4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法与流程
本发明涉及医药技术领域,特别涉及一种4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法。
背景技术:
噻唑烷酮类化合物作为一类广泛存在的具有广谱生物活性的五元杂环化合物,在医药、农药、材料等领域均有着重要的应用。特别是近些年来,研究中发现噻唑烷酮环作为一个很重要的结构单元,广泛存在于众多的天然产物和药物中,被用于β-内酰胺酶抑制剂,醛糖还原酶抑制剂,农药杀菌剂,血管紧张素受体-ⅱ拮抗剂,胰岛素增敏剂。
由于噻唑烷酮类化合物具有重要的应用价值,其合成方法一直受到广泛的关注。早期,人们使用卤代乙醛与硫代氨基甲酸为原料来合成3-甲基-4-羟基-噻唑烷-2-酮。后来人们又使用3-取代-噻唑烷二酮还原得到4-羟基-3-取代-噻唑烷-2酮类化合物。但这些制备方法的产品收率较低,仍然需要进一步改进。
技术实现要素:
本发明的主要目的是提出一种4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法,旨在解决现有的4-羟基-3-取代-噻唑烷-2-酮类化合物制备方法产品收率低的问题。
为实现上述目的,本发明提出一种4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法,所述4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法,包括以下步骤:
将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液;
将反应结束后的所述反应液过滤,取滤液并除去滤液中的杂质,蒸馏后得浓缩液;
将所述浓缩液进行硅胶柱层析,得4-羟基-3-取代-噻唑烷-2-酮。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,所述酰基叠氮类化合物的结构式为:
其中,r1为苯基、4-甲基苯基、4-氯苯基、4-溴苯基、4-三氟甲基苯基、3-硝基苯基、4-氰基苯基、2-噻吩基、3-甲氧基苯基、肉桂酸基和苯丙基中的任意一种。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,所述酰基叠氮类化合物和2,5-二羟基-1,4二噻烷的摩尔比为1:(0.4~1.0)。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,所述环合反应的温度为75℃~85℃。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,通过采用薄层色谱法检测反应液中酰基叠氮类化合物的含量来追踪环合反应进程,所述薄层色谱法中采用的展开剂为石油醚-乙酸乙酯混合溶剂,所述混合溶剂中石油醚与乙酸乙酯的体积比为(1~10):1。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,所述环合反应的反应时间为18~21h。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,所述有机溶剂的使用量为每1mmol的所述酰基叠氮类化合物配用4~6ml的溶剂。
优选地,所述将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液的步骤中,所述有机溶剂为乙腈、甲苯和1,4-二氧六环中的任意一种。
优选地,所述将反应结束后的所述反应液过滤,取滤液并除去滤液中的杂质,蒸馏后得浓缩液的步骤,具体包括:
将反应结束后的所述反应液过滤,得滤液;
向滤液中加入乙酸乙酯和水进行萃取,得有机层;
将所述有机层洗涤干燥后,蒸馏除去残余的溶剂,得浓缩液。
优选地,所述将所述浓缩液进行硅胶柱层析,得4-羟基-3-取代-噻唑烷-2-酮的步骤中,所述硅胶柱层析中采用的洗脱液为石油醚与乙酸乙酯的混合溶液,其中,石油醚与乙酸乙酯的体积比为(1~10):1。
本发明提供的4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法为一锅法,通过将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷在有机溶剂中环合得到4-羟基-3-取代-噻唑烷-2-酮,再经过除杂、纯化,即得到4-羟基-3-取代-噻唑烷-2-酮产品。由于对原料及反应条件的设计,本发明提供的制备方法原料易得,无需其他催化剂,成本低,污染小,反应条件温和,安全方便,能高效合成4-羟基-3-取代-噻唑烷-2-酮,提高了收率。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅为本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
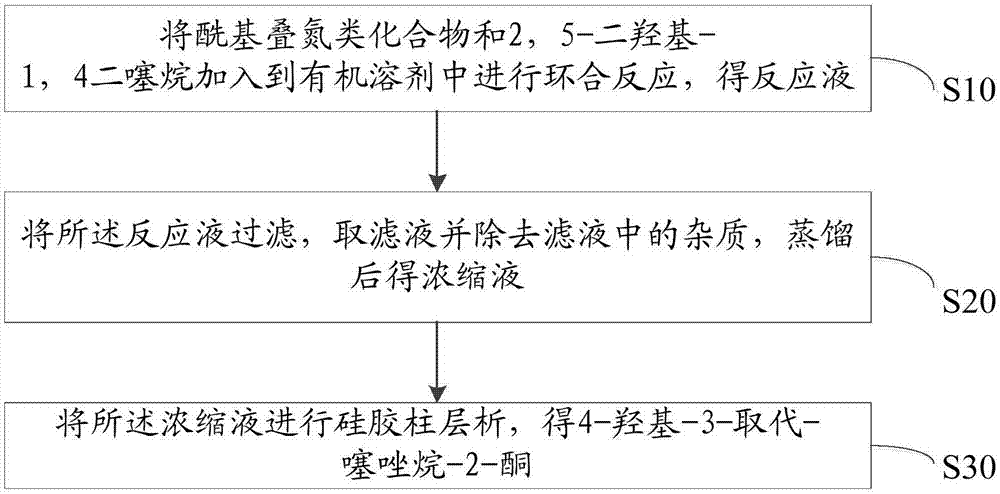
图1为本发明提供的4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法的一实施例的流程示意图。
本发明目的的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
由于噻唑烷酮类化合物具有重要的应用价值,其合成方法一直受到广泛的关注。早期,人们使用卤代乙醛与硫代氨基甲酸为原料来合成3-甲基-4-羟基-噻唑烷-2-酮。后来人们又使用3-取代-噻唑烷二酮还原得到4-羟基-3-取代-噻唑烷-2酮类化合物。但这些制备方法的产品收率刚刚达到60%,且反应所需的硫代氨基甲酸和3-取代-噻唑烷二酮都不易获得,因此,仍然需要对制备方法作进一步改进。
鉴于此,本发明提出一种4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法。结合图1所示的4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法的一实施例的流程示意图,所述4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法包括以下步骤:
步骤s10、将酰基叠氮类化合物和2,5-二羟基-1,4二噻烷加入到有机溶剂中进行环合反应,得反应液。
本发明的合成路线如下:
其中,“i”为所述酰基叠氮类化合物的结构式,r1为所需制备的4-羟基-3-取代-噻唑烷-2-酮类化合物中的3号位取代基,在本实施例中,r1优选为苯基、4-甲基苯基、4-氯苯基、4-溴苯基、4-三氟甲基苯基、3-硝基苯基、4-氰基苯基、2-噻吩基、3-甲氧基苯基、肉桂酸基和苯丙基中的任意一种,例如,当r1为苯丙基时,“i”为3-苯丙基酰基叠氮,3-苯丙基酰基叠氮和2,5-二羟基-1,4二噻烷在有机溶剂中环合反应可以生成3-苯丙基-4-羟基噻唑烷-2-酮,以此类推,本发明可适用于多种官能团取代的4-羟基-3-取代-噻唑烷-2-酮类化合物,通用性高;“ⅱ”为2,5-二羟基-1,4二噻烷的结构式,在本发明中,2,5-二羟基-1,4二噻烷作为反应原料,可以在市面上购买获得,获取难度低,而且酰基叠氮类化合物和2,5-二羟基-1,4二噻烷在一定条件下可以直接反应得到“ⅱ”(4-羟基-3-取代-噻唑烷-2-酮),步骤简单易操作,且无需添加其他催化剂,成本低,污染小。
在本实施例中,当酰基叠氮类化合物和2,5-二羟基-1,4二噻烷的加入量控制在摩尔比为1:(0.4~1.0),特别是摩尔比为1:0.55时,二者得以充分反应,合成收率增高。
为使所述环合反应顺利稳定的正向进行,需要提供一定的反应温度,在本实施例中,所述环合反应的温度为75℃~85℃,优选为80℃。为维持反应体系的稳定,保证反应顺利进行,反应体系需要的有机溶剂可以是叔丁醇、thf、乙腈、甲苯、1,4-二氧六环等任意一种溶解性能好的极性或非极性溶剂,但考虑到有机溶剂的沸点不能低于反应温度以及进一步地提高收率,在本实施例中,所述有机溶剂优选为乙腈、甲苯和1,4-二氧六环中的任意一种,所述有机溶剂的使用量为每1mmol的所述酰基叠氮类化合物配用4~6ml的溶剂。
在环合反应的过程中,为判断反应进展情况,确定反应是否结束,需要追踪反应进程,在本实施例中,通过采用薄层色谱法检测反应液中酰基叠氮类化合物的含量来追踪环合反应进程,其具体做法为:取反应溶液在薄层板上点样,以石油醚-乙酸乙酯混合溶剂为展开剂展开,然后将薄层板置于紫外灯下观察,当薄层板上不再出现酰基叠氮类化合物的斑点时,说明反应结束,其中,所述混合溶剂中石油醚与乙酸乙酯的体积比为(1~10):1。
经过发明人的多次试验,逐步统计出上述环合反应的具体时间。为进一步简化4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法,使其操作更加简单方便,在本实施例中,环合反应的时间优选为18~21h。
步骤s20、将反应结束后的所述反应液过滤,取滤液并除去滤液中的杂质,蒸馏后得浓缩液。
在具体实施时,上述步骤s20具体包括如下操作:
步骤s201、将反应结束后的所述反应液过滤,得滤液;
步骤s202、向滤液中加入乙酸乙酯和水进行萃取,得有机层;
步骤s203、将所述有机层洗涤干燥后,蒸馏除去残余的溶剂,得浓缩液。
反应结束后,反应液中除了4-羟基-3-取代-噻唑烷-2-酮,还包括固体杂质和液体杂质,通过过滤,除去固定杂质,然后加入乙酸乙酯和水充分搅拌,在静置分层后,除去水相,液体杂质中的水溶性部分溶于水相被分离,剩下的有机相部分通常会加入无水硫酸钠或者无水硫酸镁等干燥剂对有机相进行干燥,随后过滤除去干燥剂得到不含水的有机相,利用有机相中各组分沸点不同,对有机相蒸馏,即得浓缩液。蒸馏方式有多种,可以简单蒸馏或者采用旋转蒸发仪,本实施例中,优选为采用旋转蒸发仪除去残余溶剂。
步骤s30、将所述浓缩液进行硅胶柱层析,得4-羟基-3-取代-噻唑烷-2-酮。
为进一步纯化浓缩液,得到高纯度的4-羟基-3-取代-噻唑烷-2-酮,需要对浓缩液柱层析,柱层析的吸附剂和洗脱剂的选择会影响到最终的纯化效果,本实施例中,选用200-300目的硅胶作为吸附剂,石油醚与乙酸乙酯的混合溶液作为洗脱剂,其中,石油醚与乙酸乙酯的体积比为(1~10):1。
以下结合具体实施例和附图对本发明的技术方案作进一步详细说明,应当理解,以下实施例仅仅用以解释本发明,并不用于限定本发明。
实施例1
将苯酰基叠氮220.5mg(1.5mmol)加至反应瓶中,然后加入4ml乙腈于室温下搅拌溶解,随后将2,5-二羟基-1,4二噻烷125.4mg(0.825mmol)加入到反应瓶中,加入4ml乙腈,加料完毕后,于80℃温度下搅拌,期间通过tlc检测反应进程(展开剂为石油醚-乙酸乙酯(5:1,v/v)),当tlc检测反应结果为苯酰基叠氮消失时,说明反应结束。
反应结束后,过滤除去固体,再经过乙酸乙酯-水萃取,所得有机层洗涤干燥后,经过旋转蒸发仪蒸馏除去残余溶剂,得到浓缩物。
选用装有10g200-300目的硅胶作为柱层析,以石油醚:乙酸乙酯=5:1(v/v)作为洗脱剂,设定流速为3ml/min对浓缩液柱层析,收集30-60min的洗脱液,然后经旋转蒸发仪除去残余溶剂后,得到淡黄色油状产物。
产物称重234mg,可知收率为80%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
paleyellowoil1h-nmr(500mhz,cdcl3)δ7.44-7.37(m,4h),7.29-7.26(m,1h),5.61(dd,j1=5hz,j2=5hz,1h),3.79(m,2h),3.24(d,j=10hz,1h);13c-nmr(125mhz,dmso-d6)δ170.14,138.07,128.92,126.31,125.01,84.14,34.64.hrms(esi):m/zcalcdforc9h9no2s[m+h]+:196.0432,found:196.0436.
由上述分析结果可知,产物为4-羟基-3-苯基-噻唑烷-2-酮。
实施例2
将苯酰基叠氮220.5mg(1.5mmol)加至反应瓶中,然后加入3ml甲苯于室温下搅拌溶解,随后将2,5-二羟基-1,4二噻烷125.4mg(0.825mmol)加入到反应瓶中,加入3ml甲苯,加料完毕后,于80℃温度下搅拌,期间通过tlc检测反应进程(展开剂为石油醚-乙酸乙酯(1:1,v/v)),当tlc检测反应结果为苯酰基叠氮消失时,说明反应结束。
反应结束后,过滤除去固体,再经过乙酸乙酯-水萃取,所得有机层洗涤干燥后,经过旋转蒸发仪蒸馏除去残余溶剂,得到浓缩物。
选用装有10g200-300目的硅胶作为柱层析,以石油醚:乙酸乙酯=10:1(v/v)作为洗脱剂,设定流速为3ml/min对浓缩液柱层析,收集30-60min的洗脱液,然后经旋转蒸发仪除去残余溶剂后,得到淡黄色油状产物4-羟基-3苯基噻唑烷-2-酮。
产物称重219mg,可知收率为75%。
实施例3
将苯酰基叠氮220.5mg(1.5mmol)加至反应瓶中,然后加入4.5ml的1,4-二氧六环于室温下搅拌溶解,随后将2,5-二羟基-1,4二噻烷125.4mg(0.825mmol)加入到反应瓶中,加入4.5ml的1,4-二氧六环,加料完毕后,于80℃温度下搅拌,期间通过tlc检测反应进程(展开剂为石油醚-乙酸乙酯(10:1,v/v)),当tlc检测反应结果为苯酰基叠氮消失时,说明反应结束。
反应结束后,过滤除去固体,再经过乙酸乙酯-水萃取,所得有机层洗涤干燥后,经过旋转蒸发仪蒸馏除去残余溶剂,得到浓缩物。
选用装有10g200-300目的硅胶作为柱层析,以石油醚:乙酸乙酯=1:1(v/v)作为洗脱剂,设定流速为3ml/min对浓缩液柱层析,收集30-60min的洗脱液,然后经旋转蒸发仪除去残余溶剂后,得到淡黄色油状产物4-羟基-3苯基噻唑烷-2-酮。
产物称重219mg,可知收率为75%。
实施例4
将苯酰基叠氮220.5mg(1.5mmol)加至反应瓶中,然后加入4ml乙腈于室温下搅拌溶解,随后将2,5-二羟基-1,4二噻烷125.4mg(0.825mmol)加入到反应瓶中,加入4ml乙腈,加料完毕后,于85℃温度下搅拌18h,得反应液。
将反应液过滤除去固体,再经过乙酸乙酯-水萃取,所得有机层洗涤干燥后,经过旋转蒸发仪蒸馏除去残余溶剂,得到浓缩物。
选用装有10g200-300目的硅胶作为柱层析,以石油醚:乙酸乙酯=5:1(v/v)作为洗脱剂,设定流速为3ml/min对浓缩液柱层析,收集30-60min的洗脱液,然后经旋转蒸发仪除去残余溶剂后,得到淡黄色油状产物4-羟基-3苯基噻唑烷-2-酮。
产物称重204.7mg,可知收率为70%。
实施例5
将苯酰基叠氮220.5mg(1.5mmol)加至反应瓶中,然后加入4ml乙腈于室温下搅拌溶解,随后将2,5-二羟基-1,4二噻烷125.4mg(0.825mmol)加入到反应瓶中,加入4ml乙腈,加料完毕后,于75℃温度下搅拌21h,得反应液。
将反应液过滤除去固体,再经过乙酸乙酯-水萃取,所得有机层洗涤干燥后,经过旋转蒸发仪蒸馏除去残余溶剂,得到浓缩物。
选用装有10g200-300目的硅胶作为柱层析,以石油醚:乙酸乙酯=5:1(v/v)作为洗脱剂,设定流速为3ml/min对浓缩液柱层析,收集30-60min的洗脱液,然后经旋转蒸发仪除去残余溶剂后,得到淡黄色油状产物4-羟基-3苯基噻唑烷-2-酮。
产物称重228mg,可知收率为78%。
实施例6
将2,5-二羟基-1,4二噻烷的加入量改为91mg(0.6mmol),其余同实施例1。得到淡黄色油状产物4-羟基-3苯基-噻唑烷-2-酮140.4mg,收率75%。
实施例7
将2,5-二羟基-1,4二噻烷的加入量改为456mg(1.5mmol),其余同实施例1。得到淡黄色油状产物4-羟基-3苯基-噻唑烷-2-酮234mg,收率80%。
实施例8
将1.5mmol苯甲酰基叠氮换成1.5mmol3-硝基-苯基酰基叠氮,其余等同于实施例1。得到淡黄色粉末状产物。
产物称重280mg,可知收率为78%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
paleyellowpowder;1h-nmr(500mhz,dmso-d6)δ8.36(t,j=2.5hz,1h),8.10(m,1h),7.90(m,1h),7.7(t,j=8hz,1h),7.05(d,j=7.5hz,1h),5.90(m,1h),3.82(dd,j=6,12hz,1h),3.21(dd,j=1,11.5hz,1h);13c-nmr(500mhz,dmso-d6)δ170.84,148.01,139.01,130.39,130.33,120.55,118.38,83.86,34.51.hrms(esi):m/zcalcdforc9h8n2o4s[m+h]+:241.0283,found:241.0279.
由上述分析结果可知,产物为3-(3-硝基-苯基)-4-羟基-噻唑烷-2-酮。
实施例9
将1.5mmol苯甲酰基叠氮换成1.5mmol4-氯-苯基酰基叠氮,其余等同于实施例1,得到白色粉末状产物。
产物称重257.6mg,可知收率为75%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder;1h-nmr(500mhz,dmso-d6)δ7.47-7.43(m,4h),6.89(d,j=7hz,1h),5.74(m,1h),3.78(dd,j=5.5hz,11.5hz,1h),3.15(dd,j=1.5hz,12hz1h);13c-nmr(500mhz,dmso-d6)δ170.24,136.90,130.28,128.83,126.33,83.94,34.49.hrms(esi):m/zcalcdforc9h8clno2s[m+h]+:230.0037,found:230.0043.
由上述分析结果可知,产物为3-(4-氯-苯基)-4-羟基噻唑烷-2-酮。
实施例10
将1.5mmol苯甲酰基叠氮换成1.5mmol4-氰基-苯基酰基叠氮,其余等同于实施例1。得到白色粉末状产物。
产物称重244.2mg,可知收率为74%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder1h-nmr(500mhzdmso-d6)δ7.87(d,j=8.5hz,1h),7.68(d,j=9hz,1h),7.04(d,j=7.0hz,1h),5.90(t,j=5.5hz,1h),3.79(dd,j=12,6.0hz,1h),3.19(d,j=11.5hz,1h),2.49(s,1h).13c-nmr(500mhz,dmso-d6)δ170.79,142.14,133.12,123.4,118.77,107.55,83.66,34.51.hrms(esi):m/zcalcdforc10h8n2o2s[m+h]+:221.0385,found:221.0379.
由上述分析结果可知,产物为3-(4-氰基-苯基)-4-羟基-噻唑烷-2-酮。
实施例11
将1.5mmol苯甲酰基叠氮换成1.5mmol4-溴基-苯基酰基叠氮,其余等同于实施例1。得到白色粉末状产物。
产物称重319.4mg,可知收率为78%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder1hnmr(500mhz,dmso-d6)δ7.62-7.56(m,2h),7.40(dd,j=2.0,9.0hz,2h),6.90(d,j=7.0hz,1h),5.74(t,j=5.7hz,1h),3.78(dd,j=11.8,5.7hz,1h),3.15(dd,j=12,1.5hz,1h).13cnmr(500mhz,dmso-d6)δc170.6,141.6,126,126.05,124,83.8,34.6.hrms(esi):m/zcalcdforc9h8brno2s[m+h]+:273.9537,found:273.9532.
由上述分析结果可知,产物为3-(4-溴基-苯基)-4-羟基-噻唑烷-2-酮。
实施例12
将1.5mmol苯甲酰基叠氮换成1.5mmol4-甲基-苯基酰基叠氮,其余等同于实施例1,得到白色粉末状产物。
产物称重260.2mg,可知收率为83%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder1hnmr(500mhz,dmso-d6)δ7.23(dd,j=42,8hz,4h),6.80(d,j=7hz,1h),5.66(t,j=6.5hz,1h),3.77(dd,j=12,6hz,1h),3.13(d,j=12hz,1h),2.28(s,3h).13cnmr(500mhz,dmso-d6)δ170.32,136.03,135.83,129.70,125.47,84.50,34.93,21.02.hrms(esi):m/zcalcdforc10h11no2s[m+h]+:210.0589,found:210.0591.
由上述分析结果可知,产物为3-(4-甲基-苯基)-4-羟基-噻唑烷-2-酮。
实施例13
将1.5mmol苯甲酰基叠氮换成1.5mmol4-三氟甲基-苯基酰基叠氮,其余等同于实施例1,得到白色粉末状产物。
产物称重284mg,可知收率为72%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder1hnmr(500mhz,dmso-d6)δ7.79(d,j=8.5hz,2h),7.71(d,j=8.5hz,2h),7.02(d,j=7.0hz,1h),5.89(t,j=6hz,1h),3.83(dd,j=11.5,5.5hz,1h),3.21(dd,j=11.5,1.0hz,1h).13cnmr(500mhz,dmso-d6)δc170.6,137.7,132.1,127,119,84.3,34.9.hrms(esi):m/zcalcdforc10h8f3no2s[m+h]+:264.0306,found:264.0301.
由上述分析结果可知,产物为3-(4-三氟甲基-苯基)-4-羟基-噻唑烷-2-酮。
实施例14
将1.5mmol苯甲酰基叠氮换成1.5mmol2-噻吩基酰基叠氮,其余等同于实施例1,得到白色粉末状产物。
产物称重270.7mg,可知收率为89.8%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder1hnmr(500mhz,dmso-d6)δ7.19-7.15(m,2h),6.94(dd,j=5.5,4hz,1h),6.88(dd,j=3.5,1hz,1h),5.91-5.89(m,1h),3.86(dd,j=11.9,5.5hz,1h),3.22(dd,j=12,1.0hz,1h).13cnmr(125mhz,dmso-d6)δ170.08,159.57,139.13,129.60,117.07,111.67,110.75,84.16,55.32,34.61.hrms(esi):m/zcalcdforc7h7no2s2[m+h]+:201.9996,found:201.9994.
由上述分析结果可知,产物为3-(2-噻吩基)-4-羟基-噻唑烷-2-酮。
实施例15
将1.5mmol苯甲酰基叠氮换成1.5mmol3-甲氧基-苯基酰基叠氮,其余等同于实施例1,得到白色粉末状产物。
产物称重293.6mg,可知收率为85%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder,1hnmr(500mhz,dmso-d6)δ7.31(t,j=7.9hz,1h),7.02(d,j=5hz,2h),6.86(dd,j=10,20hz,2h),5.75(dd,j=10,5hz,1h),3.79(dd,j=10,5hz,1h),3.76(s,3h),3.18-3.13(m,1h).13cnmr(125mhz,dmso-d6)δ170.08,159.57,139.13,129.60,117.07,111.67,110.75,84.16,55.32,34.61.hrms(esi):m/zcalcdforc10h11no3s[m+h]+:226.0538,found:226.0538.
由上述分析结果可知,产物为3-(3-甲氧基)-4-羟基-噻唑烷-2-酮。
实施例16
将1.5mmol苯甲酰基叠氮换成1.5mmol3-肉桂酸基-苯基酰基叠氮,其余等同于实施例1,得到白色粉末状产物。
产物称重278.4mg,可知收率为84%。
对产物做核磁共振氢谱、碳谱及高分辨率质谱分析,得分析结果如下:
whitepowder,1h-nmr(500mhz,dmso-d6)δ7.38(d,j=10hz,2h),7.32(t,j=5hz,2h),7.26-7.17(m,2h),6.96(d,j=5hz,1h),6.38(d,j=15hz,1h),5.87(t,j=5hz,1h),3.78(dd,j=15,10hz,1h),3.17(d,j=10hz,1h).13c-nmr(125mhz,dmso-d6)δ169.74,136.31,128.89,126.71,125.53,122.56,112.92,80.45,34.35.hrms(esi):m/zcalcdforc11h11no2s[m+h]+:222.0589,found:22.0578.
由上述分析结果可知,产物为3-肉桂酸基-4-羟基-噻唑烷-2-酮。
综上所述,本发明提供的4-羟基-3-取代-噻唑烷-2-酮类化合物的制备方法适用于多种官能团取代的4-羟基-3-取代-噻唑烷-2-酮类化合物,通用性高,且收率均达到70%以上。
以上仅为本发明的优选实施例,并非因此限制本发明的专利范围,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!