一种硝酸酯可降解生物活性材料及其制备方法与应用与流程
本发明涉及生物活性材料领域,尤其涉及一种硝酸酯可降解生物活性材料及其制备方法与应用。
背景技术:
心血管疾病现已成为威胁人类健康的重大疾患之一,无论是发病率还是死亡率均居各类疾病之首,被医学界公认为是危害人类身体健康的“第一杀手”。据世界卫生组织预测,到2030年全世界每年死于心血管相关疾病的人数会增加到2330万。
心肌梗死是由于冠状动脉急性闭塞而导致的心脏严重而持久的缺血,局部心肌因血供和需求不平衡而导致部分心肌坏死。心肌细胞作为一种终末分化的细胞,坏死后难以再生。目前,心肌补片作为一种治疗心肌梗死的方法得到了广泛应用。心血管疾病中,当血管狭窄程度较低时,治疗手段包括血管成形术和支架植入术。研究表明血管成形术和支架植入术等介入手术通常会导致动脉内膜细胞损伤,活化的血小板聚集并与纤维蛋白一起粘附到植入装置和/或受伤部位,结果使血管内稳态被破坏。在血管损伤血栓形成后,平滑肌细胞迁移至损伤部位,其增殖导致新内膜增生,最终导致再狭窄。对于狭窄程度较高甚至堵塞的血管,血管搭桥手术是必需的。作为血管移植的黄金标准的自体血管,使用时需要二次手术,不仅造成机体损伤和额外的费用,而且有些情况下病人不能提供自体血管。随着生物医学及材料学等学科的不断进步与发展,组织工程人工血管应运而生。
许多血管疾病都与血管内皮细胞衍化释放的舒张因子一氧化氮(no)的代谢有关系。no是一种可以通过细胞膜扩散的气体自由基,其最主要的功能是作为心血管系统的生理性调节分子,具有:1)调节血管张力和心肌收缩力,参与动脉血压及器官组织血流量的调节;2)维持血管内皮完整、促进血管新生;3)抑制平滑肌细胞粘附、增殖和迁移;4)抑制血小板在局部的粘附、聚集和白细胞在血管内皮的粘附从而抑制血栓的形成等作用。一氧化氮的重要生理功能,为心血管材料的制备与修饰提供了新视角。
技术实现要素:
本发明的目的在于提供一种硝酸酯可降解生物活性材料及其制备方法与应用,本发明提供的硝酸酯可降解生物活性材料具有缓释气体小分子物质no的功能。
本发明提供了一种硝酸酯可降解生物活性材料,为端羟基可降解聚合物的端羟基共价连接m基团或n基团所得的硝酸酯聚合物;
所述m基团具有式i所示结构:
式i中,m=2、3或4;
所述n基团具有式ii所示结构:
式ii中,n=2或3;
所述端羟基可降解聚合物的数均分子量为400~10000da;
所述端羟基可降解聚合物为线性聚合物、三臂聚合物或四臂聚合物。
优选地,所述端羟基可降解聚合物包括聚己内酯、聚丙交酯、聚乙醇酸、聚丙交酯-乙醇酸、聚(丙交酯-己内酯)共聚物、聚对二氧环己酮或聚羟基脂肪酸酯。
优选地,所述硝酸酯可降解生物活性材料中硝酸酯基的个数为1~12。
优选地,所述硝酸酯可降解生物活性材料中硝酸酯基的含量为1~20wt.%。
本发明提供了上述技术方案所述硝酸酯可降解生物活性材料的制备方法,当所述端羟基可降解聚合物的端羟基共价连接m基团时,包括如下步骤:
(1)在惰性气体氛围中,将溴代酰卤加入至端羟基可降解聚合物和有机碱的二氯甲烷溶液中,进行溴代反应,得到溴代聚合物;所述溴代酰卤为3-溴丙酰氯、4-溴丁酰氯或5-溴戊酰氯;
(2)将所述溴代聚合物和硝酸银溶解于乙腈中,进行取代反应,得到硝酸酯可降解生物活性材料。
优选地,所述端羟基可降解聚合物与溴代酰卤和有机碱的摩尔比为1:1~10:1~10;
所述溴代反应包括第一阶段反应和第二阶段反应;所述第一阶段反应的温度为-5~5℃,所述第一阶段反应的时间为1~4h;所述第二阶段反应的温度为10~40℃,所述第二阶段反应的时间为20~26h。
优选地,所述端羟基可降解聚合物与硝酸银的摩尔比为1:1~10;
所述取代反应的温度为10~40℃,所述取代反应的时间为20~26h。
本发明提供了上述技术方案所述硝酸酯可降解生物活性材料的制备方法,当所述端羟基可降解聚合物的端羟基共价连接n基团时,包括如下步骤:
(1)在惰性气体氛围中,将酸酐加入至端羟基可降解聚合物的二氯甲烷溶液中,进行亲核加成反应,得到羧基化聚合物;所述酸酐为丁二酸酐或戊二酸酐;
(2)将三羟甲基氨基甲烷、乙酸和乙酸酐混合,得到酸性三羟甲基氨基甲烷溶液;在所述酸性三羟甲基氨基甲烷溶液的温度≤10℃的条件下,将质量百分浓度为85~90%的硝酸溶液滴加至酸性三羟甲基氨基甲烷溶液中,进行硝化反应,然后将硝化反应所得溶液的ph值调节至碱性,采用二氯甲烷进行萃取,得到2-氨基硝酸甘油;
(3)将羧基化聚合物、偶联剂和n,n-二甲基甲酰胺混合,进行活化,然后加入有机碱和2-氨基硝酸甘油,进行酰胺化反应,得到硝酸酯可降解生物活性材料;
所述步骤(1)和步骤(2)无时间顺序限定。
优选地,所述端羟基可降解聚合物与酸酐的摩尔比为1:2~10;
所述亲核加成反应的温度为30~60℃,所述亲核加成反应的时间为0.8~1.2h。
优选地,所述三羟甲基氨基甲烷的物质的量与乙酸、乙酸酐和硝酸溶液的体积之比为1mmol:0.45~0.55ml:0.45~0.55ml:0.25~0.35ml;
所述硝化反应包括第一阶段反应和第二阶段反应;所述第一阶段反应的温度≤10℃,所述第一阶段反应的时间为10~15min;所述第二阶段反应的温度为20~40℃,所述第二阶段反应的时间为0.8~1.2h。
优选地,所述羧基化聚合物、有机碱和2-氨基硝酸甘油的摩尔比为1:2~10:2~5;
所述酰胺化反应的温度为20~40℃,所述酰胺化反应的时间为22~26h。
本发明提供了一种心肌补片,由包括上述技术方案所述的硝酸酯可降解生物活性材料或上述技术方案所述的制备方法制备得到的硝酸酯可降解生物活性材料的原料制备而成。
本发明提供了一种人工血管,由包括上述技术方案所述的硝酸酯可降解生物活性材料或上述技术方案所述的制备方法制备得到的硝酸酯可降解生物活性材料的原料制备而成。
本发明提供了一种硝酸酯可降解生物活性材料,为端羟基可降解聚合物的端羟基共价连接具有式i所示结构的m基团或具有式ii所示结构的n基团所得的硝酸酯聚合物。本发明提供的硝酸酯可降解生物活性材料中的硝酸酯基在体内或体外的抗氧化剂的作用下能够缓释气体小分子一氧化氮(no)。实施例中试验结果表明,本发明所提供的硝酸酯可降解生物活性材料能够缓慢释放出no,由该材料所制备的心肌补片或人工血管均表现出了优异的治疗效果。
附图说明
图1为实施例1所得硝酸酯可降解生物活性材料的核磁图谱;
图2为实施例6所得硝酸酯可降解生物活性材料的核磁图谱;
图3为实施例9所得心肌补片中no含量的测定结果图;
图4为实施例10所得心脏超声检测图;
图5为实施例16所得硝酸酯人工血管中no含量的测定结果图。
具体实施方式
本发明提供了一种硝酸酯可降解生物活性材料,为端羟基可降解聚合物的端羟基共价连接m基团或n基团所得的硝酸酯聚合物;
所述m基团具有式i所示结构:
式i中,m=2、3或4;
所述n基团具有式ii所示结构:
式ii中,n=2或3;
所述端羟基可降解聚合物的数均分子量为400~10000da;
所述端羟基可降解聚合物为线性聚合物、三臂聚合物或四臂聚合物。
本发明对于所述端羟基可降解聚合物的种类没有特殊的限定,采用本领域技术人员熟知的端羟基可降解聚合物即可。在本发明中,所述端羟基可降解聚合物优选包括聚己内酯、聚丙交酯、聚乙醇酸、聚丙交酯-乙醇酸、聚(丙交酯-己内酯)共聚物、聚对二氧环己酮或聚羟基脂肪酸酯。
在本发明中,所述硝酸酯可降解生物活性材料中硝酸酯基的个数优选为1~12;所述硝酸酯可降解生物活性材料中硝酸酯基的含量优选为1~20wt.%。
本发明提供了上述技术方案所述硝酸酯可降解生物活性材料的制备方法,当所述端羟基可降解聚合物的端羟基共价连接m基团时,包括如下步骤:
(1)在惰性气体氛围中,将溴代酰卤加入至端羟基可降解聚合物和有机碱的二氯甲烷溶液中,进行溴代反应,得到溴代聚合物;所述溴代酰卤为3-溴丙酰氯、4-溴丁酰氯或5-溴戊酰氯;
(2)将所述溴代聚合物和硝酸银溶解于乙腈中,进行取代反应,得到硝酸酯可降解生物活性材料。
本发明在惰性气体氛围中,将溴代酰卤加入至端羟基可降解聚合物和有机碱的二氯甲烷溶液中,进行溴代反应,得到溴代聚合物;所述溴代酰卤为3-溴丙酰氯、4-溴丁酰氯或5-溴戊酰氯。
本发明优选先配制端羟基可降解聚合物和有机碱的二氯甲烷溶液。本发明对所述端羟基可降解聚合物和有机碱的二氯甲烷溶液的配制方法没有特殊限定,采用本领域常规的配制溶液的方式即可。在本发明实施例中,具体是先将端羟基可降解聚合物置于容器中,抽真空后通入惰性气体,在惰性气体保护条件下,通过注射器加入二氯甲烷,然后再将有机碱滴加至反应瓶中,得到惰性气体保护的端羟基可降解聚合物和有机碱的二氯甲烷溶液。
在本发明中,所述有机碱优选为吡啶、三乙胺和n,n-二异丙基乙胺中的至少一种。在本发明中,所述有机碱为缚酸剂,促进反应正向进行。
在本发明中,所述端羟基可降解聚合物与有机碱的摩尔比优选为1:1~10,更优选为1:2~8,最优选为1:4~6。
在本发明中,所述端羟基可降解聚合物的质量和二氯甲烷的体积之比优选为1g:20~30ml。在本发明中,所述二氯甲烷优选为无水二氯甲烷。
得到惰性气体保护的端羟基可降解聚合物和有机碱的二氯甲烷溶液后,本发明优选将所述端羟基可降解聚合物和有机碱的二氯甲烷溶液降温至-5~5℃,再加入溴代酰卤,进行溴代反应,得到溴代聚合物。
本发明对所述降温的速率没有特殊限定,可以为任意降温速率。在本发明中,在-5~5℃条件下加入溴代酰卤,可降低反应速率,减少副反应的发生。
在本发明中,所述惰性气体氛围能够保证反应容器中的气体不含水分,保证反应在无水氛围进行,避免水引起溴代酰卤发生水解。
在本发明中,所述溴代酰卤的加入方式优选为滴加;所述滴加的速度优选为8~12ml/h。在本发明中,在-5~5℃范围滴加溴代酰卤可降低反应速率,避免发生副反应。在本发明中,所述溴代酰卤作为溴取代试剂使用。
在本发明中,所述端羟基可降解聚合物与溴代酰卤的摩尔比优选为1:1~10,更优选为1:2~8,最优选为1:4~6。
在本发明中,所述溴代反应优选包括第一阶段反应和第二阶段反应;所述第一阶段反应的温度优选为-5~5℃,更优选为-3~3℃;所述第一阶段反应的时间优选为1~4h,更优选为2~3h;所述第二阶段反应的温度优选为10~40℃,更优选为20~30℃;所述第二阶段反应的时间优选为20~26h,更优选为22~24h。
溴代反应完成后,本发明优选对溴代反应所得产物进行后处理,得到溴代聚合物。在本发明中,所述后处理优选依次包括溶剂去除、重结晶、过滤和干燥。
在本发明中,所述溶剂去除优选采用减压蒸馏的方式;所述减压蒸馏的温度优选为室温;所述减压蒸馏的压力优选为0.04~0.06mpa;所述减压蒸馏的产物的单位质量的体积优选为1~2ml/g,或将溶剂全部蒸除,再补加二氯甲烷至单位质量的体积为1~2ml/g。
在本发明中,所述重结晶优选为将冷乙醚加入至所述溴代聚合物的二氯甲烷溶液中,析出溴代聚合物;所述冷乙醚的温度优选为0~5℃;所述冷乙醚的体积与端羟基可降解聚合物的质量之比优选为5~10ml:1g;在本发明中,所述析出过程控制温度在0~5℃,加入乙醚后,析出固体后进行过滤。
本发明对所述过滤的方式没有特殊限定,采用常规的过滤即可。
本发明对所述干燥的方式没有特殊限定,能够得到恒重的产品即可。在本发明实施例中,所述干燥优选为真空干燥。
在本发明实施例中,所述真空干燥的温度优选为20~40℃,更优选为25~35℃;所述真空干燥的压力优选为0.06~0.08mpa;所述真空干燥的时间优选为3~5h。
得到溴代聚合物后,本发明将所述溴代聚合物和硝酸银溶解于乙腈中,进行取代反应,得到硝酸酯可降解生物活性材料。
在本发明中,所述端羟基可降解聚合物和硝酸银的摩尔比优选为1:1~10,更优选为1:2~8,最优选为1:4~6。
在本发明中,所述溴代聚合物的质量与乙腈的体积之比优选为1g:5~20ml。在本发明中,所述乙腈优选为无水乙腈。
在本发明中,所述取代反应的温度优选为10~40℃,更优选为20~30℃;所述取代反应的时间优选为20~26h,更优选为22~24h。
取代反应完成后,本发明优选将取代反应所得反应液进行溶剂去除,得到硝酸酯可降解生物活性材料粗品。
在本发明中,所述取代反应所得反应液的溶剂去除优选采用减压蒸馏的方式;所述减压蒸馏的温度优选为40~50℃;所述减压蒸馏的压力优选为0.07~0.09mpa。
得到硝酸酯可降解生物活性材料粗品后,本发明优选将所述硝酸酯可降解生物活性材料粗品溶于二氯甲烷中,然后依次经洗涤、干燥和减压蒸馏,得到硝酸酯可降解生物活性材料。
在本发明中,所述硝酸酯可降解生物活性材料的质量与溶解硝酸酯可降解生物活性材料粗品用二氯甲烷的体积之比优选为1g:50~100ml,更优选为1g:70~80ml。
在本发明中,所述洗涤用洗涤剂优选为水;所述洗涤的次数优选为3~5次;所述硝酸酯可降解生物活性材料粗品的质量与每次洗涤用水的体积之比优选为1g:20~50ml,更优选为1g:30~40ml。
本发明对所述洗涤的方式没有特殊限定,采用常规的对有机相的洗涤方法进行洗涤即可。在本发明实施例中,所述洗涤的方式优选为将水和硝酸酯可降解生物活性材料粗品的二氯甲烷溶液混合后,经搅拌、静置分层,保留有机相。
洗涤完成后,本发明优选将洗涤所得有机相进行干燥。
在本发明中,所述干燥优选采用干燥剂干燥;所述干燥剂优选为无水硫酸钠。
干燥完成,本发明优选将干燥后的有机相经减压蒸馏,得到硝酸酯可降解生物活性材料。
在本发明中,对有机相的减压蒸馏的温度优选为30~40℃;所述减压蒸馏的压力优选为0.04~0.06mpa;本发明对所述减压蒸馏的时间没有特殊限定,能够减压蒸馏至无液滴馏出即可,减压蒸馏后得到液体产品。在本发明中,完成减压整流后,优选采用油泵进一步对所述液体产品进行抽干,得到蜡状固体,即为硝酸酯可降解生物活性材料。
本发明提供了上述技术方案所述硝酸酯可降解生物活性材料的制备方法,当所述端羟基可降解聚合物的端羟基共价连接n基团时,包括如下步骤:
(1)在惰性气体氛围中,将酸酐加入至端羟基可降解聚合物的二氯甲烷溶液中,进行亲核加成反应,得到羧基化聚合物;所述酸酐为丁二酸酐或戊二酸酐;
(2)将三羟甲基氨基甲烷、乙酸和乙酸酐混合,得到酸性三羟甲基氨基甲烷溶液;在所述酸性三羟甲基氨基甲烷溶液的温度≤10℃的条件下,将质量百分浓度为85~90%的硝酸溶液滴加至酸性三羟甲基氨基甲烷溶液中,进行硝化反应,然后将硝化反应所得溶液的ph值调节至碱性,采用二氯甲烷进行萃取,得到2-氨基硝酸甘油;
(3)将羧基化聚合物、偶联剂和n,n-二甲基甲酰胺混合,进行活化,然后加入有机碱和2-氨基硝酸甘油,进行酰胺化反应,得到硝酸酯可降解生物活性材料;
所述步骤(1)和步骤(2)无时间顺序限定。
本发明在惰性气体氛围中,将酸酐加入至端羟基可降解聚合物的二氯甲烷溶液中,进行亲核加成反应,得到羧基化聚合物;所述酸酐为丁二酸酐或戊二酸酐。
本发明优选配制端羟基可降解聚合物的二氯甲烷溶液,抽真空后充入惰性气体,得到惰性气体保护的端羟基可降解聚合物的二氯甲烷溶液。
得到惰性气体保护的端羟基可降解聚合物的二氯甲烷溶液后,本发明在惰性气体氛围中,将酸酐加入至端羟基可降解聚合物的二氯甲烷溶液中进行亲核加成反应,得到羧基化聚合物。在本发明中,亲核加成反应过程成中端羟基可降解聚合物进攻酸酐其中一个羰基,进行亲核加成再离去反应,端羟基可降解聚合物的端羟基与酸酐中一个羰基发生酯化反应,开环后甩出另一个羧基,得到端基为羧基的羧基化聚合物。在本发明中,所述端羟基可降解聚合物的二氯甲烷溶液的浓度优选为1:20~30g/ml。
在本发明中,所述酸酐的加入方式优选为滴加;所述滴加的速度优选为1~2ml/min。在本发明中,所述滴加的作用是降低反应速率,避免生成副产物。
本发明优选在惰性气体氛围中,将端羟基可降解聚合物的二氯甲烷溶液加热至反应所需温度,然后滴加酸酐,进行亲核加成反应。
在本发明中,所述端羟基可降解聚合物与酸酐的摩尔比优选为1:2~10,更优选为1:5~7。
在本发明中,所述亲核加成反应的温度优选为30~60℃,所述亲核加成反应的时间优选为0.8~1.2h。在本发明中,所述亲核加成反应的时间优选从酸酐滴加完成时计起。
亲核加成反应完成后,本发明优选将反应所得产物依次经减压蒸馏、结晶和干燥,得到羧基化聚合物。
在本发明中,所述减压蒸馏的压力优选为0.04~0.06mpa;所述减压蒸馏所得产物的单位质量的体积优选为1~2ml/g,或减压蒸馏至无溶剂馏出,再补加二氯甲烷至单位质量的体积为1~2ml/g。
在本发明中,所述结晶优选为将冷乙醚加入至减压蒸馏所得产物中,产生沉淀,然后过滤;所述冷乙醚的温度优选为0~5℃;所述冷乙醚的体积与端羟基可降解聚合物的质量比优选为5~10ml:1g。
在本发明中,所述干燥优选为真空干燥;所述真空干燥的温度优选为20~40℃;所述真空干燥的时间优选为3~5h。
本发明将三羟甲基氨基甲烷、乙酸和乙酸酐混合,得到酸性三羟甲基氨基甲烷溶液;在所述酸性三羟甲基氨基甲烷溶液的温度≤10℃的条件下,将质量百分浓度为85~90%的硝酸溶液滴加至酸性三羟甲基氨基甲烷溶液中,进行硝化反应,然后将硝化反应所得溶液的ph值调节至碱性,采用二氯甲烷进行萃取,得到2-氨基硝酸甘油。
本发明对所述三羟甲基氨基甲烷、乙酸和乙酸酐的混合顺序没有特殊限定,在本发明实施例中,优选将乙酸与乙酸酐混合,然后加入三羟甲基氨基甲烷。在本发明中,乙酸作为溶剂使用,乙酸酐作为吸水剂促进反应的正向进行。
在本发明中,所述三羟甲基氨基甲烷的物质的量与乙酸的体积比优选为1mmol:0.45~0.55ml,更优选为1mmol:0.5ml。在本发明中,所述三羟甲基氨基甲烷的物质的量与乙酸酐的体积比优选为1mmol:0.45~0.55ml,更优选为1mmol:0.5ml。
得到酸性三羟甲基氨基甲烷溶液后,本发明将酸性三羟甲基氨基甲烷溶液降温至10℃以下,然后维持酸性三羟甲基氨基甲烷溶液的温度≤10℃,将质量百分浓度为85~90%的硝酸溶液滴加至酸性三羟甲基氨基甲烷溶液,进行硝化反应。在本发明中,在酸性三羟甲基氨基甲烷溶液的温度≤10℃的条件,滴加硝酸溶液可减少副反应的发生。
在本发明中,所述三羟甲基氨基甲烷的物质的量与硝酸溶液的体积之比优选为1mmol:0.25~0.35ml。
在本发明中,所述硝化反应优选包括第一阶段反应和第二阶段反应;所述第一阶段反应的温度优选≤10℃,所述第一阶段反应的时间优选为10~15min;所述第二阶段反应的温度优选为20~40℃,所述第二阶段反应的时间优选为0.8~1.2h。在本发明中,在硝化反应过程中,三羟甲基氨基甲烷的羟基被硝化,生成2-氨基硝酸甘油的硝酸盐。
硝化反应完成后,本发明将硝化反应所得溶液的ph值调节至碱性,采用二氯甲烷进行萃取,得到2-氨基硝酸甘油。在本发明中,将硝化反应所得溶液的ph值调节为碱性,可将2-氨基硝酸甘油的硝酸盐转化为2-氨基硝酸甘油,然后经萃取,将2-氨基硝酸甘油提取出来,得到2-氨基硝酸甘油。
在本发明中,所述硝化反应所得溶液经调节后的ph值优选为9.5~10.5,更优选为10;所述调节硝化反应所得溶液所用碱溶液优选为饱和碳酸氢钠溶液或饱和碳酸钠溶液;所述碱溶液的温度优选≤10℃。在本发明中,低温碱溶液可避免调节ph值过程中放热导致2-氨基硝酸甘油分解。
在本发明中,所述萃取的次数优选为3~5次;每次萃取所用二氯甲烷的体积与三羟甲基氨基甲烷的物质的量之比优选为0.8~1.2ml:1mmol。
完成萃取后,本发明优选将每次萃取所得有机相合并,然后将合并后的有机相依次经干燥和溶剂去除,得到2-氨基硝酸甘油。
本发明对所述合并后的有机相的干燥方式没有特殊限定,采用常规的干燥方式即可;在本发明实施例中,所述干燥优选采用无水硫酸钠干燥。
本发明对所述溶剂去除的方式没有特殊限定,能够将溶剂去除即可;在本发明实施例中,所述溶剂去除优选为真空浓缩的方法。
得到羧基化聚合物和2-氨基硝酸甘油后,本发明将羧基化聚合物、偶联剂和n,n-二甲基甲酰胺混合,进行活化,然后依次加入有机碱和2-氨基硝酸甘油,进行酰胺化反应,得到硝酸酯可降解生物活性材料。
在本发明中,所述偶联剂优选为2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)、1-羟基苯并三唑(hobt)、o-苯并三氮唑-四甲基脲六氟磷酸酯(hbtu)或o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸(tbtu);所述羧基化聚合物与偶联剂的摩尔比优选为1:2~10,更优选为1:5~7。在本发明中,所述偶联剂可提高羧基化聚合物上的羧基的反应活性。
在本发明中,所述活化的温度优选为室温,所述活化的时间优选为25~35min。
在本发明中,所述有机碱优选为n,n-二异丙基乙胺(dipea)或三乙胺;所述羧基化聚合物与有机碱的摩尔比优选为1:2~10,更优选为1:5~7。在本发明中,所述有机碱可提供碱性环境,以利于酰胺化反应的进行。
在本发明中,所述羧基化聚合物和2-氨基硝酸甘油的摩尔比优选为1:2~5。
在本发明中,所述酰胺化反应的温度优选为室温,更优选为20~40℃;所述酰胺化反应的时间为22~26h。
酰胺化反应完成后,本发明优选将酰胺化反应所得产物依次经萃取、干燥、溶剂去除和正向柱层析分离,纯品溶液减压浓缩,得到硝酸酯可降解生物活性材料。
在本发明中,所述萃取优选包括如下步骤:
按照乙酸乙酯的体积与羧基化聚合物的质量之比为25~35ml:1g的比例混合,进行第一萃取,得到第一萃取液和第一萃余液;
将所述第一萃取液用水洗涤3~5次,并将每次洗涤所得水洗液合并为水相;将所述水相用乙酸乙酯洗涤2~4次,并将每次洗涤所得乙酸乙酯洗液合并为有机相;将洗涤后的第一萃取液和所述有机相合并,得到硝酸酯可降解生物活性材料的乙酸乙酯溶液。
在本发明中,每次洗涤第一萃取液所用水的体积与羧基化聚合物的质量之比优选为8~12ml:1g,更优选为10ml:1g。在本发明中,洗涤水相所用乙酸乙酯的体积与羧基化聚合物的质量之比优选为8~12ml:1g,更优选为10ml:1g。
萃取完成后,本发明优选将硝酸酯可降解生物活性材料的乙酸乙酯溶液进行干燥;所述干燥优选为干燥剂干燥;所述干燥剂优选为无水硫酸钠。
在本发明中,干燥后的硝酸酯可降解生物活性材料的乙酸乙酯溶液的溶剂去除方式优选为真空浓缩的方法。
在本发明中,所述正向柱层析分离所用固定相优选为正向硅胶;所述正向硅胶的粒径优选为200~300目;所述正向柱层析分离所用流动相优选为二氯甲烷。
本发明还提供了一种心肌补片,由包括上述技术方案所述硝酸酯可降解生物活性材料或上述技术方案所述制备方法制备得到的硝酸酯可降解生物活性材料的原料制备而成。
在本发明中,制备所述心肌补片的原料优选还包括可降解高分子聚合物。在本发明中,所述可降解高分子聚合物在心肌补片中可提供一定的力学支撑作用且可降解高分子聚合物能够更好适应体内的组织再生过程。
在本发明中,所述可降解高分子聚合物的数均分子量优选为20000~200000da;所述可降解高分子聚合物优选为聚己内酯、聚丙交酯、聚乙醇酸、聚(丙交酯-乙醇酸)共聚物、聚(丙交酯-己内酯)共聚物、聚羟基脂肪酸酯和聚对二氧六环己酮中的至少一种。
在本发明中,所述心肌补片的厚度优选为200~800μm。
在本发明中,所述心肌补片的制备方法优选包括如下步骤:
(1)将原料溶解于有机溶剂中,得到电纺丝溶液;
(2)将所述电纺丝溶液经静电纺丝喷射在匀速转动的接收棒上,得到心肌补片。
本发明优选将原料溶解于有机溶剂中,得到电纺丝溶液。
在本发明中,所述原料优选包括硝酸酯可降解生物活性材料和可降解高分子聚合物。在本发明中,所述硝酸酯可降解生物活性材料与可降解高分子聚合物的质量比优选为1:1~10。
在本发明中,所述有机溶剂优选为二氯甲烷、三氯甲烷、二氯乙烷、甲醇、乙醇、丙酮、三氟乙醇和六氟异丙醇中的至少一种。
在本发明中,所述电纺丝溶液中硝酸酯可降解生物活性材料和可降解高分子聚合物的总质量与有机溶剂的体积之比优选为0.1~0.25g:1ml。
得到电纺丝溶液后,本发明优选将所述电纺丝溶液经静电纺丝喷射在匀速转动的接收棒上,得到心肌补片。
在本发明中,所述静电纺丝的相对湿度优选为40~60%。
本发明对所述静电纺丝用注射器的直径、静电纺丝的溶液流速、直流电压、纺丝时间、接收棒直径、接收棒的转动速度和移动速度没有特殊限定,本领域技术人员可以根据需要进行常规调整。
在本发明中,所述静电纺丝的直流电压优选为10~20kv;所述静电纺丝的溶液流速优选为0.1~10ml/h;所述静电纺丝所用注射器渗透于接收棒的距离优选为10~15cm。
在本发明中,所述接收棒的直径优选为0.5~10mm;所述接收棒的转速优选为1~500rpm;所述接收棒的移动速度优选为1~50mm/sec。
完成静电纺丝后,本发明优选将静电纺丝所得管状材料从接收棒上取下,然后经干燥,得到心肌补片。本领域技术人员可以根据需要对所得心肌补片进行裁剪。
本发明对所述静电纺丝所得管状材料的干燥优选为真空干燥;所述真空干燥的温度优选为15~25℃;所述真空干燥的压力优选为-0.1~-0.075mpa;所述真空干燥的时间优选为3天以上。在本发明中,较长的真空干燥时间可以将制备的心肌补片内的残余有机溶剂彻底去除干净。
本发明还提供了一种人工血管,由包括上述技术方案所述的硝酸酯可降解生物活性材料或上述技术方案所述的制备方法制备得到的硝酸酯可降解生物活性材料的原料制备而成。
在本发明中,制备所述人工血管的原料优选还包括可降解高分子聚合物。本发明对人工血管中的可降解高分子聚合物的要求与前述心肌补片中的可降解高分子聚合物的要求相同,在此不再赘述。
在本发明中,所述人工血管的厚度优选为100~800μm。
在本发明中,所述人工血管的制备方法优选参照上述心肌补片的制备方法,在此不再赘述。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
硝酸酯可降解生物活性材料pcl-(ono2)2的合成方法:
将2g(1毫摩尔)聚己内酯二醇(数均分子量为2000da)置于容器中,抽真空后充入氩气,在氩气保护下,将327μl(4毫摩尔)吡啶、50ml无水二氯甲烷依次加入容器,冷却至0℃,然后将4毫摩尔的4-溴丁酰氯滴加至体系中;在0℃恒温反应4h,然后缓慢升温至室温反应24h;
反应完成后,将反应所得混合液在室温减压蒸馏,反应液浓缩至2ml/g,再按照聚己内酯二醇的质量与冷乙醚的体积之比为1g:10ml比例向减压蒸馏所得产物中加入冷乙醚(所述冷乙醚的温度为4℃),加入乙醚后析出固体即可进行抽滤,得到固体产物;将所述固体产物在25℃真空干燥4h,得到溴代聚合物;
将所述溴代聚合物溶解于30ml的无水乙腈中,然后将1.7g(10毫摩尔)的agno3加入至溴代聚合物溶液中,在室温(25℃)进行取代反应,所述取代反应的时间为24h;
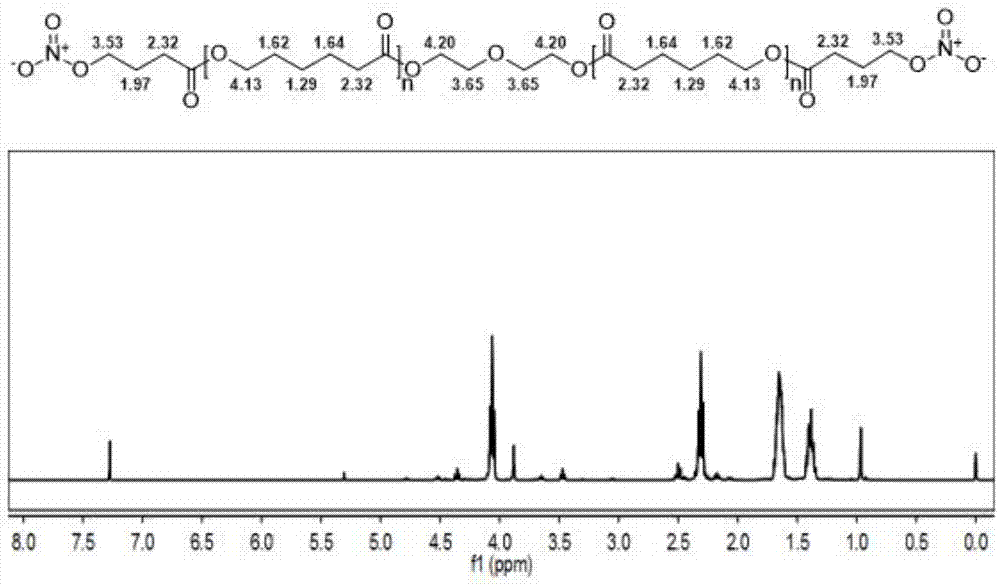
将所述取代反应所得反应液减压浓缩,去除溶剂,然后按照1g:50ml比例向减压浓缩所得产物中加入二氯甲烷,进行溶解;将溶解所得有机相用纯水洗涤3次,所述洗涤每次用纯水的量按照减压浓缩所得产品的质量与纯水的体积比为1g:50ml比例添加;将洗涤后的有机相用无水na2so4干燥后,经减压蒸馏得到硝酸酯可降解生物活性材料,即pcl-(ono2)2。本实施例所得硝酸酯可降解生物活性材料的核磁图谱如图1所示,根据图1可以确定本实施例所得产物的结构。本实施例的反应式如下式所示:
实施例2
硝酸酯可降解生物活性材料pcl-(ono2)3的合成方法:
将0.9g(1毫摩尔)聚己内酯三醇(数均分子量为900da)置于容器中,抽真空后充入氩气,在氩气保护下,将327μl(4毫摩尔)吡啶、20ml无水二氯甲烷依次加入容器,冷却至0℃,然后将4毫摩尔的5-溴戊酰氯滴加至体系中;在0℃恒温反应4h,然后缓慢升温至室温反应24h;
反应完成后,将反应所得混合液在室温减压蒸馏,反应液浓缩至2ml/g,再按照聚己内酯三醇的质量与冷乙醚的体积比为1g:10ml的比例向减压蒸馏所得产物中加入冷乙醚(所述冷乙醚的温度为4℃),加入乙醚后立即析出固体即可进行抽滤,得到固体产物;将所述固体产物在25℃真空干燥4h,得到溴代聚合物;
将所述溴代聚合物溶解于15ml的无水乙腈中,然后将0.85g(5毫摩尔)的agno3加入至溴代聚合物溶液中,在室温(25℃)进行取代反应,所述取代反应的时间为24h;
将所述取代反应所得反应液减压浓缩,去除溶剂,然后按照1g:50ml比例向减压浓缩所得产物中加入二氯甲烷,进行溶解;将溶解所得有机相用纯水洗涤3次,所述洗涤每次用纯水的量按照减压浓缩所得产品的质量与纯水的体积比为1g:50ml比例添加;将洗涤后的有机相用无水na2so4干燥后,经减压蒸馏得到硝酸酯可降解生物活性材料,即pcl-(ono2)3。本实施例的反应式如下式所示:
实施例3
硝酸酯可降解生物活性材料pla-(ono2)4的合成方法:
将3.5g(1毫摩尔)四臂聚(环氧乙烷)-block-聚丙交酯(数均分子量为3500da)置于容器中,抽真空后充入氩气,在氩气保护下,将327μl(4毫摩尔)吡啶、70ml无水二氯甲烷依次加入容器,冷却至0℃,然后将4毫摩尔的4-溴丁酰氯滴加至体系中;在0℃恒温反应4h,然后缓慢升温至室温反应24h;
反应完成后,将反应所得混合液在室温减压蒸馏,反应液浓缩至2ml/g,再按照四臂聚(环氧乙烷)-block-聚丙交酯的质量与冷乙醚的体积比为1g:8ml的比例向减压蒸馏所得产物中加入冷乙醚(所述冷乙醚的温度为4℃),加入乙醚后立即析出固体即可进行抽滤得到固体产物;将所述固体产物在25℃真空干燥4h,得到溴代聚合物;
将所述溴代聚合物溶解于30ml的无水乙腈中,然后将1.7g(10毫摩尔)的agno3加入至溴代聚合物溶液中,在室温(25℃)进行取代反应,所述取代反应的时间为24h;
将所述取代反应所得反应液减压浓缩,去除溶剂,然后按照1g:40ml比例向减压浓缩所得产物中加入二氯甲烷,进行溶解;将溶解所得有机相用纯水洗涤3次,所述洗涤每次用纯水的量按照减压浓缩所得产品的质量与纯水的体积比为1g:50ml比例添加;将洗涤后的有机相用无水na2so4干燥后,经减压蒸馏得到硝酸酯可降解生物活性材料,即pla-(ono2)4。本实施例的反应式如下式所示:
实施例4
硝酸酯可降解生物活性材料pdo-(ono2)2的合成方法:
将6.5g(1毫摩尔)双端羟基聚对二氧环己酮(数均分子量为6500da)置于容器中,抽真空后充入氩气,在氩气保护下,将327μl(4毫摩尔)吡啶、130ml无水二氯甲烷依次加入容器,冷却至0℃,然后将4毫摩尔的5-溴戊酰氯滴加至体系中;在0℃恒温反应4h,然后缓慢升温至室温反应24h;
反应完成后,将反应所得混合液在室温减压蒸馏,反应液浓缩至2ml/g,再按照双端羟基聚对二氧环己酮的质量与冷乙醚的体积比为1g:5ml的比例向减压蒸馏所得产物中加入冷乙醚(所述冷乙醚的温度为4℃),加入乙醚后立即析出固体即可进行抽滤得到固体产物;将所述固体产物在25℃真空干燥4h,得到溴代聚合物;
将所述溴代聚合物溶解于40ml的无水乙腈中,然后将1.7g(10毫摩尔)的agno3加入至溴代聚合物溶液中,在室温(25℃)进行取代反应,所述取代反应的时间为24h;
将所述取代反应所得反应液减压浓缩,去除溶剂,然后按照1g:50ml比例向减压浓缩所得产物中加入二氯甲烷,进行溶解;将溶解所得有机相用纯水洗涤3次,所述洗涤每次用纯水的量按照减压浓缩所得产品的质量与纯水的体积比为1g:20ml比例添加;将洗涤后的有机相用无水na2so4干燥后,经减压蒸馏得到硝酸酯可降解生物活性材料,即pdo-(ono2)2。本实施例的反应式如下式所示:
实施例5
硝酸酯可降解生物活性材料phb-ono2的合成方法:
将2g(1毫摩尔)聚3-羟基丁酸酯(数均分子量为2000da)置于容器中,抽真空后充入氩气,在氩气保护下,将327μl(4毫摩尔)吡啶、50ml无水二氯甲烷依次加入容器,冷却至0℃,然后将4毫摩尔的5-溴戊酰氯滴加至体系中;在0℃恒温反应4h,然后缓慢升温至室温反应24h;
反应完成后,将反应所得混合液在室温减压蒸馏,反应液浓缩至2ml/g,再按照聚3-羟基丁酸酯的质量与冷乙醚的体积比为1g:8ml的比例向减压蒸馏所得产物中加入冷乙醚(所述冷乙醚的温度为4℃),加入乙醚后立即析出固体即可进行抽滤得到固体产物;将所述固体产物在25℃真空干燥4h,得到溴代聚合物;
将所述溴代聚合物溶解于30ml的无水乙腈中,然后将1.7g(10毫摩尔)的agno3加入至溴代聚合物溶液中,在室温(25℃)进行取代反应,所述取代反应的时间为24h;
将所述取代反应所得反应液减压浓缩,去除溶剂,然后按照1g:40ml比例向减压浓缩所得产物中加入二氯甲烷,进行溶解;将溶解所得有机相用纯水洗涤3次,所述洗涤每次用纯水的量按照减压浓缩所得产品的质量与纯水的体积比为1g:50ml比例添加;将洗涤后的有机相用无水na2so4干燥后,经减压蒸馏得到硝酸酯可降解生物活性材料,即phb-ono2。本实施例的反应式如下式所示:
实施例6
硝酸酯可降解生物活性材料pcl-(ono2)6的合成方法:
将2g(1毫摩尔)的聚己内酯二醇(数均分子量为2000da)溶于50ml无水二氯甲烷中,抽真空后充入氩气,在氩气保护下,升温至50℃;维持溶液温度不变,滴加10毫摩尔的丁二酸酐,加热回流反应1h;反应结束后,将反应液室温减压浓缩至2ml/g。按照冷乙醚的体积与聚己内酯二醇的质量比为10ml:1g的比例向减压浓缩所得溶液中加入冷乙醚(所述冷乙醚的温度为4℃)使产物沉出,经过滤后,将过滤所得产物在30℃真空干燥4h,得到羧基化聚合物;
将10mmol的三羟甲基氨基甲烷溶于乙酸和乙酸酐的混合溶剂中,得到酸性三羟甲基氨基甲烷溶液,所述混合溶剂由5ml乙酸和5ml乙酸酐混合而得到;将酸性三羟甲基氨基甲烷溶液降温至10℃以下,维持温度不变,将质量百分比浓度为90%的硝酸缓慢滴加至酸性三羟甲基氨基甲烷溶液中,继续搅拌15min,然后恢复至室温,继续反应1h;反应完成后,用低于10℃的饱和碳酸氢钠溶液将所得反应液的ph值调节至10;然后二氯甲烷对反应液进行3次萃取,每次萃取所用二氯甲烷的体积为10ml,将所得有机相合并后依次经无水硫酸钠干燥和真空浓缩去除溶剂,得到2-氨基硝酸甘油;
将1mmol的羧基化聚合物和5mmol的2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)溶于2ml无水n,n-二甲基甲酰胺中,在室温活化30min后,在搅拌条件下依次加入5mmol的n,n-二异丙基乙胺(dipea)和5.0mmol的2-氨基硝酸甘油,在室温反应24h;反应完成后,将反应液转移至分液漏斗中,按照乙酸乙酯的体积与羧基化聚合物的质量之比为30ml:1g的比例将乙酸乙酯与反应液混合,进行萃取;将萃取所得萃取液用水洗涤3次,每次所用水的体积与羧基化聚合物的质量之比为10ml:1g;将洗涤所得水相用乙酸乙酯洗涤2次,每次洗涤所用乙酸乙酯的体积与羧基化聚合物的质量之比为10ml:1g;将洗涤后的萃取液和洗涤水相所得乙酸乙酯合并,然后依次进行无水硫酸钠干燥和真空浓缩,得粗品;以粒径为200~300目的正向硅胶为固定相,以二氯甲烷为流动相,对所述粗品进行正向柱层析分离,得到硝酸酯可降解生物活性材料,即pcl-(ono2)6。本实施例所得硝酸酯可降解生物活性材料的核磁图谱如图2所示,根据图2可以确定本实施例所得产物的结构(详见图2)。上述反应的化学反应式如下式所示:
实施例7
硝酸酯可降解生物活性材料plga-(ono2)3的合成方法:
将10g(1毫摩尔)聚丙交酯-乙醇酸共聚物(数均分子量为10000da)溶于200ml无水二氯甲烷中,抽真空后充入氩气,在氩气保护下,升温至45℃;维持溶液温度不变,滴加10毫摩尔的丁二酸酐,加热回流反应1h;反应结束后,将反应液室温减压浓缩至2ml/g。按照冷乙醚的体积与聚丙交酯-乙醇酸共聚物的质量比为7.5ml:1g的比例向减压浓缩所得溶液中加入冷乙醚(所述冷乙醚的温度为4℃)使产物沉出,经过滤后,将过滤所得产物在30℃真空干燥4h,得到羧基化聚丙交酯-乙醇酸共聚物;
按照实施例6所述的方法制备2-氨基硝酸甘油;
将1mmol的羧基化聚丙交酯-乙醇酸共聚物和5mmol的2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)溶于2ml无水n,n-二甲基甲酰胺中,在室温活化30min后,在搅拌条件下依次加入5mmol的n,n-二异丙基乙胺(dipea)和5.0mmol的2-氨基硝酸甘油,在室温反应24h;反应完成后,将反应液转移至分液漏斗中,按照乙酸乙酯的体积与羧基化聚丙交酯-乙醇酸共聚物的质量之比为25ml:1g的比例将乙酸乙酯与反应液混合,进行萃取;将萃取所得萃取液用水洗涤3次,每次所用水的体积与羧基化聚丙交酯-乙醇酸共聚物的质量之比为8ml:1g;将洗涤所得水相用乙酸乙酯洗涤2次,每次洗涤所用乙酸乙酯的体积与羧基化聚丙交酯-乙醇酸共聚物的质量之比为10ml:1g;将洗涤后的萃取液和洗涤水相所得乙酸乙酯合并,然后依次进行无水硫酸钠干燥和真空浓缩,得粗品;以粒径为200~300目的正向硅胶为固定相,以二氯甲烷为流动相,对所述粗品进行正向柱层析分离,得到硝酸酯可降解生物活性材料,即plga-(ono2)3。本实施例的化学反应式如下式所示:
实施例8
硝酸酯可降解生物活性材料pcl-(ono2)12的合成方法:
将2.5g(1毫摩尔)四臂聚(环氧乙烷)-block-聚己内酯(数均分子量为2500)溶于75ml无水二氯甲烷中,抽真空后充入氩气,在氩气保护下,升温至55℃;维持溶液温度不变,滴加10毫摩尔的丁二酸酐,加热回流反应1h;反应结束后,在0.05mpa的压力下减压浓缩完全除去dcm,再加入二氯甲烷,得到单位质量体积为2ml/g的溶液;按照冷乙醚的体积与四臂聚(环氧乙烷)-block-聚己内酯的质量比为10ml:1g的比例向减压浓缩所得溶液中加入冷乙醚(所述冷乙醚的温度为4℃)使产物沉出,经过滤后,将过滤所得产物在30℃真空干燥4h,得到羧基化四臂聚(环氧乙烷)-block-聚己内酯;
按照实施例6所述的方法制备2-氨基硝酸甘油;
将1mmol的羧基化四臂聚(环氧乙烷)-block-聚己内酯和5mmol的2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)溶于2ml无水n,n-二甲基甲酰胺中,在室温活化30min后,在搅拌条件下依次加入5mmol的n,n-二异丙基乙胺(dipea)和5.0mmol的2-氨基硝酸甘油,在室温反应24h;反应完成后,将反应液转移至分液漏斗中,按照乙酸乙酯的体积与羧基化四臂聚(环氧乙烷)-block-聚己内酯的质量之比为30ml:1g的比例将乙酸乙酯与反应液混合,进行萃取;将萃取所得萃取液用水洗涤3次,每次所用水的体积与羧基化四臂聚(环氧乙烷)-block-聚己内酯的质量之比为10ml:1g;将洗涤所得水相用乙酸乙酯洗涤2次,每次洗涤所用乙酸乙酯的体积与羧基化四臂聚(环氧乙烷)-block-聚己内酯的质量之比为10ml:1g;将洗涤后的萃取液和洗涤水相所得乙酸乙酯合并,然后依次进行无水硫酸钠干燥和真空浓缩,得粗品;以粒径为200~300目的正向硅胶为固定相,以二氯甲烷为流动相,对所述粗品进行正向柱层析分离,得到硝酸酯可降解生物活性材料,即pcl-(ono2)12。本实施例的化学反应式如下式所示:
实施例9
(1)将0.9g数均分子量为80000da的聚己内酯和0.1g实施例1所得硝酸酯可降解生物活性材料(pcl-(ono2)2)加入至10ml有机溶剂中,所述有机溶剂为体积比为5:1的氯仿和甲醇的混合溶剂,室温搅拌过夜,得到电纺丝溶液,即聚己内酯-硝酸酯可降解生物活性材料溶液;
(2)在室温、相对湿度为50%的条件下,将所述电纺丝溶液装入直径为14.9mm的注射器中,并将高压直流电源与注射器针头相连,调整注射器针头对准圆柱接收器的中央,设置针头与接收器之间的距离为15cm,溶液流速为2ml/h,直流电压为13kv;以直径为4mm的不锈钢圆柱为接收棒,接收棒的转速为400rpm,接收棒的移动速度为5mm/s;静电纺丝4h后,将所得管状材料从接收棒上取下,沿纵轴方向剖开后,在温度为22℃、压力为-0.075mpa的条件下真空干燥,使溶剂彻底挥发,得到心肌补片;测得心肌补片厚度为600μm。
利用化学发光技术检测本实施例所得心肌补片中no含量,具体为将心肌补片剪取2mg置于5ml的50mm的三氯化钒溶液中,采用一氧化氮分析检测仪对心肌补片中no含量进行测定。图3为本实施例制备的心肌补片中no含量的测试结果,从结果分析可知,所述心肌补片中no含量为5.5nmol/mg。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料释放no的性能进行测试,结果类似。
实施例10
建立心肌梗死模型后,根据是否植入补片及植入补片的类型将实验动物分为4组:
sham组,即假手术组,仅进行开胸手术,不缝合冠状动脉左前降支;
ami组,即单纯的心肌梗死模型组,开胸并缝合冠状动脉左前降支;
pcl补片组,即建立心肌梗死模型后,于缺血心肌外缝合单纯的聚己内酯(pcl材料)补片;
硝酸酯功能化的心肌补片组(即pcl-(ono2)2补片组),即建立心肌梗死模型,并于缺血心肌外缝合带有硝酸酯可降解生物活性材料的心肌补片(即实施例9所得心肌补片)。
各实验组大鼠在术后、术后1天和术后28天分别进行心脏超声检测。
大鼠心肌梗死模型采用如下方法建立:
(1)sd(sprague-dawley)大鼠,体重280~300g,经腹腔注射水合氯醛(10%,350mg/kg)诱导麻醉,用脱毛膏移除胸部被毛;
(2)将大鼠仰卧位固定在手术台上,经口腔进行气管插管,设置呼吸机(hallowellemc,microvent1)频率为110次/分钟,潮气量为6ml;
(3)碘伏消毒胸部皮肤,用剪刀沿剑突与腋中线起点的连线剪开皮肤,切口长度为2cm;用镊子钝性分离胸大肌和前锯肌交界处筋膜,找到肋骨,于第三肋和第四肋间切开肋间肌,打开胸腔,放置扩胸器牵开肋骨,暴露心脏;显微剪剪开心包膜,在肺动脉圆锥与左心耳交界下行2mm处用6-0丝线缝合冠状动脉左前降支。
心肌补片采用如下方法植入:
用8-0丝线将心肌补片沿梗死边界覆盖、固定、贴合在缺血心肌表面。
利用心脏超声检测心梗大鼠的心脏功能,统计分析其射血分数(ef)和缩短分数(fs)。在术后立即对各实验组大鼠做心脏超声,假手术组的ef值为63±3%,fs值为35±2%,而ami组、pcl补片组和pcl-(ono2)2补片组与假手术组相比ef值和fs值均明显降低,而三组相互之间不存在明显差异,左心室内径无明显改变。术后1天时,移植pcl-(ono2)2补片后,能够部分抑制心肌缺血对心功能的影响。到术后28天时,心脏超声数据显示,pcl补片组大鼠的ef值(22±1.2%)和fs值(12±0.3%)下降到与ami组大鼠同一水平,左心室内径也有很大程度的扩张,而pcl-(ono2)2补片组大鼠的ef值(42±0.7%)和fs值(23±0.6%)明显高于ami组和pcl组(p<0.05),左心室内径较后两组相比也明显减小(p<0.05)。相比较假手术组,其余三组心肌梗死的大鼠均表现出不同程度的左室前壁振幅减弱,室腔膨大以及室壁变薄,如图4双向箭头标示的左心室舒张末期内径,由超声检测图可观察到心梗后左室前壁的振幅明显减弱,室壁变薄。这些结果表明,植入实施例9所制备的心肌补片后,改善了心肌缺血大鼠的心功能。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例11
对心肌补片增加心梗大鼠心肌血流灌注的情况进行检测:
按照实施例10所述的分组方式分组,采用实施例10所述的方法建立心梗模型和植入补片。在术后1天,利用荧光微球法测定心肌层血液灌注程度。采用如下方法向心内注射荧光微球:
(1)经腹腔注射10%水合氯醛(350mg/kg)麻醉大鼠,经口气管插管;
(2)沿剑突和腋中线起点的连线剪开皮肤,并分离肌肉,打开胸腔,暴露心脏;
(3)用2.5ml注射器将直径为15μm、红色荧光标记的聚苯乙烯微球(106/ml)缓慢注射至左心室内,注射体积为1ml;
(4)45min后,进行心内灌注和取材,并冷冻切片。
利用荧光微球法测定心肌层血液灌注程度,硝酸酯功能化的心肌补片组与ami组和pcl补片组相比,在缺血边界区域有更多的荧光微球分布,提示该区域有更多的血流灌注。对荧光微球的量化分析显示,以假手术组的血流灌注量为基准(计为100%),移植实施例9所得心肌补片后,心肌缺血大鼠在缺血边界区的血流灌注量恢复到假手术组的30%以上,ami组和pcl补片组分别在缺血边界区的血流灌注降低到假手术组的12%和15%左右。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例12
心肌补片减轻心肌缺血损伤的测试:
采用实施例10所述的方法进行分组、建立模型和植入补片。在大鼠手术后1天,收集血清和心脏样本,利用nagor-olsen染色观察心肌缺血损伤的范围和程度,用以评价实施例9所述心肌补片能够减轻心肌缺血损伤的作用。
利用nagor-olsen染色直接显示缺血受损的心肌细胞,在pcl-(ono2)2心肌补片组,红色着染的心肌缺血损伤的组织面积约为20±0.3%(占样品总面积的百分比),与ami组的缺血面积(35±1.8%)和pcl补片组(30±1.5%)相比明显减少(p<0.05),说明实施例9所得心肌补片的植入能够减轻心肌缺血损伤和减缓心肌梗死区和缺血边界区的细胞凋亡。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例13
心肌补片移植后心肌梗死体积的测试:
采用实施例10所述方法进行分组、建立模型和植入补片。各组大鼠在术后28天取材,得到的大鼠心脏从心尖到心底以1mm厚度间隔切成5块,然后分别包埋制成蜡块,石蜡切片。
masson染色是参照masson三色染色试剂盒(北京雷根生物技术有限公司)进行染色,染色的切片利用虚拟切片扫描显微镜(motic,ba600)扫描得到组织全图。
利用masson染色显示纤维化瘢痕组织,并计算心肌梗死体积,统计结果显示,pcl-(ono2)2补片组心肌梗死体积百分比为20%,而ami组心肌梗死体积百分比为39%,pcl组心肌梗死体积百分比为34%。结果表明,实施例9所制备的pcl-(ono2)2补片组大鼠的心肌梗死范围与ami组和pcl组相比明显减少(p<0.05)。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例14
心肌补片调节心肌缺血后的炎症反应测试:
采用实施例10所述的方法进行分组、建立模型和植入补片。在术后28天采用巨噬细胞浸润免疫荧光染色的方法测试炎症反应。
巨噬细胞浸润免疫荧光染色的具体步骤:
(1)心肌冷冻切片室温晾干2h后,用流水冲洗5min,之后再用pbs漂洗2次,每次5min;
(2)视具体染色情况做通透性处理:将切片浸泡于0.5%tritonx-100中,室温放置10min,之后用pbs洗3次,每次5min;
(3)组织表面滴加封闭用山羊血清,将切片置于湿盒中,室温放置30min;
(4)一抗(cd68)孵育:甩去组织表面的封闭血清,不经pbs洗涤直接滴加抗体工作液,切片置于湿盒中,4℃孵育过夜(约14h);
(5)复温和二抗孵育:取出切片复温,pbs洗3次,每次5min,在组织表面滴加二抗工作液,避光、室温反应2h,之后再用pbs洗3次,每次5min;
(6)dapi封片,荧光显微镜下观察,取图。
通过免疫荧光检测并计数缺血后心肌内巨噬细胞浸润情况,数据分析显示,植入实施例9所得心肌补片后,在缺血边界区,cd68标记的浸润的巨噬细胞的数量与ami组和pcl组相比,明显减少(p<0.001)。
通过免疫荧光检测并计数缺血后心肌内巨噬细胞浸润,植入实施例9所得心肌补片后,在缺血边界区,ami组和pcl组相比,cd68标记的浸润的巨噬细胞数量为30.57±5.61/hpf和32.79±0.64/hpf;pcl-(ono2)2补片组,cd68标记的浸润的巨噬细胞数量为8.62±0.74/hpf,浸润的巨噬细胞的数量明显减少(p<0.001)。
血清mpo活性的测定:利用髓过氧化物酶(mpo)测试盒(南京建成生物工程研究所,货号a044)测定血清mpo的含量。
血清mpo含量能够反映中性粒细胞的浸润程度,同时是导致氧化应激损伤的重要酶类。建立ami模型后,血清mpo的活性显著提高,而pcl-(ono2)2补片组大鼠血清mpo的活性为12u/l,ami组和pcl组的血清mpo的活性分别为18u/l和16u/l,以上结果提示实施例9所提供的补片能够调节心肌梗死后的炎症反应,显著降低氧化应激损伤。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例15
心肌补片促进心肌细胞增殖的测试:
采用实施例10所述的方法进行分组、建立模型和植入补片。
brdu是胸腺嘧啶的类似物,会在细胞增殖时整合进入细胞dna分子中,从而使增殖的细胞被brdu标记,而后利用brdu抗体进行免疫染色(同实施例14),以显示增殖的细胞。在大鼠在术后第10天开始经腹腔注射brdu,连续注射7天后,收集大鼠心脏,经4%多聚甲醛固定后,进行冷冻切片,切片厚度为5μm;之后,利用免疫荧光双重染色技术,将brdu和心肌细胞的标记物α-sa(α-sarcomericactin)进行共染,荧光显微镜下观察,双阳性细胞即为增殖的心肌细胞。在梗死边界取图,并统计brdu/α-sa双阳性细胞数量。在进行抗体孵育前,需用2n的盐酸处理,使dna双链破坏,从而便于brdu抗体的结合,处理条件为37℃浸泡20min,之后用ph值为8.0的硼酸中和10min,pbs洗涤后滴加抗体孵育。
移植pcl-(ono2)2补片组的心梗大鼠,增殖的心肌细胞密度为13±3/mm2,与ami组(5±1/mm2)和pcl组(6±2/mm2)相比,显著增多(p<0.001),说明实施例9所得心肌补片能促进心肌细胞增殖。
按照上述方法对实施例2~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例16
(1)将2.25g聚(丙交酯-己内酯)共聚物(plcl)(丙交酯和己内酯的单体比例为1:1)和0.25g实施例7所得硝酸酯可降解生物活性材料(plga-(ono2)3)加入至10ml有机溶剂中,所述有机溶剂为六氟异丙醇,室温搅拌过夜,得到电纺丝溶液,即聚(丙交酯-己内酯)共聚物-硝酸酯可降解生物活性材料溶液;
(2)在室温、相对湿度为50%的条件,将所述电纺丝溶液装入直径为14.9mm的注射器中,并将高压直流电源与注射器针头相连,调整注射器针头对准圆柱接收器的中央,设置针头与接收器之间的距离为12cm,溶液流速为8ml/h,直流电压为13kv;以直径为2mm的不锈钢圆柱为接收棒,接收棒的转速为400rpm,接收棒的移动速度为5mm/s;静电纺丝8min后,将所得管状材料从接收棒上取下,沿纵轴方向剖开后,在温度为22℃、压力为-0.075mpa的条件下真空干燥,使溶剂彻底挥发,得到厚度为450μm的硝酸酯功能化人工血管。
利用化学发光技术检测本实施例所得硝酸酯功能化人工血管中no含量,具体为将硝酸酯功能化人工血管剪取2mg置于5ml的50mm的三氯化钒溶液中,采用一氧化氮分析检测仪对硝酸酯功能化人工血管中no含量进行测定。图5为本实施例制备的硝酸酯功能化人工血管中no含量的测试结果,从结果分析可知,硝酸酯功能化人工血管中no含量为7.2nmol/mg。
按照按上述方法对实施例1~6和实施例8所得硝酸酯可降解生物活性材料释放no的性能进行测试,结果类似。
实施例17
对大鼠腹主动脉移植人工血管改善血管通畅性的测试:
按照如下方法分别对大鼠腹主动脉原位移植实施例16所得硝酸酯功能化人工血管和单纯的plcl人工血管(记为对照组):
截取长度为1.0cm的硝酸酯功能化人工血管,利用大鼠腹主动脉移植模型,用9-0尼龙缝合线通过端端吻合术将人工血管植入自体大鼠的腹主动脉中,具体如下:
(1)sd(sprague-dawley)大鼠,体重280-300g,经腹腔注射水合氯醛(10%,350mg/kg)诱导麻醉,腹部备皮;
(2)沿腹部中线剪开皮肤和肌肉层,将腹主动脉剥离,结扎其分支,用动脉夹夹住腹主动脉的两端,然后从中间剪开,用9-0带针缝合线原位缝合人工血管,每端8针;待两端缝好后,缓慢摘除动脉夹,恢复血流;
(3)用硫酸庆大霉素冲洗腹腔,将胃肠等器官复位,用3-0缝合线缝合肌肉层和皮肤,碘伏消毒;
(4)术后4和12周观察,处死前利用多普勒超声检测植入血管的通畅情况,取材后通过体式显微镜对移植血管内外进行整体观测。
通过彩色多普勒超声发现,植入4周和12周后,实施例16所得硝酸酯功能化人工血管通畅良好,没有发现渗血和动脉瘤;通过超声结果观察到实施例16所得硝酸酯功能化人工血管移植后没有再狭窄,而单纯plcl人工血管的对照组的血流速度为168±5.6mm/s,植入实施例16所得硝酸酯功能化人工血管人工血管12周后的血流速度为226±3mm/s,更接近天然颈动脉血流速度(227±6mm/s)。
按照上述方法对实施例1和3~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例18
实施例16所得硝酸酯功能化人工血管移植对组织再生的影响:
人工血管移植方法同实施例17,移植后取材分析:
(1)大鼠腹主动脉移植后术后12周的时候,将大鼠麻醉,尾静脉注射肝素进行全身肝素化,腹主动脉备皮,打开皮肤肌肉层,再用生理盐水心脏灌注,然后剥离植入的人工血管,进行取材;
(2)将人工血管两端天然血管用3-0缝合线结扎,剪下人工血管,迅速用50u/ml肝素钠生理盐水溶液冲洗血管内腔,并浸泡于50u/ml肝素钠生理盐水溶液中,用显微剪小心剥离血管外面的结蹄组织和脂肪层;
(3)取材后进行体视显微镜观察并冷冻切片后进行组织学分析。
采用h&e染色、masson染色、verhoeff-vangieson组织学染色,观察血管组织再生情况。结果显示,实施例16所得硝酸酯功能化人工血管移植后通过vvg染色观察弹性蛋白在人工血管支架管壁的沉积,弹性蛋白分布的区域和平滑肌细胞一致;植入12周后,大量弹性纤维分布于新生内膜层和血管中层。通过h&e染色表明实施例16所得硝酸酯功能化人工血管材料具有的纤维结构和孔结构有利于细胞向材料内部迁移,从而促进血管再生与重构,能够利用植入部位微环境实现拟天然再生。
按照上述方法对实施例1和3~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
实施例19
对人工血管平滑肌及内皮再生情况的测试:
按照实施例17的方法分别对大鼠腹主动脉原位移植实施例16所得硝酸酯功能化人工血管和聚己plcl人工血管(对照组)。在术后28天,对原位移植的人工血管取材分析,利用免疫荧光染色,其中一抗分别用cd31抗体和α-sma抗体分别观察人工血管内皮和平滑肌再生,免疫荧光染色步骤同实施例14。
α-sma免疫荧光染色观察平滑肌再生情况,实施例16所得硝酸酯功能化人工血管的外侧和中层分布有大量成肌纤维细胞(α-sma+),平滑肌的厚度为89±2μm,常规plcl人工血管(对照组)只有外侧有一些从周围组织迁移过来的成肌纤维细胞,平滑基层的厚度为51±3μm。通过cd31免疫荧光染色观察内皮的形成,实施例16所得硝酸酯功能化人工血管的内皮覆盖率在2周和4周时分别为27.2±11.5%和51.1±6.4%,显著高于plcl对照组的内皮化(2周1.8±1.1%,4周11.5±3.2%)。通过cd31染色观察到4周后实施例16制备的硝酸酯功能化人工血管外侧形成新生微血管密度显著高于plcl人工血管对照组。
按照上述方法对实施例1和3~8所得硝酸酯可降解生物活性材料进行测试,结果类似。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!