一种酵母中高效克隆载体的构建方法及应用与流程
本发明属于生物工程技术领域,尤其是一种酵母中高效克隆载体的构建方法及应用。
背景技术
在分子生物学实验中,载体的构建是研究基因功能的基础性工作。大部分目的基因都是通过设计不同需求的特异性引物进行pcr扩增,再连接到相应的载体上。在连接方式上,有in-fusion,gateway等同源重组技术的连接,以及通过dna连接酶介导的粘性末端及平末端的连接。同源重组方式连接价格较为昂贵,针对不同的目的片段进行克隆连接效率波动也较大;酶切连接的方式较为简单,然而酶切位点具有选择局限性,要通过含有相应酶切位点的特异性引物扩增克隆目的基因再进行下一步扩增,操作增加了繁琐。
线性化的t载体在其两端各具有一个突出的dt,通过高保真酶扩增后的pcr产物经过加a反应后在其两端各会产生一个突出的da,pcr产物与线性化的t载体中突出的dt和da互补配对,在连接酶的作用下完成克隆连接。在ta克隆过程中,线性化载体不会对目的基因进行选择,因此对于大量的、各不相同的目的基因同时进行ta克隆连接,会大大增加克隆基因的数量。通过ta克隆的方式进行目的基因与载体的连接,其成本较低,操作简单,连接效率高,可进行高通量的基因克隆。
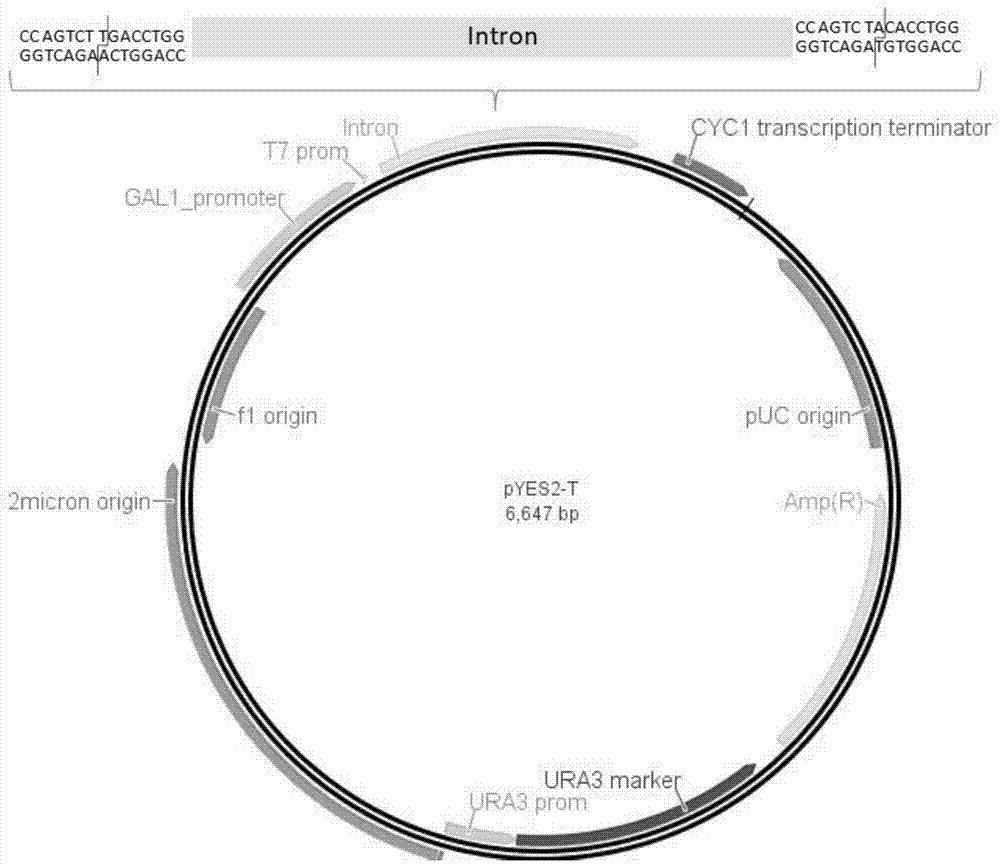
pyes2酵母表达载体是一种穿梭质粒,具有原核复制子和真核复制子,可以存在于酿酒酵母和大肠杆菌两种宿主菌内,可同时进行基因操作以及真核表达。pyes2具有gal1半乳糖诱导的真核启动子,因此在半乳糖的诱导下,一方面可以使目的基因在酵母中稳定表达,通过不同的酵母培养基底物对基因功能进行验证,另一方面在ta克隆过程中存在的反向连接可以得到有效筛选。
通过检索,尚未发现与本发明专利申请相关的专利公开文献。
技术实现要素:
本发明的目的在于克服现有技术的不足之处,提供一种酵母中高效克隆载体的构建方法及应用,该方法构建的酵母表达载体使用方便,效率高,成本低,应用前景广泛,该方法可以在酿酒酵母中诱导表达目的基因的表达以及高通量的筛选目的基因。
本发明解决其技术问题是采取以下技术方案实现的:
一种酵母中高效克隆载体的构建方法,所述载体为pyes2-t,所述方法中使用了一对特异添加的接头引物以及含有pyes2-t的宿主细胞。
而且,步骤如下:
⑴通过定点突变将pyes2载体ura3中原有的xcmⅰ酶切位点进行定点同义突变;
⑵通过含有xcmⅰ酶切位点的一对特异性引物以pfgc5941载体为模板,利用dna聚合酶进行pcr扩增得到含有761bp的intron;
⑶用scaⅰ和bamhⅰ对步骤⑴所得载体进行酶切,电泳,并胶回收线性化载体;
⑷通过同源重组技术将扩增的xcmⅰ-intron-xcmⅰ片段连接到线性化的载体中,即得酵母中高效克隆载体。
而且,所述步骤⑴中对酵母表达载体pyes2载体骨架中尿嘧啶筛选标记基因含有的xcmⅰ位点进行点突变;
或者,所述步骤⑷中同源重组技术为利用酶切位点,通过同源重组酶或者核酸外切酶3介导的同源重组技术。
而且,在半乳糖诱导gal1启动子后构建xcmⅰ酶切位点以及xcmⅰ酶切位点之间761bp的intron,intron的序列为seqidno.1所示。
而且,所述步骤⑵中dna聚合酶为高保真dna聚合酶;或者,一对特异性引物为含有xcmⅰ酶切位点的一对特异性引物。
而且,所述方法在pyes2-t多克隆位点位置保留了pyes2的所有多克隆位点;
或者,通过t4dna连接酶对由pcr扩增得到的平端pcr片段加a后或对添加有a的黏性末端进行ta克隆连接。
而且,所述一对接头引物为含有scaⅰ和bamhⅰ酶切位点的一对接头引物。
而且,所述宿主细胞是酿酒酵母细胞inscv1或大肠杆菌。
而且,具体步骤如下:
⑴原始载体pyes2中xcmⅰ的定点突变
将ura3元件上的xcmⅰ酶切位点进行同义点突变,使用正向引物seqidno:1及反向引物seqidno:2,使用dna聚合酶以pyes2为模板进行反向扩增,扩增体系为:模板50ng,10mm正向引物和反向引物各1μl,dna聚合酶buffer4μl,10mmol/ldntps0.4μl,dna聚合酶0.2μl,dmso0.6μl,共20μl体系,剩余用水补齐;
pcr扩增程序为:98℃30s,30个循环的程序为:98℃10s,62℃30s,68℃3min,最后68℃5min;pcr产物使用t4连接酶连接,体系为:pcr产物5μl,10xt4dnaligasebuffer5μl,t4dnaligase5μl,ddh2o5μl,22℃孵育1小时;转化大肠杆菌并挑单克隆测序;
⑵设计含有xcmⅰ酶切位点的特异性引物扩增intron
利用特异性引物以pfgc5941为模板扩增intron,所述特异性引物为:正向引物:seqidno:3,反向引物:seqidno:4,得到xcmⅰ-intron-xcmⅰ元件;扩增体系为:模板50ng,10mm正向引物反向引物各1μl,dna聚合酶buffer4μl,10mmol/ldntps0.4μl,dna聚合酶0.2μl,共20μl体系,剩余用水补齐;
pcr扩增程序为:98℃30s,30个循环的程序为:98℃10s,60℃30s,72℃1min,最后72℃5min;pcr产物电泳切胶保存;
⑶scaⅰ和bamhⅰ酶切载体、同源重组连接及转化
对pyes2进行双酶切,双酶为scaⅰ和bamhⅰ,酶切体系为:saci1μl,bamhⅰ1μl,10×bufffer2.5μl,pyes25μl,ddh2o18μl;
使用正向引物seqidno:5,反向引物seqidno:6对步骤⑵中的pcr产物进行扩增,扩增体系为:模板1μl,10mm正向引物反向引物各1μl,dna聚合酶buffer4μl,10mmol/ldntps0.4μl,dna聚合酶0.2μl,共20μl体系,剩余用水补齐;
pcr扩增程序为:98℃30s,30个循环的程序为:98℃10s,60℃30s,72℃1min,最后72℃5min;pcr产物电泳切胶回收;回收产物通过dna重组酶与酶切后的载体进行同源重组连接,连接体系为:线性化载体pyes23μl;pcr产物5μl;dna重组酶2μl;50℃,温育15min,然后置于冰上;将连接好的产物通过热激转化的方法转入大肠感受态细胞top10中;转化大肠杆菌并挑单克隆测序;正确克隆即为酵母中高效克隆载体,将正确克隆置于-80℃保存。
如上所述的酵母中高效克隆载体的构建方法所构建的载体在高通量地对cdna文库进行目的基因的筛选方面的应用。
本发明取得的优点和积极效果是:
1、本发明方法通过构建酵母中应用的高效克隆载体,当xcmⅰ酶切后使载体产生两个突出的t末端,一方面可以通过pcr产物加a的方式直接进行ta克隆,所述载体保留了原载体的多克隆位点,方便下游实验的使用;另一方面利用在半乳糖诱导启动子的调控下,对ta克隆后的基因在酵母中诱导表达。利用本改造载体,可以高通量地对大量cdna文库进行目的基因的筛选;本发明方法构建的酵母表达载体使用方便,效率高,成本低,应用前景广泛。
2、本发明方法在xcmⅰ酶切位点之间增加了761bp的intron,无外源基因效应的同时可显著提高酶切效率及回收效率。
3、本发明方法可以快速地通过ta克隆,转化酿酒酵母来对目的基因进行功能验证。
4、本发明方法可以通过半乳糖诱导表达有效方便快捷地筛选出ta克隆存在的反向克隆以及无效克隆。
5、本发明方法保留了原载体多克隆位点,可将xcmⅰ-intron-xcmⅰ元件盒快速克隆到其他载体中。
附图说明
图1为本发明中pyes2-t前载体图;
图2为本发明中xcmⅰ酶切pyes2-t前载体电泳图;
图3为本发明中pyes2-t载体构建完成测序图;
图4为本发明中pyes2-t-egfp载体图;
图5为本发明pyes2-t-egfp载体构建完成测序图。
具体实施方式
下面结合通过具体实施例对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围。
本发明中所使用的原料,如无特殊说明,均为常规的市售产品;本发明中所使用的方法,如无特殊说明,均为本领域的常规方法。
实施例1
一种酵母中高效克隆载体的构建方法,所述载体为pyes2-t,所述方法中使用了一对特异添加的接头引物以及含有pyes2-t的宿主细胞。
较优地,步骤如下:
⑴通过定点突变将pyes2载体ura3中原有的xcmⅰ酶切位点进行定点同义突变;
⑵通过含有xcmⅰ酶切位点的一对特异性引物以pfgc5941载体为模板,利用dna聚合酶进行pcr扩增得到含有761bp的intron;
⑶用scaⅰ和bamhⅰ对步骤⑴所得载体进行酶切,电泳,并胶回收线性化载体;
⑷通过同源重组技术将扩增的xcmⅰ-intron-xcmⅰ片段连接到线性化的载体中,即得酵母中高效克隆载体。
较优地,所述步骤⑴中对酵母表达载体pyes2载体骨架中尿嘧啶筛选标记基因含有的xcmⅰ位点进行点突变;
或者,所述步骤⑷中同源重组技术为利用酶切位点,通过同源重组酶或者核酸外切酶3介导的同源重组技术。
较优地,在半乳糖诱导gal1启动子后构建xcmⅰ酶切位点以及xcmⅰ酶切位点之间761bp的intron,intron的序列为seqidno.1所示。
较优地,所述步骤⑵中dna聚合酶为高保真dna聚合酶;或者,一对特异性引物为含有xcmⅰ酶切位点的一对特异性引物。
较优地,所述方法在pyes2-t多克隆位点位置保留了pyes2的所有多克隆位点;
或者,通过t4dna连接酶对由pcr扩增得到的平端pcr片段加a后或对添加有a的黏性末端进行ta克隆连接。
较优地,所述一对接头引物为含有scaⅰ和bamhⅰ酶切位点的一对接头引物。
较优地,所述宿主细胞是酿酒酵母细胞inscv1或大肠杆菌。
如上所述的酵母中高效克隆载体的构建方法所构建的载体在高通量地对cdna文库进行目的基因的筛选方面的应用。
实施例2
一种酵母中高效克隆载体的构建方法,步骤如下:
(1)通过定点突变将pyes2载体尿嘧啶缺陷筛选基因ura3中原有的xcmⅰ酶切位点的进行定点同义突变,不改变氨基酸序列。
(2)通过含有xcmⅰ酶切位点的一对特异性引物以pfgc5941为模板利用高保真phusion酶进行pcr扩增得到含有761bp的intron。
(3)用scaⅰ和bamhⅰ对(1)所得载体进行酶切,电泳,并胶回收线性化载体。
(4)通过dna同源重组酶介导的同源重组方式将扩增的xcmⅰ-intron-xcmⅰ片段连接到线性化的载体中,得到pyes2-t前载体,即得酵母中高效克隆载体。
上述酵母中高效克隆载体的构建方法构建的高效克隆载体在大肠杆菌top10中增殖,构建过程中所用大肠杆菌为top10。
所述一种酵母表达pyes2-t前载体通过xcmⅰ酶切后电泳,选择目的片段胶回收即可获得线性化pyes2-t载体。通过t4连接酶可将经加“a”反应后的待鉴定功能的pcr产物与所述pyes2-t载体连接。
实施例3
一种酵母中高效克隆载体的构建方法,步骤如下:
1、原始载体pyes2中xcmⅰ的定点突变
将ura3元件上的xcmⅰ酶切位点进行同义点突变,使用正向引物seqidno:1及反向引物seqidno:2,使用dna聚合酶以pyes2为模板进行反向扩增,扩增体系为:模板50ng,10mm正向引物和反向引物各1μl,dna聚合酶buffer4μl,10mmol/ldntps0.4μl,dna聚合酶0.2μl,dmso0.6μl,共20μl体系,剩余用水补齐。pcr扩增程序为:98℃30s,30个循环的程序为98℃10s,62℃30s,68℃3min,最后68℃5min。pcr产物使用t4dna连接酶(连接,体系为:pcr产物5μl,10xt4dnaligasebuffer5μl,t4dnaligase5μl,ddh2o5μl,22℃孵育1小时。转化大肠杆菌并挑单克隆测序。
2、设计含有xcmⅰ酶切位点的特异性引物扩增intron
利用特异性引物(正向引物:seqidno:3,反向引物:seqidno:4)以pfgc5941为模板扩增intron,得到xcmⅰ-intron-xcmⅰ元件。扩增体系为:模板50ng,10mm正向引物反向引物各1μl,dna聚合酶buffer4μl,10mmol/ldntps0.4μl,dna聚合酶0.2μl,共20μl体系,剩余用水补齐。pcr扩增程序为:98℃30s,30个循环的程序为98℃10s,60℃30s,72℃1min,最后72℃5min。pcr产物电泳切胶保存。
3、scaⅰ和bamhⅰ酶切载体、同源重组连接及转化
对pyes2进行双酶切(scaⅰ和bamhⅰ购自neb),酶切体系为:saci1μl,bamhⅰ1μl,限制性内切酶缓冲液2.5μl,pyes25μl,ddh2o18μl。
使用正向引物seqidno:5,反向引物seqidno:6对步骤2中的pcr产物进行扩增,扩增体系为:模板1μl,10mm正向引物反向引物各1μl,dna聚合酶buffer4μl,10mmol/ldntps0.4μl,dna聚合酶0.2μl,共20μl体系,剩余用水补齐。pcr扩增程序为:98℃30s,30个循环的程序为98℃10s,60℃30s,72℃1min,最后72℃5min。pcr产物电泳切胶回收。回收产物通过dna重组酶与酶切后的载体进行同源重组连接,连接体系为:线性化载体pyes23μl;pcr产物5μl;dna同源重组酶2μl。50℃,温育15min,然后置于冰上。将连接好的产物通过热激转化的方法转入大肠感受态细胞top10中。转化大肠杆菌并挑单克隆测序。将正确克隆置于-80℃保存。
4、测试酵母表达载体实施效率
使用dna聚合酶扩增egfp基因,成功通过ta克隆方式构建到酵母表达pyes2-t载体,得到pyes2-t-egfp载体,通过转化大肠杆菌top10,挑取10个单克隆测序,全部转化成功。
核苷酸序列
seqidno:1
tcaactaattgcagtaattccttggtggtac
seqidno:2
agcattaggtcccaaaatttgtttac
seqidno:3
ccagtcttgacctggagcgta
seqidno:4
ccaggtgtagactggtcctg
seqidno:5
ttaagcttggtaccgagctcccagtcttgacctggagcgta
seqidno:6
cgttactagtggatccccaggtgtagactggtcctg
尽管为说明目的公开了本发明的实施例和附图,但是本领域的技术人员可以理解:在不脱离本发明及所附权利要求的精神和范围内,各种替换、变化和修改都是可能的,因此,本发明的范围不局限于实施例和附图所公开的内容。
序列表
<110>天津农学院
<120>一种酵母中高效克隆载体的构建方法及应用
<160>6
<170>siposequencelisting1.0
<210>1
<211>31
<212>dna
<213>正向引物(unknown)
<400>1
tcaactaattgcagtaattccttggtggtac31
<210>2
<211>26
<212>dna
<213>反向引物(unknown)
<400>2
agcattaggtcccaaaatttgtttac26
<210>3
<211>21
<212>dna
<213>特异性引物的正向引物(unknown)
<400>3
ccagtcttgacctggagcgta21
<210>4
<211>20
<212>dna
<213>特异性引物的反向引物(unknown)
<400>4
ccaggtgtagactggtcctg20
<210>5
<211>41
<212>dna
<213>正向引物(unknown)
<400>5
ttaagcttggtaccgagctcccagtcttgacctggagcgta41
<210>6
<211>36
<212>dna
<213>反向引物(unknown)
<400>6
cgttactagtggatccccaggtgtagactggtcctg36
- 还没有人留言评论。精彩留言会获得点赞!