一种蒽醌衍生物的制备方法与流程
本发明涉及荧光检测领域,具体涉及一种蒽醌衍生物及合成方法和应用。
背景技术
工业污染导致hg2+在环境中的污染已经变得十分普遍。hg2+是具有严重生理毒性的过渡金属离子之一,一旦进入到海洋中,无机的hg2+会在细菌的作用下转变为危害更大的甲基汞并进入到食物链中,甲基汞对人体的危害十分显著,它能轻易的被人体吸收并突破人体的血脑屏障,然后直接作用于我们的中枢神经系统,对人体造成巨大伤害。因此,开发和研究新型的汞离子及汞化合物检测方法有着重要的意义。
技术实现要素:
本发明的目的可以通过以下技术方案实现:
一种蒽醌衍生物,该衍生物的结构式如下所示:
一种上述的蒽醌衍生物的制备方法,该方法反应路线如下:
上述制备方法包括以下步骤:
1)化合物ⅰ在氧化剂的作用下进行氧化反应,制备得到化合物ⅱ;
2)在酸性环境下,化合物ⅱ与乙醇进行酯化反应,得到化合物ⅲ;
3)在加热回流的条件下,化合物ⅲ与水合肼进行反应,得到化合物ⅳ;
4)在惰性气体保护的环境下,化合物ⅳ与异硫氰酸苯酯进行反应,得到化合物ⅴ。
本发明技术方案中:步骤1)中所述的氧化即为三氧化铬、高锰酸钾或次氯酸钠;氧化反应所用的溶剂为乙醇或冰醋酸。
本发明技术方案中:步骤2)中酸性环境所用的酸性试剂为为硫酸、磷酸或者硼酸,酯化反应的温度为加热回流温度。
本发明技术方案中:步骤3)反应所用的溶剂为甲醇、乙醇或乙腈。
本发明技术方案中:步骤4)反应的温度为0~100℃,反应所用的溶剂为甲醇、乙醇、乙腈或二氯甲烷。
本发明技术方案中:所述的蒽醌衍生物作为检测汞离子的应用。
本发明技术方案中:所述的蒽醌衍生物在环境中作为检测汞离子的应用。
本发明的有益效果:
本发明所提供的制备方法简单,易于工业化生产。且制备得到的多信号探针对汞离子的检出限低且选择性高。
附图说明
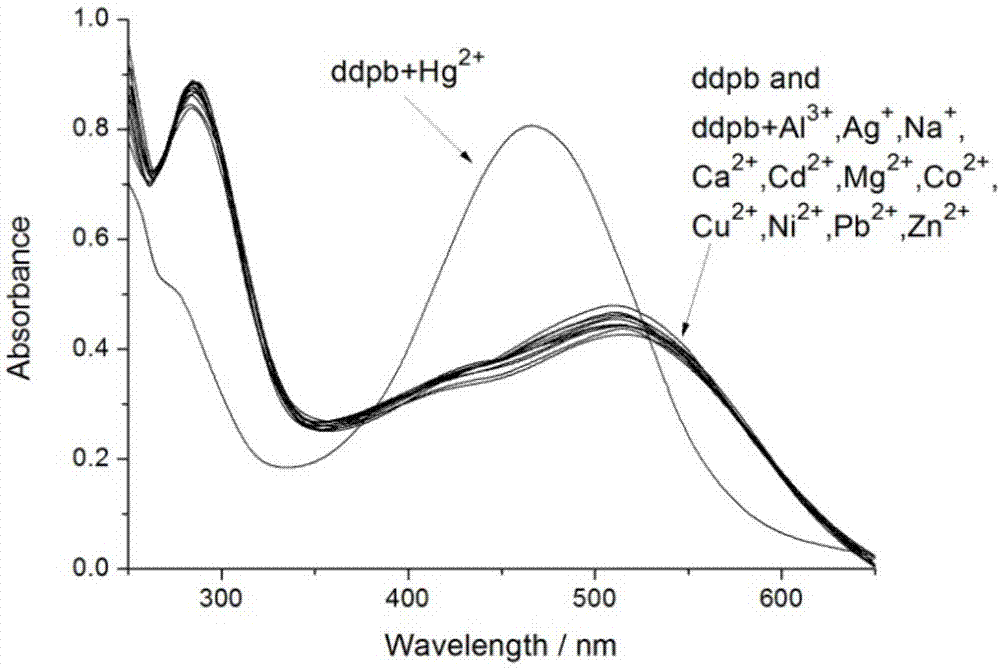
图1是探针分子ddpb对hg2+的选择性吸收光谱识别。
图2为hg2+对探针分子ddpb的吸收光谱滴定图。
图3是探针分子ddpb对hg2+的选择性荧光光谱识别。
图4为hg2+对探针分子ddpb的荧光光谱滴定图。
图5为hg2+与探针分子ddpb反应时间对溶液荧光强度的影响图。
图6为当溶液中有其它共存金属离子时对探针ddpb选择性识别hg2+的影响图。
图7为hg2+浓度与荧光强度的线性关系图。
具体实施方式
下面结合实施例对本发明做进一步说明,但本发明的保护范围不限于此:
实施例1
在100ml冰醋酸中加入1,4-二甲基蒽醌(10mmol,2.36g)和三氧化铬(100mmol,10g),在搅拌的条件下加热回流10小时。反应结束后冷却至室温,抽滤,将所得固体产物溶于10%的热氢氧化钠溶液中,趁热过滤,所得滤液冷却后用浓盐酸调节溶液ph值到2左右,抽滤,所得固体用丙酮洗涤,真空干燥,得淡黄色化合物ⅱ2.77g,产率:93.6%,纯度:99.54%。
元素分析:(%)forc16h8o6:计算值:c64.87;h2.72,实测值:c65.18;h2.85。
ir(kbr),ν,cm-1:3088,1780,1693,1674,1588,1373,1286,1257,1203,897,814,747,683。
1hnmr(500mhz,cdcl3,tms):δ=7.81(d,j=6.8,2h),8.34(d,j=6.8,2h),8.19(s,2h),12.97(s,2h)ppm.
在100ml无水乙醇中加入化合物ⅱ(10mmol,2.96g)和5ml浓硫酸,加热回流6小时。反应结束后,旋转蒸干乙醇,所得固体依次用5%的碳酸钠溶液、水洗至中性,再用丙酮洗,真空干燥,得黄色化合物ⅲ3.37g,产率:95.6%,纯度:99.28%。
元素分析:(%)forc20h16o6:计算值:c68.18;h4.58,实测值:c68.73;h4.37。
ir(kbr),ν,cm-1:3072,1732,1691,1578,1532,1497,1217,1138,961,819,767。
1hnmr(500mhz,cdcl3,tms):δ=1.33(t,j=7.0,6h),4.36(q,j=7.0,4h),7.83(d,j=6.8,2h),8.32(d,j=6.8,2h),8.57(s,2h).
在100ml无水乙醇中加入化合物ⅲ(10mmol,3.52g),在加热回流的条件下用恒压漏斗缓慢的滴加10ml水合肼,滴加完成后继续反应6小时。反应结束后冷却至室温,将反应液倒入100ml的水中,然后用乙酸乙酯萃取(50ml×3),合并有机相,无水硫酸镁干燥过夜,过滤,旋转蒸干溶剂后得黄色化合物ⅳ3.07g,产率:94.7%,纯度:99.19%。元素分析:(%)forc16h12n4o4:计算值:c59.26;h3.73;n17.28,实测值:c59.17;h3.41;n17.46。
ir(kbr),ν,cm-1:3378,3284,3068,1697,1688,1642,1516,1482,1286,1329,1157,932,827,715,6741hnmr(500mhz,cdcl3,tms):δ=3.87(d,j=7.2,4h),7.71(d,j=7.0,2h),8.12(d,j=6.8,2h),8.38(d,j=6.8,2h),10.51(br,2h).
在通入n2保护的条件下,在100ml无水乙腈中加入化合物ⅳ(10mmol,3.24g)和异硫氰酸苯酯(22mmol,2.97g),在室温下反应24小时。反应结束后旋转蒸干溶剂,将所得固体产物过硅胶柱(乙酸乙酯:己烷=1:3),旋转蒸除溶剂的橘黄色化合物ⅴ(ddpb)5.53g,产率:93.1%,纯度:99.47%。
元素分析:(%)forc30h22n6o4s2:计算值:c60.59;h3.73;n14.13,实测值:c61.13;h3.84;n13.91。
ir(kbr),ν,cm-1:3267,3216,3011,1721,1683,1632,1421,1207,1189,1157,878,704,682。
1hnmr(500mhz,cdcl3,tms):δ=3.67-3.71(m,4h),4.38(s,2h),6.95-7.03(m,6h),7.16(d,j=6.8,4h),7.82(d,j=6.8,2h),8.03(d,j=6.8,2h),8.33(d,j=6.8,2h).
实施例2
在100ml50%的乙醇溶液中加入1,4-二甲基蒽醌(10mmol,2.36g)和高锰酸钾(100mmol,15.8g),在搅拌的条件下加热回流12小时。反应结束后冷却至室温,抽滤,将所得固体产物溶于10%的热氢氧化钠溶液中,趁热过滤,所得滤液冷却后用浓盐酸调节溶液ph值到2左右,抽滤,所得固体用丙酮洗涤,真空干燥,得淡黄色化合物ⅱ2.64g,产率:89.1%,纯度:99.17%。
在100ml无水乙醇中加入化合物ⅱ(10mmol,2.96g)和5ml磷酸,加热回流8小时。反应结束后,旋转蒸干乙醇,所得固体依次用5%的碳酸钠溶液、水洗至中性,再用丙酮洗,真空干燥,得黄色化合物ⅲ3.24g,产率:92.1%,纯度:98.17%。
在100ml无水甲醇中加入化合物ⅲ(10mmol,3.52g),在加热回流的条件下用恒压漏斗缓慢的滴加15ml水合肼,滴加完成后继续反应8小时。反应结束后冷却至室温,将反应液倒入100ml的水中,然后用乙酸乙酯萃取(50ml×3),合并有机相,无水硫酸镁干燥过夜,过滤,旋转蒸干溶剂后得黄色化合物ⅳ3.01g,产率:92.8%,纯度:99.17%。
在通入n2保护的条件下,在100ml无水乙醇中加入化合物ⅳ(10mmol,3.24g)和异硫氰酸苯酯(25mmol,3.38g),在室温下反应30小时。反应结束后旋转蒸干溶剂,将所得固体产物过硅胶柱(乙酸乙酯:己烷=1:3),旋转蒸除溶剂的橘黄色化合物ⅴ(ddpb)5.37g,产率:90.3%,纯度:98.16%。
实施例3
在100ml50%的乙醇溶液中加入1,4-二甲基蒽醌(10mmol,2.36g),加热回流搅拌。然后用恒压漏斗缓慢滴加300ml10%的次氯酸钠溶液。溶液滴加完成后继续在搅拌的条件下加热回流15小时。反应结束后冷却至室温,用浓盐酸调节溶液ph值到2左右,抽滤,所得固体用丙酮洗涤,真空干燥,得淡黄色化合物ⅱ2.51g,产率:84.7%,纯度:98.12%。
在100ml无水乙醇中加入化合物ⅱ(10mmol,2.96g)和5ml硼酸,加热回流10小时。反应结束后,旋转蒸干乙醇,所得固体依次用5%的碳酸钠溶液、水洗至中性,再用丙酮洗,真空干燥,得黄色化合物ⅲ3.15g,产率:89.4%,纯度:99.26%。
在100ml无水乙腈中加入化合物ⅲ(10mmol,3.52g),在加热回流的条件下用恒压漏斗缓慢的滴加20ml水合肼,滴加完成后继续反应10小时。反应结束后冷却至室温,将反应液倒入100ml的水中,然后用乙酸乙酯萃取(50ml×3),合并有机相,无水硫酸镁干燥过夜,过滤,旋转蒸干溶剂后得黄色化合物ⅳ2.94g,产率:90.6%,纯度:99.09%。
在通入n2保护的条件下,在100ml二氯甲烷中加入化合物ⅳ(10mmol,3.24g)和异硫氰酸苯酯(30mmol,4.06g),在室温下反应48小时。反应结束后旋转蒸干溶剂,将所得固体产物过硅胶柱(乙酸乙酯:己烷=1:3),旋转蒸除溶剂的橘黄色化合物ⅴ(ddpb)5.29g,产率:89.0%,纯度:98.16%。
性质部分
1、吸收光谱实验
蒽醌衍生物ddpb对hg2+的吸收光谱识别
图1是探针分子ddpb对hg2+的选择性吸收光谱识别。在10ml浓度为0.1mmol/l探针分子ddpb溶液中分别加入10μl浓度为0.2mol/l(2倍摩尔量)的金属离子溶液(al3+、ag+、na+、ca2+、cd2+、hg2+、mg2+、co2+、k+、cu2+、ni2+、pb2+、zn2+)。实验中所使用的溶液体系均为乙腈/水(1:1,v:v)的混合溶液,吸收光谱在岛津uv-2450型紫外分光光度计上测定。
由图1可以看出探针分子在乙腈/水(1:1,v:v)的混合溶液中自身的吸收在510nm左右,当我们向探针分子溶液中加入过量的金属离子后,我们发现只有在加入hg2+后,溶液的吸收蓝移至465nm左右,溶液的颜色也由紫色变为黄色,而当在探针分子溶液中加入其它金属离子时,则没有这一现象的发生,这说明该探针分子的吸收光谱对hg2+有着独特的响应。
图2为hg2+对探针分子ddpb的吸收光谱滴定图。在10ml浓度为0.1mmol/l探针fcl溶液中依次加入0.2、0.4、0.6、0.8、1.0、1.2、1.4、1.6、1.8、2.0、2.5、3.0倍摩尔量的hg2+。实验中所使用的溶液体系均为乙腈/水(1:1,v:v)的混合溶液,吸收光谱在岛津uv-2450型紫外分光光度计上测定。由图2可以看出,随着hg2+的加入,溶液的吸收波长逐渐由510nm蓝移至465nm,当hg2+加入量达到探针分子2倍摩尔量后,溶液的吸收波长不再移动,且峰的强度基本不变。这说明探针分子ddpb与hg2+是1:2配位的。
2、荧光光谱实验
蒽醌衍生物ddpb对hg2+的荧光识别
图3是探针分子ddpb对hg2+的选择性荧光光谱识别。将探针分子ddpb溶于乙腈/水(1:1,v:v)的混合溶液中,配制成浓度为10μmol/l的溶液,在此溶液中分别加入2倍摩尔量的金属离子(al3+、ag+、na+、ca2+、cd2+、hg2+、mg2+、co2+、k+、cu2+、ni2+、pb2+、zn2+)。激发波长为470nm,测定溶液的荧光光谱。从图3中可以看出,探针分子溶液在525nm处有一个弱荧光发射峰,在加入hg2+后,溶液在525nm处弱荧光发射峰消失,而在582nm处出现一个很强的荧光发射峰,而加入其它金属离子则没有这一现象,这说明该探针分子对hg2+表现出非常强的荧光选择识别性。实验中所使用的溶液体系均为乙腈/水(1:1,v:v)的混合溶液,荧光光谱在amincobowmanseries2荧光光谱仪上测得。
图4为hg2+对探针分子ddpb的荧光光谱滴定图。在10μmol/l的探针分子ddpb的乙腈/水(1:1,v:v)的混合溶液中,分别加入0.2、0.4、0.6、0.8、1.0、1.2、1.4、1.6、1.8、2.0、2.5、3.0倍摩尔量的hg2+。在470nm处激发,测量溶液的发射光谱,如图所示随着hg2+的浓度增加,525nm处弱荧光发射峰逐渐减弱最终消失,而在582nm处出现一个新的荧光发射峰,并且在hg2+加入量达到2倍摩尔量后582nm处的发射峰强度基本不再增加。
图5为hg2+与探针分子ddpb反应时间对溶液荧光强度的影响图。在10μmol/l的探针分子ddpb的乙腈/水(1:1,v:v)的混合溶液中,加入2倍摩尔量的hg2+。在激发波长470nm,发射波长525nm处,分别在0、1、2、3、4、5、6、7、8分钟时记录溶液的荧光强度。如图所示,在探针分子ddpb溶液中加入hg2+5分钟后,荧光强度达到最大值,且随着时间延长基本保持不变。
图6为当溶液中有其它共存金属离子时对探针ddpb选择性识别hg2+的影响图。在10μmol/l的探针分子ddpb的乙腈/水(1:1,v:v)的混合溶液中,分别加入溶有10倍摩尔量的金属离子(al3+、ag+、na+、ca2+、cd2+、hg2+、mg2+、co2+、k+、cu2+、ni2+、pb2+、zn2+),在激发波长470nm,发射波长525nm处,测量溶液的荧光强度,然后再在上述溶液中加入10倍摩尔量的hg2+,在激发波长470nm,发射波长525nm处,测量溶液的荧光强度,从图6中可以看出,当溶液中大量存在其他金属离子时,探针分子ddpb对hg2+的选择性识别并不受影响。
图7为hg2+浓度与荧光强度的线性关系图。在1μmol/l的探针分子ddpb的乙腈/水(1:1,v:v)的混合溶液中,分别加入0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8倍摩尔量的hg2+在激发波长470nm,发射波长525nm处,测量溶液的荧光强度。从图中可以看出当hg2+浓度在0.1-0.8μmol/l范围内呈现出良好的线性关系(r2=0.9963),使用3σiupac标准计算所得的检测限为2.45×10-8mol/l。
- 还没有人留言评论。精彩留言会获得点赞!