一种用于检测靶基因是否发生融合突变的方法、引物组合、试剂盒及其应用与流程
本发明涉及基因检测领域,具体而言,涉及一种用于检测靶基因是否发生融合突变的方法、引物组合、试剂盒及其应用。
背景技术:
融合基因(fusiongene)属于基因突变的一种,是指两个基因的全部或一部分序列相互融合形成的一个新基因,其有可能是染色体易位、中间缺失或染色体倒位所致的结果。大多数情况下,融合基因可以导致异常序列或功能的蛋白质的产生,或者某些基因表达的失调,从而导致或者促进肿瘤的发生。自1973年发现费城染色体后,随着检测技术的发展,尤其是测序技术的发展,科学家陆续在白血病等血液肿瘤以及肺癌、前列腺癌、乳腺癌、卵巢癌、尤文肉瘤、滑膜肉瘤等实体瘤中发现大量融合基因的存在,且研究结果表明,融合基因作为肿瘤驱动基因,在肿瘤的诊断、预后、分层、治疗以及药物研发中具有重要意义。
以alk融合基因为例,alk融合是非小细胞肺癌(nsclc)的关键驱动机制之一,在nsclc患者中的发生频率约为4-7%左右。针对alk融合的抑制剂克唑替尼和色瑞替在治疗alk融合阳性的nsclc患者中都取得了巨大成功。因此,对融合基因进行准确检测无论是对药物研发还是对精准医疗都具有重要意义。
目前,基因突变的主要检测方式为扩增子测序(ampliconsequencing)。扩增子测序是一种高靶向性测定方法,其根据特定基因组区域在其两端设计引物,在pcr过程中靶向捕获目标区域,可同时对16~1536个靶点进行检测。配合以二代测序的高深度和umi策略,辅以合理的生信分析流程,扩增子测序可有效地识别低频突变并对其进行特征分析。
尽管具有诸多优点,传统的扩增子测序方法在检测基因融合时仍存在较多不足,具体表现为:(1)由于无法获得未知融合基因的伙伴基因信息与断点位置,传统的扩增子测序方法难于对未知断点融合基因进行检测,现有技术中少有基于扩增子测序的试剂盒对dna样本尤其血液类样本进行融合检测,国内更是少见。(2)靶向捕获区域两端的引物序列固定,对于高度片段化的dna片段,如ffpe、cfdna等样本的捕获效率较低,可能导致假阴性结果。目前,尚缺乏能很好地检测断裂点、融合方向未知、模板高度片段化的基因融合突变的技术。
为填补该领域技术空白,特提出本发明。
技术实现要素:
本发明的目的在于提供一种设计用于检测靶基因是否发生融合突变的引物组合的方法,根据所述方法设计所得的引物组合、试剂盒,以及使用前述引物组合的dna建库方法、融合基因检测方法,实现对发生未知融合突变的靶基因,尤其是来自高度片段化的dna样本,进行富集和检测的目的。
本发明的目的还在于提供一套检测肺癌基因(包括alk基因、ntrk1基因、ret基因、ros1基因、fgfr3基因和ntrk3基因的融合突变)融合突变的引物组合,该引物组合经过多轮检测和优化,其对测序数据覆盖的均一性达85%以上,捕获效率达92%以上,成功实现对多种肺癌基因未知融合突变的有效检测。
为了实现本发明的上述目的,特采用以下技术方案:
一种设计用于检测靶基因是否发生融合突变的引物组合的方法,所述方法包括以下步骤:
(1)选择可能与其他基因发生融合的靶基因,确定所述靶基因上可能发生断裂融合的dna区域;
(2)预判断裂融合发生后所述其他基因与所述靶基因的相对位置关系,根据所述相对位置关系设计引物组:
a、如果融合后,所述其他基因位于所述靶基因的5’端,则沿所述dna区域的正义链的3’端至5’端方向,依次设计多条3’端与所述正义链反向互补配对的引物,组成引物组1,其中,每条引物中与所述正义链反向互补配对的区域为靶基因结合区,相邻两条引物的靶基因结合区在所述正义链上间隔约0~130bp(例如,0bp,5bp,10bp,15bp,20bp,25bp,30bp,35bp,40bp,45bp,50bp,55bp,60bp,65bp,70bp,75bp,80bp,85bp,90bp,95bp,100bp,105bp,110bp,115bp,120bp,125bp,130bp);
b、如果融合后,所述其他基因位于所述靶基因的3’端,则沿所述dna区域的反义链的3’端至5’端方向,依次设计多条3’端与所述反义链反向互补配对的引物,组成引物组2,其中,每条引物中与所述反义链反向互补配对的区域为靶基因结合区,相邻两条引物的靶基因结合区在所述正义链上的间隔约0~130bp(例如,0bp,5bp,10bp,15bp,20bp,25bp,30bp,35bp,40bp,45bp,50bp,55bp,60bp,65bp,70bp,75bp,80bp,85bp,90bp,95bp,100bp,105bp,110bp,115bp,120bp,125bp,130bp);
c、如果无法预判断裂融合发生后所述其他基因与所述靶基因的相对位置关系,则根据所述步骤a和所述步骤b设计所述引物组1和引物组2。
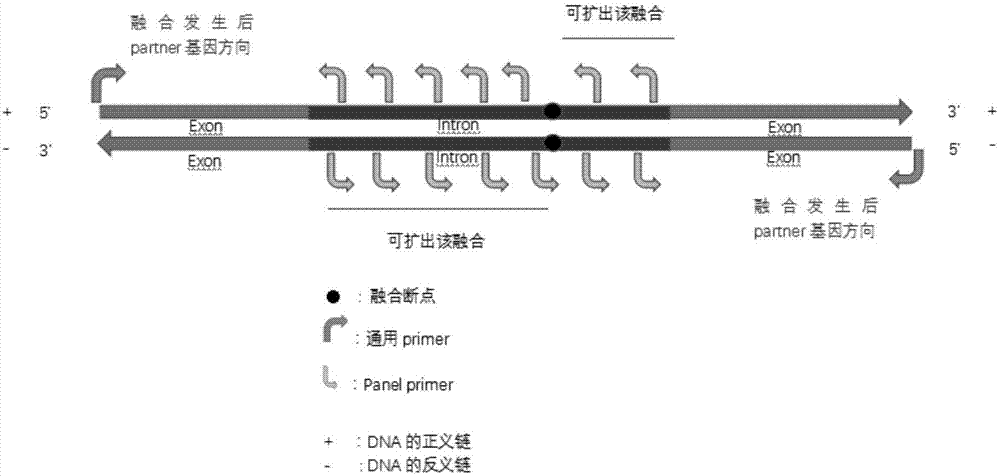
如图1所示,本发明所述方法在靶基因的潜在融合断裂区域沿可能的基因融合发生方向依次设计多条单向引物,组成引物组合,所述引物沿基因融合发生的方向延伸,能够在融合断裂点未知的情况下,对靶基因进行定向富集,克服现有扩增子测序方法无法靶向富集未知融合基因的缺点,有利于未知的基因融合类型的发现和检测。其次,本发明所述方法的步骤c同时设计引物组1和引物组2,可实现融合方向未知情况下靶基因的定向富集。再者,本发明所述引物组合中相邻两个引物之间的间隔约为0~130bp,对高度片段化的样本(如cfdna、ffpe样本)的富集效果好。
在一些具体的实施方式中,所述方法还包括以下步骤:(3)设计通用引物,所述通用引物与所述引物组1和/或引物组2组成用于靶向扩增的引物对,优选地,所述通用引物与接头序列中的至少一部分反向互补配对;优选地,所述步骤(2)与所述步骤(3)无先后顺序;
优选地,所述方法还进一步包括以下步骤:(4)设计用于对靶向扩增产物进行再次扩增的上、下游引物;
优选地,所述上游引物与所述通用引物相同,所述下游引物包括位于5’端的测序区、barcode区和位于3’端的延伸区,所述测序区用于引发测序引物延伸,所述延伸区与所述引物组1和/或所述引物组2中的每条引物的5’端反向互补配对。
在一些具体的实施方式中,所述靶基因包括alk基因、ntrk1基因、ret基因、ros1基因、fgfr3基因和ntrk3基因中的一种或多种。
优选地,所述靶基因包括alk基因,所述alk基因上可能发生断裂融合的dna区域为:第2号染色体上第29446208~29448431位核苷酸。
优选地,所述靶基因包括ntrk1基因,所述ntrk1基因上可能发生断裂融合的dna区域为:第1号染色体上第156843425~156846364位核苷酸。
优选地,所述靶基因包括ret基因,所述ret基因上可能发生断裂融合的dna区域为:第10号染色体上第43606649~43607677位核苷酸,和/或第10号染色体上第43609004~43612179位核苷酸。
优选地,所述靶基因包括ros1基因,所述ros1基因上可能发生断裂融合的dna区域为:第6号染色体上第117641031~117658508位核苷酸。
优选地,所述靶基因包括fgfr3基因,所述fgfr3基因上可能发生断裂融合的dna区域为:第4号染色体上第1808273~1810599位核苷酸。
优选地,所述靶基因包括ntrk3基因,所述ntrk3基因上可能发生断裂融合的dna区域为第15号染色体上第88472421~88472665位核苷酸、第88476242-88476415位核苷酸,第88483853-88483984位核苷酸、第88522575~88522694位核苷酸、第88524456-88524591位核苷酸、第88576087~88576276位核苷酸,和/或第88423501-88423659位核苷酸。
本发明还涉及根据前述方法设计得到的引物组合,所述引物组合至少包括引物组1和/或引物组2,优选地,所述引物组合还包括通用引物,更优选地,所述引物组合还包括所述上、下游引物。
在一些具体的实施方式中,所述靶基因包括alk基因,用于检测所述alk基因是否发生融合突变的引物组包括27条引物,其引物序列分别如seqidno:1~27所示。
在一些具体的实施方式中,所述靶基因包括ntrk1基因,用于检测所述ntrk1基因是否发生融合突变的引物组包括31条引物,其引物序列分别如seqidno:28~58所示。
在一些具体的实施方式中,所述靶基因包括ret基因,用于检测所述ret基因是否发生融合突变的引物组包括48条引物,其引物序列分别如seqidno:59~106所示。
在一些具体的实施方式中,所述靶基因包括ros1基因,用于检测所述ros1基因是否发生融合突变的引物组包括170条引物,其引物序列分别如seqidno:107~276所示。
在一些具体的实施方式中,所述靶基因包括fgfr3基因,用于检测所述fgfr3基因是否发生融合突变的引物组包括30条引物,其引物序列分别如seqidno:277~306所示。
在一些具体的实施方式中,所述靶基因包括ntrk3基因,用于检测所述ntrk3基因是否发生融合突变的引物组包括23条引物,其引物序列分别如seqidno:307~329所示。
在一些具体的实施方式中,所述引物组合包括通用引物,所述通用引物的核苷酸序列如seqidno:330所示。
在一些具体的实施方式中,所述上游引物的核苷酸序列如seqidno:330所示,所述下游引物的核苷酸序列如seqidno:331所示。
本发明还涉及一种用于检测靶基因是否发生融合突变的试剂盒,所述试剂盒包括前述的引物组合和其他检测试剂。
在一些具体的实施方式中,所述其他检测试剂包括用于测序前构建文库的试剂和/或测序用试剂,优选地,所述其他检测试剂包括用于扩增子建库的试剂。
本发明还涉及一种测序前构建dna文库的方法,所述方法包括以下步骤:(1)提取dna样本,片段化并加接头;(2)使用前述引物组1和/或引物组2,以及通用引物,进行pcr反应,靶向富集所述dna样本;(3)使用前述上游引物和下游引物对靶向富集的所述dna样本进行pcr扩增,获得可用于测序的dna文库。
在一些具体的实施方式中,步骤(2)所述pcr的反应体系包括:dna样本9~10μl,5×缓冲液4~5μl,引物组1和/或引物组2共4.5~5.5μl,通用引物共0.5~1μl,以及dna聚合酶0.5~1μl;优选地,步骤(2)所述pcr反应体系包括:dna样本9.4μl,5×缓冲液4μl,引物组1和/或引物组2共5μl,通用引物共0.8μl,以及dna聚合酶0.8μl。
在一些具体的实施方式中,步骤(2)所述pcr的扩增程序包括:步骤a、预变性:先在94~96℃下变性12~14min,再在97~99℃下变性1~3min,优选先在95℃下变性13min,再在98℃下变性2min;步骤b、循环扩增:先在97~99℃下变性10~20s,再在67~69℃下延伸7~12min,循环数为5~10,优选地,先在98℃下变性15s,再在68℃下延伸10min,循环数为8。
在一些具体的实施方式中,步骤(3)所述pcr的反应体系包括;靶向富集的dna样本13~14μl,5×缓冲液4~5μl,dna聚合酶0.7~1.2μl,上游引物和下游引物共0.7~1.2μl;优选地,步骤(3)所述pcr的反应体系包括;靶向富集的dna样本13.4μl,5×缓冲液4μl,dna聚合酶1μl,上游引物和下游引物共1.6μl。
在一些具体的实施方式中,步骤(3)反应测序包括:步骤a、预变性:先在94~96℃下变性12~14min,再在97~99℃下变性1~3min,优选先在95℃下变性13min,再在98℃下变性2min;步骤b、循环扩增:先在97~99℃下变性10~20s,再在59~61℃下延伸1.5~2.5min,循环数为20~24,优选地,先在98℃下变性15s,再在60℃下延伸2min,根据dna样本的来源选择循环数:当dna样本来自外周血基因组dna、福尔马林固定的石蜡组织、或者循环游离dna时,其循环数分别为20、24和22。
在一些具体的实施方式中,所述dna样本选自高度片段化的dna样本,例如循环游离dna、福尔马林固定的石蜡组织。
本发明还涉及一种检测融合基因的方法,所述方法使用上述方法构建dna文库,并进行测序,获得融合基因的检测结果。在一些具体的实施方式中,所述方法为非诊断目的。
与现有技术相比,本发明的有益效果为:
(1)本发明所述方法沿可能的基因融合发生方向依次设计多条引物,组成引物组合,所述引物组合的引物沿基因融合发生的方向延伸,能够对发生未知融合的靶基因进行定向富集,克服现有扩增子测序方法难于靶向富集未知融合基因的缺点,有利于未知基因融合类型的发现和检测。本发明所述方法的步骤c同时设计引物组1和引物组2,可实现融合方向未知情况下靶基因的定向富集。本发明所述引物组合中相邻两个引物之间的间隔约为0~130bp,对高度片段化的样本(如cfdna、ffpe样本)的富集效果好。
(2)本发明所述方法还包括设计与所述引物组1、引物组2配合使用的通用引物,形成引物对,通过pcr反应获得包含融合突变的双链dna,以利于后续的建库和测序,所述通用引物与接头序列中的至少一部分反向互补配对,因此,样品dna经过加接头即可用于基于前述引物对的pcr反应,扩增获得双链dna,用于后续的测序。优选地,本发明还包括设计对靶向扩增产物进行再次扩增的上、下游引物,并对上、下游引物进行特殊设计,使得靶向扩增产物在经过再次扩增之后即完成建库,可用于测序,简化建库的操作流程。
(3)本发明所述方法还对靶基因以及可能发生断裂融合的dna区域进行优化,其中所述靶基因属于频繁发生断裂融合且具有重大检测价值的靶基因,所述可能发生断裂融合的区域是在考量融合断裂发生概率、扩增效率以及pcr反应的复杂程度后,最终优化获得的dna区域,既保证对可能发生的基因断裂融合的覆盖率,又保证检测效率。
(4)在针对肺癌检测alk基因、ntrk1基因、ret基因、ros1基因、fgfr3基因和ntrk3基因的融合突变时,疑似发生断裂融合dna区域的范围广,涉及的引物众多,反应体系复杂,出现测序数据覆盖的均一性和捕获效率下降的问题,为解决该问题,本发明对所述引物组合的具体序列进行多轮检测和优化,最终所得引物组合对测序数据的覆盖的均一性达到85%以上,捕获效率达到92%以上。另外,本发明还对建库过程中靶向富集以及进一步扩增的pcr反应体系和程序做出优化,有效提高pcr反应的扩增效率和均一性。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明检测未知融合基因的引物设计原理图;
图2为检测alk基因融合情况的引物组相关信息;
图3为检测ntrk1基因融合情况的引物组相关信息;
图4~图5为检测ret基因融合情况的引物组相关信息;
图6~图11为检测ros1基因融合情况的引物组相关信息;
图12为检测fgfr3基因融合情况的引物组相关信息;
图13为检测ntrk3基因融合情况的引物组相关信息;
图14为通用引物(上游引物)和下游引物的相关信息。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市场购买获得的常规产品。
实施例1
本实施例设计能够用于检测alk融合基因、ntrk1融合基因、ret融合基因、ros1融合基因、fgfr3融合基因和ntrk3融合基因的引物组合,其设计原则如下所示:
(1)选择可能与其他基因发生融合的靶基因,确定所述靶基因上可能发生断裂融合的dna区域;
(2)预判断裂融合发生后,所述其他基因与所述靶基因的相对位置关系,根据所述相对位置关系设计引物组:
a、如果融合后,所述其他基因位于所述靶基因的5’端,则沿所述dna区域的正义链的3’端至5’端方向,依次设计多条3’端与所述正义链反向互补配对的引物,组成引物组1,其中,每条引物中与所述正义链反向互补配对的区域为靶基因结合区,相邻两条引物的靶基因结合区在所述正义链上间隔约0~130bp;
b、如果融合后,所述其他基因位于所述靶基因的3’端,则沿所述dna区域的反义链的3’端至5’端方向,依次设计多条3’端与所述反义链反向互补配对的引物,组成引物组2,其中,每条引物中与所述反义链反向互补配对的区域为靶基因结合区,相邻两条引物的靶基因结合区在所述正义链上的间隔约0~130bp;
c、如果无法预判断裂融合发生后所述其他基因与所述靶基因的相对位置关系,则根据所述步骤a和所述步骤b设计所述引物组1和引物组2。
(3)设计通用引物,所述通用引物能够与所述引物组1和/或引物组2中的引物形成引物对,所述通用引物与接头序列中的至少一部分反向互补配对。
(4)设计用于对靶向富集产物进行再次扩增的上、下游引物,所述上游引物与所述通用引物相同,所述下游引物包括位于5’端的测序区、barcode区和位于3’端的延伸区,所述测序区用于引发测序引物延伸,所述延伸区与所述引物组1和/或所述引物组2中的每条引物的5’端反向互补配对。
本实施例在获得能够检测alk融合基因等引物组合后,经过十余轮测试验证和优化,最终获得表用于检测alk、ntrk1、ret、ros1、fgfr3和ntrk3的引物组合,其中具体的引物信息参见说明书附图图2~图14。经检测发现,图2~图14所示引物组合对测试数据的覆盖的均一性达到85%以上,捕获效率达到92%以上。
表1靶基因的潜在断裂融合区域及相关信息
实施例2
采用上述表1所述引物组合建立用于测序的扩增文库,具体建库方法如下所示,图2~图13所示引物组合委外合成,命名为建库引物组,图14所示通用引物/上游引物委外合成,建库使用的其他试剂均购买自凯杰公司:
1、片段化、末端修复和“a”连接
1.1、根据样本类型和样本质量,取20-80ng的dna用于后续的建库操作,其中cfdna样本建议投入20-40ng,ffpe样本建议投入40-80ng。在冰上,根据表2配制片段化、末端修复和“a”连接混合液。瞬时离心,然后用移液器吹吸7-8次混匀并再次瞬时离心。
表2片段化、末端修复和“a”连接反应混合液
1.2、加5μlfragmentationenzymemix到每个反应管中。瞬时离心,然后用移液器吹吸7–8次混匀(勿振荡)并再次瞬时离心。在整个反应过程中,保持反应管/板在冰上。
1.3、根据表3编排pcr仪运行程序:使用仪器热盖82℃;将反应管/板置于pcr仪前,开始运行程序。当pcr仪温度达到4℃时,暂停程序。pcr仪必须预冷并暂停在4℃;将准备好的反应管/板置于已预冷的pcr仪上,恢复循环程序;完成时,pcr仪温度返回到4℃;把样品放在冰上,然后立即进行后续的“接头连接”步骤。
表3片段化、末端修复和“a”连接的循环条件
2、接头连接
2.1、根据表4配制接头连接混合液。瞬时离心,然后用移液器吹吸10-12次混匀并再次瞬时离心。注意:勿将移液吸头外壁粘上连接溶液,否则将过多加入连接溶液。
2.2、将一个pcr仪温度程序设为20℃,并孵育反应15分钟,完成时,将反应管/板置于冰上,进行adapter-ligateddna的纯化处理,或者将样本置于–20℃恒温冰箱中,可保存3天。注意:在孵育过程中勿使用热盖。
表4接头连接反应混合液
2.3纯化接头连接dna
2.3.1、如果是外周血gdna/ffpe样本,加入50μl无核酸酶水将每个样本体积补至100μl。如果是cfdna样本,加入30μl无核酸酶水将每个样本体积补至80μl。
2.3.2、如果是外周血gdna/ffpe样本,加入100μlqiaseq磁珠。如果是cfdna样本,加入112μlqiaseq磁珠。使用移液器上下吹打几次混匀。室温孵育5min。
2.3.3、将管子/板子置于磁力架上10min。待澄清后,小心地弃去上清。注意:不要丢掉结合有目的dna的磁珠。
2.3.4、保持磁珠在吸附状态,加入200μl80%乙醇。转动管子(2-3次)或使板子在磁力架两列间移动以清洗磁珠。小心地弃去清洗液。
2.3.5、重复使用乙醇清洗一次。注意:第二次清洗后需完全弃去残余乙醇。先使用200μl移液器,然后使用10μl移液器去除所有剩余残液。
2.3.6、使磁珠保持在磁力架上,室温干燥5min。注意:肉眼检查磁珠是否完全干燥。
2.3.7、将磁珠从磁力架上取下,加入52μl无核酸酶水将dna从磁珠上溶解下来。使用移液器吹打混匀完全。将管子/板子放回到磁力架直到液体澄清。转移50μl上清到另外一个干净管子/板子。
2.3.8、如果是外周血gdna/ffpe样本,加入50μlqiaseq磁珠;如果是cfdna样本,加入70μlqiaseq磁珠,使用移液器上下吹打混匀数次。室温孵育5min。
2.3.9、将管子/板子置于磁力架上5min。待溶液澄清后,将磁珠保持在磁力架上,小心地弃去上清。注意:不要丢掉结合有目的dna的磁珠。
2.3.10、保持磁珠在吸附状态,加入200μl80%乙醇。转动管子(2-3次)或使板子在磁力架两列间移动以清洗磁珠。小心地弃去清洗液。
2.3.11、重复使用乙醇清洗一次。注意:第二次清洗后需完全弃去残余乙醇。先使用200μl移液器,然后使用10μl移液器去除所有剩余残液。
2.3.12、使磁珠保持在磁力架上,室温干燥5min。注意:肉眼检查磁珠是否完全干燥。如果残余乙醇进入靶向富集pcr步骤会影响富集pcr的效率。
2.3.13、将磁珠从磁力架上取下,加入12μl无核酸酶水将dna从磁珠上溶解下来。使用移液器吹打混匀完全。将管子/板子放回到磁力架直到液体澄清。转移9.4μl上清到另外一个干净管子/板子。进行下面的“靶向富集”步骤。如果不马上进行下面的实验,可以将样本储存在-20℃恒温冰箱最多3天。
3、靶向富集
3.1、根据表5准备靶向富集混合液。瞬时离心,移液器上下混匀7-8次并再次瞬时离心。在pcr仪上根据表6所列条件设置程序。将靶向富集反应液置于pcr仪中并开始运行程序。反应完成后,将反应液置于冰上并进行下面的“靶向富集纯化”步骤。如果不马上进行下面的实验,可以将样本储存在-20℃恒温冰箱最多3天。
表5靶向富集反应混合液
表6靶向富集循环条件
3.2、靶向富集纯化
3.2.1、如果是外周血gdna/ffpe样本,加入80μl无核酸酶水将每个样本体积补至100μl。如果是cfdna样本,加入70μl无核酸酶水将每个样本体积补至90μl。
3.2.2、如果是外周血gdna/ffpe样本,加入100μlqiaseq磁珠;如果是cfdna样本,加入108μlqiaseq磁珠,使用移液器上下吹打混匀数次。室温孵育5min。
3.2.3、将管子/板子置于磁力架上5min。待溶液澄清后,将磁珠保持在磁力架上,小心地弃去上清。注意:不要丢掉结合有目的dna的磁珠。
3.2.4、不离开磁力架,加入200μl80%乙醇。旋转两到三次(管子)或在磁力架两列间移动(96孔板)来清洗磁珠。小心吸取并弃去上清。重复清洗一次。注意:第二次清洗后要完全弃去管中的乙醇。先用200μl移液器吸出并弃去乙醇,之后用10μl移液器仔细弃去残余的乙醇。
3.2.5、不离开磁力架,室温下干燥5min。注意:外观检查确认磁珠已干燥,残余的乙醇会影响后续pcr反应效率。
3.2.6、移开磁力架,加入16μl无核酸酶水以洗去dna。吸打混匀。放回磁力架,静置至溶液澄清。移取13.4μl上清液到干净的管子中。进行接下来的“universalpcr”步骤。安全终止点:-20℃恒温冰箱能保存至多3天。
4、universalpcr
4.1、根据表7配制pcr反应体系。瞬时离心,吸打7-8次,再次瞬时离心。根据表8中的参数设定pcr反应条件。反应结束后,将反应产物放在冰上保存。安全终止点:-20℃恒温冰箱能保存至多3天。
表7universalpcr反应体系
表8universalpcr循环条件
4.2、cleanupofuniversalpcr
4.2.1、外周血gdna/ffpe:加入80μl无核酸酶水使每个样品体积增至100μl。cfdna:加入70μl无核酸酶水使每个样品体积增至90μl。
4.2.2、外周血gdna/ffpe:加入100μlqiaseq磁珠;cfdna:加入108μlqiaseq磁珠。吹打若干次使溶液充分混匀。室温下孵育5min。置于磁力架上5min,分离磁珠。溶液澄清后立即小心吸取弃去上清。注意:不要丢掉磁珠。
4.2.3、仍置于磁力架上,加入200μl80%乙醇。旋转两到三次(管子)或在磁力架两列间移动(96孔板)来清洗磁珠。小心吸取并弃去上清。重复一次。注意:第二次清洗后要完全弃去管中的乙醇。先用200μl移液器吸出并弃去乙醇,之后用10μl移液器仔细弃去残余的乙醇。
4.2.4、不离开磁力架,室温下干燥5min。注意:外观检查确认磁珠已干燥,残余的乙醇会影响后续测序反应效率。
4.2.5、移开磁力架,加入30μl无核酸酶水以洗脱dna。吸打混匀。放回磁力架,静置至溶液澄清。移取28μl上清液到干净的管子中。
4.2.6、使用thermoqubittmdsdnahsassaykit对文库浓度进行测定,使用agilent4200d1000试剂对文库片段大小进行测定。制备的文库可以在-20℃下保存。扩增文库在-20℃条件下可以稳定保存数月。
实验例1
本实验例首先使用国际上被广泛认可的horizon标准品对本发明实施例1最终优化得到的引物组合及建库方法进行性能验证。对其进行建库并使用illumina的nextseq550ar测序仪对构建的文库进行测序并分析下机数据,统计检出的突变结果与厂商标记的结果是否一致。其次,除了标准品外,我们还对市场上收集到的经其它检测平台验证过的阴、阳性临床样本进行验证,统计其结果的一致性。另外,考虑到现有技术中少有基于扩增子测序试剂盒在dna层面检测融合突变,本实验例选择一种凯杰商品化的常规非特异性针对融合突变的基因突变检测试剂盒(lungcancerpanel),作为对照。实验设计如下:
1.对于ffpe样本,阳性标准品采用horizon的hd796(含有eml4-alk、ccdc6-ret、slc34a2-ros1、tpm3-ntrk1、etv6-ntrk3五种类型的融合基因)和hd784(含有eml4-alk、ccdc6-ret、slc34a2-ros1三种类型的融合基因),结果参见表9。
2.对于外周血gdna样本,阳性标准品采用horizon的hd753(ccdc6-ret、slc34a2-ros1两种类型的融合基因),结果参见表10。
3.对于cfdna样本,阳性标准品采用horizon的hd786(含有ccdc6-ret、slc34a2-ros1两种类型的融合基因),结果参见表11。
4.收集临床阳性样本进行检测,与其他检测方法的一致性结果如表12所示。
5.对于阴性样本,采用coriellinstitute的na12878和收集到的各种类型的阴性临床样本进行检测,结果参见表13。
6.使用作为对照的基因突变检测试剂盒对前述ffpe样本、外周血gdna样本、cfdna样本、临床阳性样本和阴性样本进行检测。
实验样本:本实验例一共测试92例各种类型的样本,其中的14例阳性ffpe样本中,有7例阳性标准品和7例阳性临床样本;26例阳性外周血gdna样本中,有23例阳性标准品和3例阳性临床样本;11例阳性cfdna样本中,有10例阳性标准品和1例阳性临床样本;41例阴性样本中,有15例阴性组织标准品和16例阴性临床组织样本以及10例阴性临床cfdna样本。
实验结果:本实验的方法在54例阳性样本中50例与原始已知结果相符,仅4例与原始已知结果不符,灵敏度达到92.6%;41例阴性样本全部与原始已知结果相符,特异性达到100%;总体的准确度达到95.8%。而用于对照的lungcancerpanel产品无法用于检测融合突变,即使偶尔出现极低频的突变检出,在生信分析过程中由于达不到阈值,数据即被生信流程过滤。
实验结果表明,本发明使用方法能够有效地在dna层面检测融合基因,检测结果可以指导患者精准用药应用于商业化。而用于对照的常规扩增子法基因突变检测试剂盒(lungcancerpanel))无法实现对融合基因的检测。
表9ffpe样本,阳性标准品的检测结果
表10外周血gdna样本,阳性标准品的检测结果
表11cfdna样本,阳性标准品的检测结果
表12临床阳性样本进行检测结果
表13阴性样本检测结果
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!