险的试剂盒,其特征在于,包括用于检测3’UTR信号的试剂,所述3’UTR信号选自如下基因中的一种或多种的3’UTR信号:
SMAD6基因、CXCL8基因、CLIC2基因、PRCKB基因、RTN1基因、ZCCHC14基因、PPIC基因、SIK3基因、UBE2G2基因、SCL2A3基因、SYNGR1基因、NPL基因、PRSS12基因、ADGRL2基因、ZER1基因、WDHD1基因、N4BP2L2基因。
在本发明一些实施方式中,所述乳腺癌选自三阴性乳腺癌。
本发明第四方面提供一种评价三阴性乳腺癌风险评估方法,包括:
S1:获取样本的SMAD6基因、CXCL8基因、CLIC2基因、PRCKB基因、RTN1基因、ZCCHC14基因、PPIC基因、SIK3基因、UBE2G2基因、SCL2A3基因、SYNGR1基因、NPL基因、PRSS12基因、ADGRL2基因、ZER1基因、WDHD1基因、N4BP2L2基因的3’UTR的ERI值;
S2:根据各基因的3’UTR的ERI值,获取患者的risk score。
在本发明一些实施方式中,各基因的3’UTR的ERI值的计算方法如下:
其中,es表示样本中3’UTR的short form的表达量之和;
eL表示样本中3’UTR的long form的表达量之和;
αS表示样本中5’探针组与short forms的亲和情况之和;
αL表示样本中5’探针组与long forms的亲和情况之和;
βS表示样本中3’探针组与short forms的亲和情况之和;
βL表示样本中3’探针组与long forms的亲和情况之和;
在本发明一些实施方式中,所述risk score的计算方法如下:
risk score=-0.104×ERISMAD6–0.125×ERICXCL8–0.174×ERICLIC2–0.213×ERIPRCKB-0.232×ERIRTN1–0.292×ERIZCCHC14–0.292×ERIPPIC+0.090×ERISIK3+0.119×ERIUBE2G2+0.175×ERISCL2A3+0.188×ERISYNGR1+0.213×ERINPL+0.271×ERIPRSS12+0.304×ERIADGRL2+0.364×ERIZER1+0.453×ERIWDHD1+0.527×ERIN4BP2L2。
上述公式中,ERISMAD6、ERICXCL8、ERICLIC2、ERIPRCKB、ERIRTN1、ERIZCCHC14、ERIPPIC、ERISIK3、ERIUBE2G2、ERISCL2A3、ERISYNGR1、ERINPL、ERIPRSS12、ERIADGRL2、ERIZER1、ERIWDHD1、ERIN4BP2L2分别表示其下标所标注的基因的ERI值。
在本发明一些实施方式中,由样本的microarry数据获取所述es、eL、αS、αL、βS、βL。
在本发明一些实施方式中,所述样本的microarry数据由RMA(robust multi-array average)矫正,矫正后的microarry数据,可以提取出信号强度高的5’和3’探针组的数据。
在本发明一些实施方式中,所述样本的microarry数据由single Affymetrix platform处理获得。
在本发明一些实施方式中,microarry数据中,short long form和long form的分类标准如下:将各探针所针对的3’UTR序列总碱基数之和除以探针数量得到均值(均值=各探针所针对的3’UTR序列总碱基数之和÷探针数量),利用均值将探针所针对的3’UTR分为short form与long form。其中,大于均值的3’UTR归为long form,小于等于均值的3’UTR定义为short form。
在本发明一些实施方式中,根据risk score判断患者是否属于高风险复发或转移,当risk score越高,复发或转移的风险越大,当risk score越低,复发或转移的风险越小。
在本发明一些实施方式中,所述评价三阴性乳腺癌风险评估方法还包括:根据risk score的threshold判断患者是否属于高风险复发或转移,当risk score>threshold时,将患者归类为高风险复发或转移,当risk score≤threshold时,将患者归类为低风险复发或转移。
当通过risk score的threshold判断患者是否属于高风险复发或转移时,通常需要一定数量的样本以构成样本组,从而可以获得合理的threshold值。例如,样本可以来源于一定数量的三阴性乳腺癌患者。
在本发明一些实施方式中,通过X-tile获取样本组的risk score的threshold值,样本数量不少于50个。
在本发明一些实施方式中,样本组中各样本来源于不同的个体。
在本发明一些实施方式中,所述评价三阴性乳腺癌风险评估方法还包括:根据基因的ERI值的threshold判断患者是否属于高风险复发或转移;
对于N4BP2L2基因、WDHD1基因、ZER1基因、ADGRL2基因、PRSS12基因、NPL基因、SYNGR1基因、SCL2A3基因、UBE2G2基因、或SIK3基因,当该基因的ERI值>threshold时,将患者归类为高风险复发或转移,当该基因的ERI值≤threshold时,将患者归类为低风险复发或转移;
对于PPIC基因、ZCCHC14基因、RTN1基因、PRCK8基因、CLIC2基因、CXCL8基因、或SMAD6基因,当该基因的ERI值<threshold时,将患者归类为高风险复发或转移,当该基因的ERI值≥threshold时,将患者归类为低风险复发或转移。
当通过各基因的ERI值的threshold判断患者是否属于高风险复发或转移时,通常需要一定数量的样本以构成样本组,从而可以获得合理的各基因的ERI值所对应的threshold值。例如,样本可以来源于一定数量的三阴性乳腺癌患者。
在本发明一些实施方式中,通过X-tile获取样本组的ERI值的threshold值,样本数量不少于50个。
在本发明一些实施方式中,样本组中各样本来源于不同的个体。
本发明第五方面提供一种用于评价三阴性乳腺癌风险的装置,所述装置包括用于接收数据的计算机,所述计算机被编程以执行如上所述的评价三阴性乳腺癌风险评估方法。
本发明第六方面提供一种存储有计算机程序的计算机可读介质,所述计算机程序被执行时可以实现如上所述的评价三阴性乳腺癌风险评估方法。
本发明发明人发现通过17个基因的3’UTR构建的预后模型来预测三阴性乳腺的复发和转移风险,其精确程度要高于通过普通临床因素(年龄,淋巴结状态,肿瘤尺寸等)的预测结果,并可根据上述3’UTR的shortening及lengthening这两类分类,进一步预测样本是否属于高风险、预后差或低风险、预后较好的类型。此外,经327例三阴性乳腺癌病人样本的microarry数据和其他相关数据分析证实,通过上述3’UTR构建的模型,能够有效地预测出高风险、预后差的样本,可见其在预测及其他关于预后的用途中具有高度产业利用价值。
附图说明
图1显示为通过X-tile选择threshold得到的图及其生存分析。左图颜色由暗(黑)到亮表示其关联程度越高,红色表示risk scores和生存情况呈现负相关,绿色表示正相关。中图为所有样本的柱状图显示,右图为生存分析图,可以看到根据上述预后模型分类得到的类群显示出较为准确的结果。
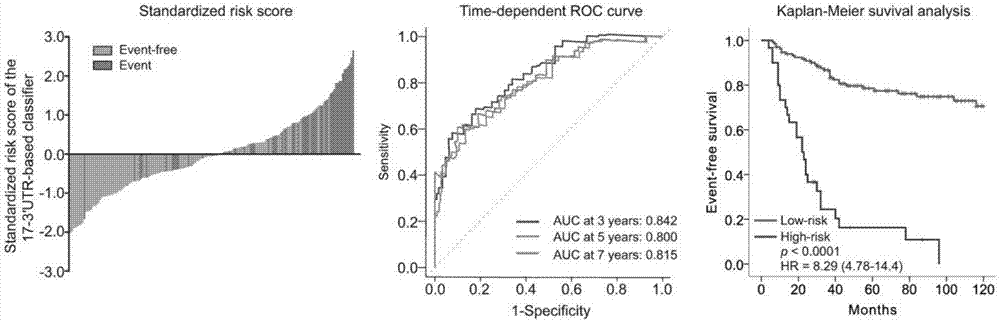
图2显示为预后模型的risk score,time-dependent ROC曲线及生存曲线。可以看到通过模型得到的结果具有较高的精确性。
图3显示为10个shortening表现为较差预后的3’UTR(N4BP2L2,WDHD1,ZER1,ADGRL2,PRSS12,NPL,SYNGR1,SCL2A3,UBE2G2,SIK3)的X-tile plot。
图4显示为7个lengthening表现为较差预后的3’UTR(PPIC,ZCCHC14,RTN1,PRCK8,CLIC2,CXCL8,SMAD6)的X-tile plot。
图5显示为比较17个3’UTR混合分类模型及单独的3’UTR在327个三阴性乳腺癌样本中的精确度的ROC曲线。
具体实施方式
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围;在本发明说明书和权利要求书中,除非文中另外明确指出,单数形式“一个”、“一”和“这个”包括复数形式。
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
除非另外说明,本发明中所公开的实验方法、检测方法、制备方法均采用本技术领域常规的分子生物学、生物化学、染色质结构和分析、分析化学、细胞培养、重组DNA技术及相关领域的常规技术。这些技术在现有文献中已有完善说明,具体可参见Sambrook等MOLECULAR CLONING:A LABORATORY MANUAL,Second edition,Cold Spring Harbor Laboratory Press,1989and Third edition,2001;Ausubel等,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John Wiley&Sons,New York,1987 and periodic updates;the series METHODS IN ENZYMOLOGY,Academic Press,San Diego;Wolffe,CHROMATIN STRUCTURE AND FUNCTION,Third edition,Academic Press,San Diego,1998;METHODS IN ENZYMOLOGY,Vol.304,Chromatin(P.M.Wassarman and A.P.Wolffe,eds.),Academic Press,San Diego,1999;和METHODS IN MOLECULAR BIOLOGY,Vol.119,Chromatin Protocols(P.B.Becker,ed.)Humana Press,Totowa,1999等。
实施例1
样本来自327个三阴性乳腺癌患者,microarry数据来自single Affymetrix platform,包括HG-U133A和HG-U133plus 2.0。3’UTR的分析包含从GEO(Gene Expression Omnibus)获得的公共数据(GSE31519,GSE29690,GSE2603,GSE2034,GSE5327,GSE11121,GSE7390,和GSE21653)。
利用R包“ERI-expr”来分析microarrys(参见https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3275581/#pone.0031129.s005),ERI-expr含有自定义的针对HG-U133A,HG-U133B和UG-U133plus的chip description files(CDF)。经过RMA(robust multi-array average,http://www.molmine.com/magma/loading/rma.htm)矫正,提取出信号强度高的5’(S5’)和3’(S3’)探针组。通过ERI(expression ratio index)来定义5’和3’探针组的APA位点的信号比:
其中es(eL)表示3’UTR的short(long)form的表达量(expression),αS,L(βS,L)表示5’(α)和3’(β)探针组与short(S)或long(L)forms的亲和情况(亲和情况即CEL文件(探针检测后的结果文件,包括五列,分别为在探针上的横坐标X和纵坐标Y,灰度平均值,灰度标准差及用了多少像素来求这个平均值)的灰度标准差)。此处的short和long forms的分类通过设计探针时每段序列总碱基数除以探针数量得到均值,利用均值将序列分为short与long forms。其中,大于均值定义为long forms,小于等于均值定义为short forms。ERI是一个关于3’UTR shortening form和lengthening form表达定量的线性函数。将各基因所对应的ERI值带入预后模型:
risk score=-0.104×ERISMAD6–0.125×ERICXCL8–0.174×ERICLIC2–0.213×ERIPRCKB-0.232×ERIRTN1–0.292×ERIZCCHC14–0.292×ERIPPIC+0.090×ERISIK3+0.119×ERIUBE2G2+0.175×ERISCL2A3+0.188×ERISYNGR1+0.213×ERINPL+0.271×ERIPRSS12+0.304×ERIADGRL2+0.364×ERIZER1+0.453×ERIWDHD1+0.527×ERIN4BP2L2
standardized risk score=(risk score–0.625)/0.578
通过X-tile软件选择threshold,具体步骤为:第一步将每个样本的Risk score添加入excel表格的最后一列,前面的信息至少包括生存时间(DeadOfDisease),添加后将excel复制入txt文本文件;第二步打开X-tile软件,点击Analyze进入分析界面,点击File-open打开刚建立的txt文本;第三步进行设置,最左边一栏显示的是所有列的标题,中间一栏是选中列的具体数值,最右一栏为设置界面,将生存时间列添加入最右一栏的Survival Time,将Riskscore添加入Marker1,Marker2没有则不填;最后一步点击左上角的do,选择Kaplan-Meier——Marker1,则可得出结果。鼠标点击2Pop X-tile Plot所指的矩形图则可找到最佳cutoff值,柱状图横坐标数值即为最佳cutoff值。最终得到threshold为risk score 1.146(standardized risk score 0.903)。当risk scores>1.146,则视为有高风险复发或转移;risk scores<1.146,则认为有较低的风险复发或转移。实施例1中所得实验结果如图1和图2所示。图1中图为所有样本的柱状图显示,横坐标为riskscore,纵坐标为在该riskscore下的样本数量,横坐标两个颜色交界的地方即为threshold,最右的327个样品缩对应的患者的生存分析图,按照risk score及threshold将患者对应分为低风险组和高风险组,可以看到根据上述预后模型分类得到的类群显示出较为准确的结果。图2左显示为预后模型的risk score示意图,图2中显示为time-dependent ROC曲线、图2右显示为327个样品缩对应的患者的生存分析图,按照risk score及threshold将患者对应分为低风险组和高风险组。可以看到通过模型得到的结果具有较高的精确性。
实施例2
根据实施例一计算出17个基因(SMAD6,CXCL8,CLIC2,PRCKB,RTN1,ZCCHC14,PPIC,SIK3,UBE2G2,SCL2A3,SYNGR1,NPL,PRSS12,ADGRL2,ZER1,WDHD1,N4BP2L2)3’UTR的ERI值,将其上传入X-tile,软件使用方法同实施例一,Marker1选择基因的ERI值,根据ERI的值得到3’UTR的threshold,定义当3’UTR的ERI超过threshold时为shortening,低于threshold时为lengthening,根据此种方法将每个基因的3’UTR分类为shortening和lengthening。
最终得到17个基因的3’UTR中有10个基因的3’UTR shortening(N4BP2L2,WDHD1,ZER1,ADGRL2,PRSS12,NPL,SYNGR1,SCL2A3,UBE2G2,SIK3)与较差的预后有关,如图3的生存分析(图3a对应ADGRL2基因,图3b对应N4BP2L2基因,图3c对应NPL基因,图3d对应PRSS12基因,图3e对应SCL2A3基因,图3f对应SIK3基因,图3g对应SYNGR1基因,图3h对应UBE2G2基因,图3i对应WDHD1基因,图3j对应ZER1基因;图3中,各组基因所对应的附图中,左图部分颜色由暗(黑)到亮表示其关联程度越高,红色表示ERI值和生存情况呈现负相关,绿色表示正相关,左图部分的横坐标为low population,纵坐标为high population,比例尺代表明暗程度,0是明度为0(即为黑色),10表示明度为最大,即为亮红或亮绿;中图部分为所有样本的柱状图显示,横坐标为ERI的risk score,纵坐标为在该riskscore下的样本数量,横坐标两个颜色交界的地方即为threshold,图3a中risk score最大值为2.3,threshold为1.375,图3b中risk score最大值为2.5,threshold为1.233,图3c中risk score最大值为2.5,threshold为1.124,图3d中risk score最大值为2.4,threshold为1.384,图3e中risk score最大值为2.6,threshold为1.438,图3f中risk score最大值为2.5,threshold为1.467,图3g中risk score最大值为2.8,threshold为1.426,图3h中risk score最大值为2.7,threshold为1.677,图3i中risk score最大值为2.7,threshold为1.399,图3j中risk score最大值为2.9,threshold为1.372;右图为生存分析图,横坐标为生存时间,坐标中每格代表18个月,纵坐标为存活百分比,坐标中每格代表50%,蓝色表示3’UTR lengthening,灰色表示3’UTR shortenning,图3a中p值为0.0021,图3b中p值为0.0006,图3c中p值为0.0128,图3d中p值为0.1003,图3e中p值<0.0001,图3f中p值为0.0062,图3g中p值为0.0077,图3h中p值为0.0289,图3i中p值为0.0001,图3j中p值为0.0019,可以看到这十个3’UTR的shortening比lengthening的生存情况差。剩下的7个(PPIC,ZCCHC14,RTN1,PRCK8,CLIC2,CXCL8,SMAD6)其lengthening表现出较差的预后,如图4的生存分析(图4a对应CLIC2基因,图4b对应CXCL8基因,图4c对应PPIC,基因,图4d对应PRCK8基因,图4e对应RTN1,基因,图4f对应SMAD6基因,图4g对应ZCCHC14基因;图4中,各组基因所对应的附图中,左图部分颜色由暗(黑)到亮表示其关联程度越高,红色表示ERI值和生存情况呈现负相关,绿色表示正相关,左图部分的横坐标为low population,纵坐标为high population,比例尺同图3;中图为所有样本的柱状图显示,横坐标为ERI的risk score,纵坐标为在该riskscore下的样本数量,横坐标两个颜色交界的地方即为threshold,图4a中risk score最大值为2.5,threshold为0.865,图4b中risk score最大值为2.4,threshold为1.039,图4c中risk score最大值为2.5,threshold为0.993,图4d中risk score最大值为2.8,threshold为1.231,图4e中risk score最大值为2.5,threshold为0.739,图4f中risk score最大值为2.4,threshold为0.825,图4g中risk score最大值为2.8,threshold为0.973;右图为生存分析图,横坐标为生存时间,坐标中每格代表18个月,纵坐标为存活百分比,坐标中每格代表50%,蓝色表示3’UTR lengthening,灰色表示3’UTR shortenning,图4a中p值为0.0102,图4b中p值为0.0003,图4c中p值为0.0001,图4d中p值为0.0001,图4e中p值为0.0143,图4f中p值为0.0040,图4g中p值为0.0006,可以看到这七个3’UTR的lengthening比shortening的生存情况更差。
实施例3
利用R包‘pROC’绘制17个3’UTR混合分类模型及单独的3’UTR在327个三阴性乳腺癌样本中的精确度的ROC曲线。第一步,制作需要的文件格式。文件分为两列。第一列为label,具体分别为17个基因的名字及17个基因混合在一起的分类器,即SMAD6,CXCL8,CLIC2,PRCKB,RTN1,ZCCHC14,PPIC,SIK3,UBE2G2,SCL2A3,SYNGR1,NPL,PRSS12,ADGRL2,ZER1,WDHD1,N4BP2L2,17-3’UTR-based-classifier。第二列为score,具体的值为risk score的计算方法,label为哪一个基因则用哪个基因来计算,例如SMAD6,risk score=-0.104x SMAD6–0.125x 1–0.174x 1–0.213x 1-0.232x 1–0.292x 1–0.292x 1+0.090x 1+0.119x 1+0.175x 1+0.188x 1+0.213x 1+0.271x 1+0.304x 1+0.364x 1+0.453x 1+0.527x 1,以此类推。两列间以逗号分隔,保存为文本文档(txt)。第二步,打开R,载入pROC包,利用roc()函数计算各ROC值,将其合并为一组数据后作图,利用R包“ggplot2”,将各ROC曲线会画在一张图上。
综上所述,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!