鸭瘟病毒gE基因ET区域无痕缺失株DPVCHv-gEΔET及其构建方法与流程
本发明属于基因工程技术领域,具体涉及一种鸭瘟病毒ge基因胞外域(et)无痕缺失株dpvchv-geδet及其构建方法。
背景技术:
细菌人工染色体(bacterialartificialchromosome,bac)是一种新发展起来的dna载体系统,它具有容量大、遗传特性稳定、易于操作等优点,在基因文库构建和基因功能分析等方面有广泛的应用。将完整病毒基因组dna分子插入bac载体,利用该载体编码的最小生育力因子复制子(minimalfertilityfactorreplicon,mini-f)获得分子克隆化病毒,并结合大肠杆菌中成熟的基因定位修饰技术,从而实现在原核系统中病毒基因的缺失及外源基因的插入。目前较为常用的大肠杆菌基因定位修饰技术主要包括red/et介导的同源重组技术、reca蛋白介导的同源重组技术、cre/loxp介导的同源重组技术以及tn转座子介导的随机插入和突变技术。利用分子克隆化技术手段现已获得成熟的细菌人工染色体鸭瘟病毒拯救系统平台,同时利用大肠杆菌基因定位修饰red/et介导的同源重组技术可在细菌人工染色体鸭瘟病毒拯救系统平台上进行鸭瘟病毒基因的缺失和外源基因的插入,该成果极大的推动了鸭瘟病毒基因功能的研究进程。
red/et介导的同源重组技术是基于λ-噬菌体red操纵子(redα/redβ/redγ)和rac噬菌体rece/rect同源重组酶的同源重组打靶技术。该技术操作简单、快速、高效,广泛应用于基因缺失、突变工作。但是利用该操作技术进行基因缺失、突变会在缺失或突变位点残留约80bp左右外源碱基序列(frt位点),该位点的残留将影响基因功能的准确分析。minif元件作为维持bac载体复制的最小生育力因子复制子,主要由调控bac复制起点(oris)的repe、repf基因、调控复制子分配的sopa、sopb基因以及编码着丝粒区域的sopc基因构成。为完成细菌抗性筛选及bac标记筛选,minif元件加入了抗性筛选基因和荧光标记基因。minif元件插入病毒基因组,增加基因组长度,对病毒复制产生不确定影响。同时minif元件作为细菌序列在病毒基因组上残留,不利于减毒活疫苗的开发和许可。因此解决碱基残留问题并去除minif元件获得无痕基因缺失株,已成为探究鸭瘟病毒基因缺失方法的重点。
鸭瘟(duckplague,dp)是由α-疱疹病毒亚科中鸭瘟病毒(duckplaguevirus,dpv)引起的鸭、鹅等水禽的急性接触性高度致死性传染病。该病首先由荷兰报道,随即在我国华南、华中和华东等养鸭业较为发达的地区流行,给我国的养鸭业造成了严重的经济损失。因此深入了解鸭瘟病毒基因功能、加强对鸭瘟疫病的研究对确保我国养鸭业健康、可持续发展尤为重要。
鸭瘟病毒dpv-chv株基因组dna全长162175bp,包含78个开放阅读框,可编码参与鸭瘟病毒生命周期的结构蛋白和非结构蛋白,其中结构蛋白主要包括衣壳蛋白、皮层蛋白和囊膜蛋白。囊膜蛋白为糖基化蛋白,包括gb、gc、gd、ge、gg、gh、ge、gj、gk、gl、gm、gn十二种。糖蛋白具有介导病毒吸附、进入敏感细胞以及促进病毒在细胞间传播的功能,同时携带抗原决定簇,可诱导动物机体免疫系统对病毒的识别并造成组织的病理损伤,因此探究囊膜糖蛋白在鸭瘟病毒生命周期中的作用对深入探究鸭瘟病毒基因功能及开展鸭瘟疫病防治工作至关重要。
现有技术中利用以bac为平台分子克隆化病毒的技术,将鸭瘟病毒基因组重组到含有bac的病毒转移载体中,构建细菌人工染色体重组鸭瘟病毒拯救系统平台dpvchv-bac-g。同时结合red/et修饰技术,在原核系统中通过成熟的基因操作手段完成鸭瘟病毒基因缺失和外源基因插入。但利用red/et修饰技术在细菌人工染色体重组鸭瘟病毒拯救系统平台上对鸭瘟病毒基因缺失后,会在缺失基因处残余两处frt位点,同时残留minif元件。frt外源位点和minif元件的残留对基因功能的探究、减毒活疫苗的开发和许可存在影响。
技术实现要素:
针对现有技术中存在的上述问题,本发明提供一种鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet及其构建方法,该构建方法可有效解决缺失鸭瘟病毒基因时在缺失位点残留碱基,同时残留minif元件的问题。
为实现上述目的,本发明解决其技术问题所采用的技术方案是:
鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet,其构建方法包括以下步骤:
(1)将pbac-dpv质粒转化到gs1783大肠杆菌感受态中,获得gs1783-pbac-dpv菌株,然后制备gs1783-pbac-dpv感受态;
(2)以pepkan-s为模板,以gs1783-bac-geδet-f和gs1783-bac-geδet-r作为引物,通过pcr方法扩增包含i_scei酶切位点和kana元件的碱基片段以及ge基因et区域上游和下游各40bp同源臂的打靶片段i_scei-kana-et,切胶回收获得i_scei-kana-et片段;
(3)将i_scei-kana-et片段转化到gs1783-pbac-dpv感受态中,经抗生素筛选和pcr鉴定,获得阳性克隆子gs1783-pbac-dpv-et-kana;
(4)将阳性克隆子gs1783-pbac-dpv-et-kana中的i_scei-kana片段去掉,制备gs1783-pbac-dpv-geδet感受态;
(5)以pepkan-s为模板,以gs1783-minif-f和gs1783-minif-r作为引物,通过pcr方法扩增包含i_scei酶切位点和kana元件的碱基片段、位于minif元件ori2基因下游240bp处和位于minif元件ori2基因下游290bp处的同源臂片段i_scei-kana-minif,切胶回收获得i_scei-kana-minif片段;
(6)以chv基因组为模板,以chv-ul23-f和chv-ul23-r作为引物,通过pcr方法扩增包含ul23基因、i_scei-kana-minif下游同源臂重叠25bp和位于minif元件ori2基因下游180bp处的同源臂片段,切胶回收获得ul23-minif片段;
(7)以i_scei-kana-minif片段和ul23-minif片段为模板,进行pcr融合反应,然后以融合片段为模板,以gs1783-minif-f和chv-ul23-r作为引物进行pcr扩增获得i_scei-kana-minif-ul23打靶片段;
(8)将i_scei-kana-minif-ul23打靶片段转化到gs1783-pbac-dpv-geδet感受态中,经抗生素筛选和pcr鉴定,获得阳性克隆子gs1783-pbac-dpv-geδet-ul23-kana;
(9)将阳性克隆子gs1783-pbac-dpv-geδet-ul23-kana中的i_scei-kana片段去掉,获得阳性克隆子gs1783-pbac-dpv-geδet-ul23;
(10)从阳性克隆子gs1783-pbac-dpv-geδet-ul23中提取pbac-dpv-geδet-ul23质粒,将pbac-dpv-geδet-ul23质粒转染def细胞,通过克隆筛选,获得的ge基因et区域无痕缺失株dpvchv-geδet。
进一步地,步骤(2)、步骤(5)和步骤(6)中pcr扩增体系为:ddh2o22μl、
进一步地,步骤(2)中引物序列为:
gs1783-bac-geδet-f:5’-ctgccggccagactacggaacctcaacaattggtacgatgtagggataacagggtaatcgattt-3’;
gs1783-bac-geδet-r:5’-taatagtcacgacccctagtactccgagaccgactacaaacatcgtaccaattgttgaggttccgtagtctggccggcaggccagtgttacaaccaat-3’。
进一步地,步骤(5)中引物序列为:
gs1783-minif-f:5’-ttattaatctcaggagcctgtgtagcgtttataggaagtagtgttctgtcatgatgcctgcaagcggtaacgaaaacgattgttacaaccaattaacc-3’;
gs1783-minif-r:5’-atcgttttcgttaccgcttgcaggcatcatgacagaacactacttcctattagggataacagggtaatcgat-3’。
进一步地,步骤(6)中引物序列为:
chv-ul23-f:5’-gcctgcaagcggtaacgaaaacgattcaattaattgtcatctcgg-3’;
chv-ul23-r:5’-ccgctccacttcaacgtaacaccgcacgaagatttctattgttcctgaaggcatattcaacggacatattaaaaattga-3’。
进一步地,步骤(7)中引物序列为:
gs1783-minif-f:5’-ttattaatctcaggagcctgtgtagcgtttataggaagtagtgttctgtcatgatgcctgcaagcggtaacgaaaacgattgttacaaccaattaacc-3’;
chv-ul23-r:5’-ccgctccacttcaacgtaacaccgcacgaagatttctattgttcctgaaggcatattcaacggacatattaaaaattga-3’。
进一步地,步骤(7)中pcr融合体系为:ddh2o8μl、
进一步地,步骤(7)中pcr扩增体系为:融合模板20μl、上游引物gs1783-minif-f0.5μl、下游引物chv-ul23-r0.5μl;pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min。
本发明提供的鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet及其构建方法,具有以下有益效果:
为获得无外源碱基和minif元件残留的鸭瘟病毒基因缺失株,本发明在细菌人工染色体重组鸭瘟病毒拯救系统平台的基础上,利用red-based修饰技术,即利用含有可编码red操纵子和i_scei酶基因序列的gs1783大肠杆菌菌株及含有编码卡纳抗性及i_scei酶切位点的质粒pepkan-s,首先在细菌人工染色体重组鸭瘟病毒拯救系统平台上经两次同源重组缺失鸭瘟病毒ge基因et区域,再经细胞内自发同源重组方法缺失minif元件,首次完成了无外源碱基和minif元件残留的鸭瘟病毒无痕缺失株的构建。本发明技术方案解决了缺失鸭瘟病毒基因时在缺失位点残留碱基的问题并缺失了minif元件,为准确探究鸭瘟病毒基因功能及减毒活疫苗的构建提供了充分的技术支持。
附图说明
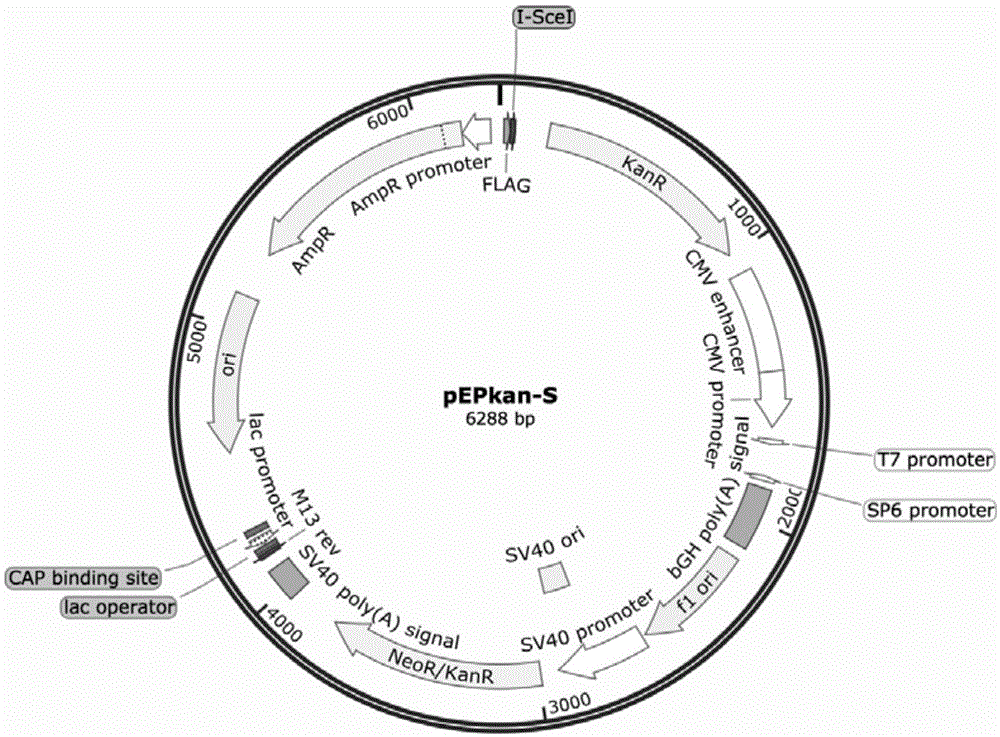
图1为pepkan-s质粒图谱。
图2为利用red-based修饰技术在细菌人工染色体重组鸭瘟病毒拯救系统平台上进行基因缺失的操作流程图。
图3为利用red-based修饰技术和细胞内自发同源重组技术对minif元件进行缺失的操作流程图。
图4为dpvchv-geδet无痕缺失病毒株病毒拯救后图片。
具体实施方式
鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet,构建过程所用材料及试剂如下:
1、实验材料
(1)细胞、菌株、病毒毒株、质粒
原代鸭胚成纤维细胞由10-11日龄非免疫受精鸭胚按常规方法制备;gs1783菌株由四川农业大学实验室保存;pbac-dpv质粒由四川农业大学实验室构建并保存;pepkan-s质粒由四川农业大学实验室保存。
2、分子生物学试剂
质粒小提试剂盒购自tiangen公司;qiagenplasmidmidikit购自qiagen公司;普通琼脂糖凝胶dna回收试剂盒购自tiangen公司;
3、实验所用溶液及其配制
lb液体培养基:称取tryptone10g、yeastextract5g、氯化钠10g溶于800ml去离子水中,充分搅拌,定容至ll,高温高压灭菌。
lb固体培养基:在定容至1l的lb液体培养基中加入15g琼脂粉,高温高压灭菌后,冷却至60℃左右,加入1.5ml氯霉素(储存浓度25mg/ml)或1.5ml卡那霉素(储存浓度50mg/ml),铺制平板,待凝固后,4℃保存。
mem:将9.6gmem干粉和2.2g碳酸氢钠溶于800ml去离子水,充分搅拌,调节ph值至7.4,定容至ll,过滤除菌,4℃保存。
实施例1制备鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet
鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet,其构建方法包括以下步骤:
1、制备gs1783电转感受态,电转化pbac-dpv质粒
(1)复苏带pbac-dpv质粒的大肠杆菌于含有氯霉素的lb固体培养基中,37℃培养过夜;挑单菌落接种于含有氯霉素的lb液体培养基中,37℃培养过夜;
(2)按照qiagenplasmidmidikit操作说明提取pbac-dpv质粒;
(3)复苏gs1783冻存菌于lb固体培养基中,30℃培养过夜;
(4)挑取gs1783单菌落接种于5mllb液体培养基中,30℃培养过夜,获得种子液;
(5)将5ml种子液加入100mllb液体培养基中,30℃摇至od600值在0.5~0.7之间;
(6)将步骤(5)所得菌液立即放入冰水混合物中冷却20min;
(7)取步骤(6)所得菌液,4℃、4500×g离心10min去上清;
(8)用预冷超纯水在冰上反复清洗步骤(7)菌体沉淀;
(9)向步骤(8)所得菌体中加入超纯水将菌液定容至500μl,每管100μl分装至预冷ep管,获得gs1783电转感受态;
(10)在100μlgs1783电转感受态中加入20ngpbac-dpv质粒,混匀后将感受态和质粒一起加入2mm预冷的电击杯底部,15kv/cm条件下进行电击;
(11)取100μllb液体培养基重悬电击后菌体,30℃摇菌1h后,4500×g离心菌体2min,弃上清,采用200μllb液体培养基悬浮沉淀,涂布在含有氯霉素的lb固体培养基上,30℃培养24h,获得gs1783-pbac-dpv菌株。
2、扩增i_scei-kana-et打靶片段
(1)复苏带有pepkan-s质粒的大肠杆菌于含有卡那霉素的lb固体培养基中,37℃培养过夜;挑单菌落接种于含有卡那霉素的lb液体培养基中,37℃培养过夜,采用质粒小提试剂盒提取pepkan-s质粒(pepkan-s质粒图谱见图1);
(2)以pepkan-s质粒为模板,gs1783-bac-geδet-f和gs1783-bac-geδet-r为引物,扩增i_scei-kana-et打靶片段,采用普通琼脂糖凝胶dna回收试剂盒回收扩增片段;
gs1783-bac-geδet-f:5’-ctgccggccagactacggaacctcaacaattggtacgatgtagggataacagggtaatcgattt-3’(seqidno:1)
gs1783-bac-geδet-r:5’-taatagtcacgacccctagtactccgagaccgactacaaacatcgtaccaattgttgaggttccgtagtctggccggcaggccagtgttacaaccaat-3’(seqidno:2)
pcr扩增体系为:ddh2o22μl、
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
利用red-based修饰技术在细菌人工染色体重组鸭瘟病毒拯救系统平台上进行基因缺失的操作流程图见图2,具体过程包括如下的3和4两个过程。
3、制备gs1783-pbac-dpv电转感受态进行打靶片段打靶
(1)复苏gs1783-pbac-dpv冻存菌于含有氯霉素的lb固体培养基中,30℃培养过夜;
(2)挑取gs1783-pbac-dpv单菌落接种于5ml含氯霉素的lb液体培养基中,30℃培养过夜,获得种子液;
(3)将5ml种子液加入100ml含氯霉素的lb液体培养基中,置于30℃摇至od600值在0.5~0.7之间;
(4)将步骤(3)所得菌液于42℃培养15min后立即放入冰水混合物中冷却20min;
(5)取50ml步骤(4)所得菌液,于4℃4500×g离心10min,去上清;
(6)用预冷超纯水在冰上反复清洗步骤(5)菌体沉淀;
(7)向步骤(6)所得菌体中加入超纯水将菌液定容至500μl,每管100μl分装至预冷ep管,获得gs1783-pbac-dpv电转感受态;
(8)在100μl电转感受态中加入200ngi_scei-kana-et打靶片段,混匀后将感受态和打靶片段一起加入2mm预冷的电击杯底部,15kv/cm条件下进行电击;
(9)取100μllb液体培养基重悬电击后菌体,30℃摇菌1h后,于4500×g离心菌体2min,弃上清,200μllb液体培养基悬浮沉淀,涂布于含有卡那霉素和氯霉素双抗生素抗性的lb固体培养基,于30℃培养48h;
(10)pcr鉴定步骤(9)所得单菌落,获得阳性菌落gs178-pbac-dpv-et-kana,以步骤(9)中生长的单菌落重悬液为模板,利用扩增i_scei-kana-et打靶片段上游引物gs1783-bac-geδet-f和鉴定ge基因et区域下游引物ge-r鉴定阳性菌落,获得阳性克隆子gs1783-pbac-dpv-et-kana;
gs1783-bac-geδet-f:5’-ctgccggccagactacggaacctcaacaattggtacgatgtagggataacagggtaatcgattt-3’(seqidno:1)
ge-r:5’-agcgagtacttctctgcgtc-3’(seqidno:3)
pcr扩增体系为:ddh2o22μl、
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
4、去掉i_scei-kana片段
(1)挑取gs1783-pbac-dpv-et-kana单菌落接种于2ml含氯霉素的lb液体培养基中,30℃培养过夜,获得种子液;
(2)取10μl步骤(1)所得种子液接种于2ml含氯霉素的lb液体培养基中,30℃培养2h至菌液呈现轻微云雾状;
(3)向步骤(2)所得菌液中加入1ml含氯霉素的lb液体培养基和5m终浓度为2%的l-阿拉伯糖,30℃培养1h;
(4)将步骤(3)所得菌液立即放入42℃水浴中培养30min;
(5)将步骤(4)所得菌液置于30℃培养2h后,取1μl菌液加入200μllb液体培养基中混匀后涂布到含有氯霉素的lb固体培养基,30℃培养24h~48h;
(6)挑取步骤(5)中所得单菌落在含氯霉素和卡纳霉素双抗性lb固体培养基和氯霉素单抗性lb固体培养基上进行平行筛选,将氯霉素和卡纳霉素双抗性lb固体培养基不生长,氯霉素单抗性lb固体培养基生长的菌落通过pcr方法利用ge基因et区域鉴定引物鉴定,获得阳性克隆子gs1783-pbac-dpv-geδet。
ge-f:5’-tctcaagacgctctggaatc-3’(seqidno:4)
ge-r:5’-agcgagtacttctctgcgtc-3’(seqidno:3)
pcr扩增体系为:ddh2o22μl、
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
5、扩增i_scei-kana-minif片段
(1)复苏带有pepkan-s质粒的大肠杆菌于含有卡那霉素的lb固体培养基中,37℃培养过夜;挑单菌落接种于含有卡那霉素的lb液体培养基中,37℃培养过夜,质粒小提试剂盒提取pepkan-s质粒;
(2)以pepkan-s质粒为模板,gs1783-minif-f和gs1783-minif-r为引物,扩增i_scei-kana-minif打靶片段,普通琼脂糖凝胶dna回收试剂盒回收扩增片段;
gs1783-minif-f:5’-ttattaatctcaggagcctgtgtagcgtttataggaagtagtgttctgtcatgatgcctgcaagcggtaacgaaaacgattgttacaaccaattaacc-3’(seqidno:5)
gs1783-minif-r:5’-atcgttttcgttaccgcttgcaggcatcatgacagaacactacttcctattagggataacagggtaatcgat-3’(seqidno:6)
pcr扩增体系为ddh2o22μl、
pcr扩增条件为98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
6、扩增ul23-minif片段
(1)制备鸭胚成纤维细胞(def)并接种于25t细胞瓶,37℃,5%co2培养24h后,接种5moidpvchv,37℃,5%co2培养48h后收获病毒,反复冻融2次后,按照takaraminibestviralrna/dnaextractionkitver.5.0说明书提取dpvchv基因组;
(2)dpvchv基因组为模板,chv-ul23-f和chv-ul23-r为引物,扩增ul23-minif打靶片段,普通琼脂糖凝胶dna回收试剂盒回收扩增片段;
chv-ul23-f:5’-gcctgcaagcggtaacgaaaacgattcaattaattgtcatctcgg-3’(seqidno:7)
chv-ul23-r:5’-ccgctccacttcaacgtaacaccgcacgaagatttctattgttcctgaaggcatattcaacggacatattaaaaattga-3’(seqidno:8)
pcr扩增体系为ddh2o22μl、
pcr扩增条件为98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
7、融合i_scei-kana-minif片段和ul23-minif片段获得i_scei-kana-minif-ul23打靶片段
(1)以i_scei-kana-minif片段和ul23-minif片段为模板,融合i_scei-kana-minif片段和ul23-minif片段;
pcr扩增体系为:ddh2o8μl、
pcr扩增条件为:95℃预变性5min、95℃变性15s、55℃退火5s、72℃延伸1min,共5个循环。
(2)向步骤(1)所得的融合模板中加入引物gs1783-minif-f和chv-ul23-r,扩增i_scei-kana-minif-ul23打靶片段,普通琼脂糖凝胶dna回收试剂盒回收扩增片段;
gs1783-minif-f:5’-ttattaatctcaggagcctgtgtagcgtttataggaagtagtgttctgtcatgatgcctgcaagcggtaacgaaaacgattgttacaaccaattaacc-3’(seqidno:5)
chv-ul23-r:5’-ccgctccacttcaacgtaacaccgcacgaagatttctattgttcctgaaggcatattcaacggacatattaaaaattga-3’(seqidno:8)
pcr扩增体系为:融合模板20μl、上游引物gs1783-minif-f0.5μl、下游引物chv-ul23-r0.5μl;
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
利用red-based修饰技术和细胞内自发同源重组技术对minif元件进行缺失的操作流程图见图3,具体过程包括如下的8、9和10。
8、制备gs1783-pbac-dpv-geδet电转感受态进行打靶片段打靶
(1)复苏gs1783-pbac-dpv-geδet冻存菌于含有氯霉素的lb固体培养基中,30℃培养过夜;
(2)挑取gs1783-pbac-dpv-geδet单菌落接种于5ml含氯霉素的lb液体培养基中,30℃培养过夜,获得种子液;
(3)将5ml种子液加入100ml含氯霉素的lb液体培养基中,于30℃摇至od600值在0.5~0.7之间;
(4)将步骤(3)所得菌液于42℃培养15min后立即放入冰水混合物中冷却20min;
(5)取50ml步骤(4)所得菌液,于4℃4500×g离心10min,去上清;
(6)用预冷超纯水在冰上反复清洗步骤(5)所得菌体沉淀;
(7)加入超纯水将步骤(6)所得菌液定容至500μl,每管100μl分装至预冷ep管,获得gs1783-pbac-dpv-geδet电转感受态;
(8)在100μl电转感受态中加入200ngi_scei-kana-minif-ul23打靶片段,混匀后将感受态和打靶片段一起加入2mm预冷的电击杯底部,15kv/cm条件下进行电击;
(9)取100μllb液体培养基重悬电击后菌体,30℃摇菌1h后,4500×g离心菌体2min,弃上清,200μllb液体培养基悬浮沉淀,涂布含有卡那霉素和氯霉素双抗生素抗性的lb固体培养基,于30℃培养48h;
(10)pcr鉴定步骤(9)所得单菌落,获得阳性菌落gs1783-pbac-dpv-geδet-ul23-kana,以步骤(9)中生长的单菌落重悬液为模板,利用鉴定引物minif-f和minif-r鉴定阳性菌落;
minif-f:5’-gttatccactgagaagcgaacg-3’(seqidno:9)
minif-r:5’-ggctgtaaaaggacagaccaca-3’(seqidno:10)
pcr扩增体系为:ddh2o22μl、
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸15s,共30个循环,最后72℃延伸10min,于16℃保存。
9、去掉i_scei-kana片段
(1)挑取gs1783-pbac-dpv-geδet-ul23-kana单菌落接种于2ml含氯霉素的lb液体培养基中,30℃培养过夜,获得种子液;
(2)取10μl步骤(1)所得种子液接种于2ml含氯霉素的lb液体培养基中,30℃培养2h至菌液呈现轻微云雾状;
(3)向步骤(2)所得菌液中加入1ml含氯霉素的lb液体培养基和5m终浓度为2%的l-阿拉伯糖30℃,培养1h;
(4)将步骤(3)所得菌液立即放入42℃水浴中培养30min;
(5)将步骤(4)所得菌液置于30℃培养2h后,取1μl菌液加入200μllb液体培养基中混匀后涂布到含有氯霉素的lb固体培养基,30℃培养24h~48h;
(6)挑取步骤(5)中所得单菌落在含氯霉素和卡纳霉素双抗性lb固体培养基和氯霉素单抗性lb固体培养基上进行平行筛选,将氯霉素和卡纳霉素双抗性lb固体培养基不生长,氯霉素单抗性lb固体培养基生长的菌落通过pcr方法利用minif基因鉴定引物鉴定,获得阳性克隆子gs1783-pbac-dpv-geδet-ul23。
minif-f:5’-gttatccactgagaagcgaacg-3’(seqidno:9)
minif-r:5’-ggctgtaaaaggacagaccaca-3’(seqidno:10)
pcr扩增体系为:ddh2o22μl、
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
10、拯救dpvchv-geδet病毒
(1)复苏gs1783-pbac-dpv-geδet-ul23冻存菌于含有氯霉素的lb固体培养基中,30℃培养过夜;
(2)按照qiagenplasmidmidikit操作说明提取pbac-dpv-geδet-ul23质粒;
(3)制备鸭胚成纤维细胞(def)并接种于12孔板,37℃,5%co2培养24h后,按照lipofectamine3000操作说明转染pbac-dpv-geδet-ul23质粒,96h后观察荧光斑,收集病毒,反复冻融2次后接种于长满def的6孔板;
(4)重复步骤(3)三次;
(5)步骤(4)获得的病毒反复冻融2次后,10倍倍比稀释,接种于长满def的6孔板中,37℃,5%co2孵育2h后,弃孵育液,加入1%甲基纤维素固定细胞,37℃,5%co2培养120h后,挑取无荧光病变细胞,反复冻融2次后重新接种于def中,获得dpvchv-geδet-q;
(6)按照takaraminibestviralrna/dnaextractionkitver.5.0说明书提取dpvchv-geδet-q病毒基因组,利用minif鉴定引物minif-f和minif-r,鉴定缺失的minif元件,最终获得无minif元件残留,缺失基因处无碱基残留的无痕缺失病毒株dpvchv-geδet阳性病毒株,见图4。
minif-f:5’-gttatccactgagaagcgaacg-3’(seqidno:9)
minif-r:5’-ggctgtaaaaggacagaccaca-3’(seqidno:10)
pcr扩增体系为:ddh2o22μl、
pcr扩增条件为:98℃预变性2min、98℃变性10s、55℃退火15s、72℃延伸5s,共30个循环,最后72℃延伸10min,于16℃保存。
实施例2无痕缺失株dpvchv-geδet生长曲线的测定
1、一步生长曲线的测定
将亲本病毒dpvchv和ge基因et区域无痕缺失株dpvchv-geδet分别以2moi接种def细胞,接毒后6h、12h、18h、24h收集上清和细胞,每个时间点做三次重复。待收集完全后,反复冻融2次,于96孔板检测病毒滴度,结果表明ge基因et区域的缺失部分影响dpvchv病毒的复制。
2、多步生长曲线的测定
将亲本病毒dpvchv和ge基因et区域无痕缺失株dpvchv-geδet分别以0.01moi接种def细胞,接毒后12h、24h、48h、72h收集上清和细胞,每个时间点做三次重复。待收集完全后,反复冻融2次,于96孔板检测病毒滴度,结果表明ge基因et区域的缺失显著影响dpvchv病毒的增殖情况。
实施例3无痕缺失株dpvchv-geδet空斑形成实验
将亲本病毒dpvchv和ge基因et区域无痕缺失株dpvchv-geδet分别以0.01moi接种布满def细胞的6孔板,37℃、5%co2吸附2h后,去上清,加入2ml1%甲基纤维素,37℃、5%co2培养48h后,去1%甲基纤维素,pbs洗涤3次,4%多聚甲醛4℃固定过夜,pbs洗涤3次,加入h2o2和甲醇以体积比为1:50混合的混合液,室温孵育30min,蒸馏水洗涤3次,加入5%bsa封闭液,室温孵育30min,加入兔抗dpv,4℃孵育过夜,pbs洗涤3次,加入生物素化山羊抗兔igg,37℃孵育30min,pbs洗涤3次,滴加sabc底物,37℃孵育30min,pbs洗涤3次,dab显色液避光显色,封片,结果表明ge基因et区域的缺失可能影响dpvchv病毒在细胞内增殖及传播情况。
此外,本发明还对鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet做了遗传稳定性实验和免疫原性实验。
遗传稳定性:鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet在def细胞上传代20代,均出现蚀斑,表明所获得的鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet在def中稳定遗传。
免疫原性:将10只4周龄dpv抗体阴性和pcr检测阴性鸭,随机分成2组,每组5只。第1组肌肉注射dpvchv-geδet,第2组注射等剂量的mem,作为对照组,每组单独隔离饲养。在免疫后15天采血分离血清,利用纯化的dpv作为包被抗原进行间接elisa检测,结果可检测到注射dpvchv-geδet鸭血清中的抗体,说明dpvchv-geδet具有良好的免疫原性。
序列表
<110>四川农业大学
<120>鸭瘟病毒ge基因et区域无痕缺失株dpvchv-geδet及其构建方法
<160>10
<170>siposequencelisting1.0
<210>1
<211>64
<212>dna
<213>人工序列(artificialsequence)
<400>1
ctgccggccagactacggaacctcaacaattggtacgatgtagggataacagggtaatcg60
attt64
<210>2
<211>98
<212>dna
<213>人工序列(artificialsequence)
<400>2
taatagtcacgacccctagtactccgagaccgactacaaacatcgtaccaattgttgagg60
ttccgtagtctggccggcaggccagtgttacaaccaat98
<210>3
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>3
agcgagtacttctctgcgtc20
<210>4
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>4
tctcaagacgctctggaatc20
<210>5
<211>98
<212>dna
<213>人工序列(artificialsequence)
<400>5
ttattaatctcaggagcctgtgtagcgtttataggaagtagtgttctgtcatgatgcctg60
caagcggtaacgaaaacgattgttacaaccaattaacc98
<210>6
<211>72
<212>dna
<213>人工序列(artificialsequence)
<400>6
atcgttttcgttaccgcttgcaggcatcatgacagaacactacttcctattagggataac60
agggtaatcgat72
<210>7
<211>45
<212>dna
<213>人工序列(artificialsequence)
<400>7
gcctgcaagcggtaacgaaaacgattcaattaattgtcatctcgg45
<210>8
<211>79
<212>dna
<213>人工序列(artificialsequence)
<400>8
ccgctccacttcaacgtaacaccgcacgaagatttctattgttcctgaaggcatattcaa60
cggacatattaaaaattga79
<210>9
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>9
gttatccactgagaagcgaacg22
<210>10
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>10
ggctgtaaaaggacagaccaca22
- 还没有人留言评论。精彩留言会获得点赞!