用于提高聚酰亚胺树脂的粘合特性的化合物和使用其制备的聚酰亚胺共聚物的制作方法
本申请要求于2017年9月28日提交的韩国专利申请第10-2017-0125671号和于2018年6月25日提交的韩国专利申请第10-2018-0072773号的优先权的权益,其全部公开内容通过引用并入本文。
本发明涉及能够改善聚酰亚胺树脂的粘合性的新的化合物和使用其制备的聚酰亚胺共聚物。
背景技术:
近年来,在显示器领域中产品的轻量化和小型化受到重视。然而,玻璃基底重且脆,并且难以应用于连续过程。因此,积极地进行研究以用塑料基底代替玻璃基底,所述塑料基底具有轻质、柔性并且能够在诸如移动电话、笔记本电脑、pda等的设备中连续处理的优点。
特别地,聚酰亚胺(pi)树脂具有以下优点:易于合成,可以以薄膜的形式制备并且不需要用于固化的交联基团。出于这些原因,根据近来电子产品的趋势(例如轻量化和精细化),许多研究已经尝试将pi作为lcd、pdp等的半导体中的集成用材料用于具有轻质和柔性特性的柔性塑料显示板。
通过由聚酰亚胺树脂形成膜而制备的聚酰亚胺(pi)膜通常通过以下过程制备:进行芳族二酐和芳族二胺或芳族二异氰酸酯的溶液聚合以制备聚酰胺酸衍生物的溶液,将溶液涂覆在硅晶片或玻璃上,并通过热处理固化。
为了通过使用聚酰亚胺树脂制备电路板、半导体基底、柔性显示器基底等,除了诸如耐热氧化性、耐热性、耐辐射性、耐低温性、耐化学性等的物理特性之外,还需要对硅晶片、玻璃或金属具有优异的粘合。
通常,使用粘合促进剂(例如硅烷化合物)来改善聚酰亚胺膜与玻璃或金属表面之间的粘合力。当将粘合促进剂施加至表面以改善粘合力时,其充当异物。因此,可能无法获得基底的光滑表面。此外,应在施加过程之后再一次重复涂覆过程,这是不经济有效的。
当将粘合促进剂直接添加到聚酰胺酸中时,可以使由施加过程引起的问题最小化。然而,硅烷化合物的氨基可以与聚酰胺酸的羧基反应以产生盐,使得可能在基底上形成异物。
因此,需要开发用于聚酰亚胺树脂的粘合促进剂,所述粘合促进剂通过省略向聚酰亚胺树脂赋予粘合力的步骤而能够改善生产率和过程效率,并且能够在确保优异的机械特性的同时显著改善表面粘合,而不使最终产品的外观特性劣化。
技术实现要素:
技术问题
本发明要解决的问题是提供可用作用于聚酰亚胺树脂的粘合促进剂的新的化合物。
本发明还提供了包含所述新的化合物作为用于聚酰亚胺树脂的粘合促进剂的聚酰亚胺共聚物。
本发明还提供了使用聚酰亚胺共聚物制备的聚酰亚胺膜。
技术方案
为了解决本发明的问题,提供了具有由下式(1a)或(1b)表示的结构的化合物。
[式1a]
[式1b]
在式(1a)和(1b)中,
在式(1a)和(1b)中,
x1和x2各自独立地为经取代或未经取代的具有1至30个碳原子的三价有机基团,或者各自独立地为经取代或未经取代的具有3至30个碳原子的四价有机基团使得x1和x2彼此键合,
x3和x4各自独立地为经取代或未经取代的具有1至30个碳原子的二价有机基团,或者各自独立地为经取代或未经取代的具有3至30个碳原子的三价有机基团使得x3和x4彼此键合,
r1和r3各自独立地为具有1至5个碳原子的烷基,
r2和r4各自独立地为氢原子、或具有1至5个碳原子的烷基,
a和b各自独立地为1至3的整数,以及
n和m各自独立地为0至3的整数。
根据一个实施方案,式(1a)或(1b)的化合物可以为式(2a)或(2b)的化合物。
[式2a]
[式2b]
在式(2a)和(2b)中,
r1和r3各自独立地为具有1至5个碳原子的烷基,
r2和r4各自独立地为氢原子、或具有1至5个碳原子的烷基,
a和b各自独立地为1至3的整数,
n和m各自独立地为0至3的整数,以及
虚线(------)表示成键或不成键。
根据一个实施方案,式(2a)的化合物可以选自下式(3a)至(3f)的化合物。
根据一个实施方案,式(2b)的化合物可以选自下式(4a)至(4f)的化合物。
为了解决本发明的其他问题,提供了聚酰亚胺共聚物,其通过使包含以下的聚酰亚胺前体组合物聚合和固化来制备:
作为聚合组分的酸二酐、二胺和二甲基硅氧烷(dms)-二苯基硅氧烷(dps)低聚物;
具有在25℃下的正分配系数(logp)的溶剂;和
式(1a)或(1b)的化合物。
根据一个实施方案,尺寸为50nm或更小的dms-dps低聚物的区域均匀地分布在聚酰亚胺基体中,并且dms-dps区域占据的体积可以为总体积的15体积%至30体积%。
根据一个实施方案,dms-dps区域的尺寸可以为1nm至50nm。
根据一个实施方案,dms-dps低聚物可以具有以下结构:
[式6]
其中,p和q为摩尔分数,以及设p+q=100,则p为70至90且q为10至30。
根据一个实施方案,相对于100重量份的聚酰亚胺前体,用于聚酰亚胺树脂的粘合促进剂以0.1重量份至10重量份的量包含在内。
根据一个实施方案,用于聚酰亚胺树脂的粘合促进剂以每摩尔酸二酐0.001摩尔至0.5摩尔的量包含在内。
根据一个实施方案,具有式(6)的结构的二胺化合物的分子量可以为4000g/mol或更大。
根据一个实施方案,具有正分配系数(logp)的溶剂可以为基于酰胺的溶剂。
根据一个实施方案,基于酰胺的溶剂可以为选自以下的至少一者:二甲基丙酰胺(dmpa)、二乙基丙酰胺(depa)、n,n-二乙基乙酰胺(deac)、n,n-二乙基甲酰胺(def)和n-乙基吡咯烷酮(nep)。
本发明还提供了由聚酰亚胺共聚物制备的聚酰亚胺膜。
根据一个实施方案,聚酰亚胺膜的延迟可以为-500nm至500nm。
根据一个实施方案,聚酰亚胺膜与载体基底之间的粘合力可以为至少5gf/in。
发明效果
本发明提供了具有芴骨架的新的用于聚酰亚胺树脂的粘合促进剂,由此包含所述粘合促进剂的聚酰亚胺共聚物不表现出延迟的增加,同时保持优异的现有特性例如耐热性和机械特性,并且即使在高温过程中也保持与载体基底的粘合特性。
附图说明
图1是合成例1中制备的化合物的1hnmr谱。
图2是根据合成例1的化合物的1h-1htocsy(全相关谱)谱。
图3示出了aptes(3-氨基丙基三乙氧基硅烷)和根据合成例1的化合物的1hnmr谱的比较。
具体实施方式
由于可以在本发明中做出各种修改和变化,因此在附图中示出并在详细描述中详细地描述了特定实施方案。然而,应理解,本发明不旨在限于特定实施方案,而是包括落入本发明的精神和范围内的所有修改、等同方案和替代方案。在本发明的以下描述中,如果确定已知功能的详细描述可能使本发明的主旨模糊,则将省略所述已知功能的详细描述。
在本公开内容中,除非另外说明,否则所有化合物或有机基团可以为经取代或未经取代的。在本文中,术语“取代”意指化合物或有机基团中包含的至少一个氢被选自以下的取代基取代:卤素原子、具有1至10个碳原子的烷基、卤代烷基、具有3至30个碳原子的环烷基、具有6至30个碳原子的芳基、羟基、具有1至10个碳原子的烷氧基、羧基、醛基、环氧基、氰基、硝基、氨基、磺酸基、或其衍生物。
本发明提供了具有由下式(1a)或(1b)表示的结构的化合物。
[式1a]
[式1b]
在式(1a)和(1b)中,
x1和x2各自独立地为经取代或未经取代的具有1至30个碳原子的三价有机基团,或者各自独立地为经取代或未经取代的具有3至30个碳原子的四价有机基团使得x1和x2彼此键合,
x3和x4各自独立地为经取代或未经取代的具有1至30个碳原子的二价有机基团,或者各自独立地为经取代或未经取代的具有3至30个碳原子的三价有机基团使得x3和x4彼此键合,
r1和r3各自独立地为具有1至5个碳原子的烷基,
r2和r4各自独立地为氢原子、或具有1至5个碳原子的烷基,
a和b各自独立地为1至3的整数,以及
n和m各自独立地为0至3的整数。
根据优选实施方案,其可以为具有由式(2a)或(2b)表示的结构的化合物。
[式2a]
[式2b]
在式(2a)和(2b)中,
r1、r2、r3、r4、a、b、n和m与以上限定的相同,虚线(------)表示成键或不成键。
对于用作常规柔性显示器基底的高耐热性聚酰亚胺,已经使用了以下方法:将粘合促进剂施加至作为载体基底的玻璃或其上沉积有无机层的玻璃基底以改善对玻璃的粘合,然后成膜。然而,这样的常规粘合促进剂具有的问题在于:由于施加粘合促进剂而产生异物,或者需要另外的涂覆过程,从而具有低的经济效率。此外,即使在将粘合促进剂直接添加到聚酰亚胺前体中时,氨基也与聚酰胺酸的羧基反应以形成盐,导致粘合性降低。
还存在其中可以将粘合促进剂直接添加到聚酰亚胺前体中以改善粘合性的常规方法。然而,厚度方向上的延迟值增加。因此,尽管粘合特性增加,但是存在所得聚酰亚胺膜的物理特性可能受到影响的问题。这是因为现有技术的粘合促进剂由通常包含两个或更多个芳族结构的二酐制备,其由于相对刚性结构而在固化之后在该部分处导致相位延迟现象。
或者,当使用包含柔性结构的粘合促进剂例如odpa(4,4'-氧双邻苯二甲酸酐)时,由于结构的柔性,延迟值可能不增加,但tg趋于降低。
因此,本发明人对以下粘合促进剂进行了研究,所述粘合促进剂在与聚酰亚胺前体混合时不发生盐析,使得异物的产生可以最小化并且对基底的粘合优异,并且其不影响作为通过施加和固化而制备的聚酰亚胺膜的光学特性的厚度方向上的延迟值。
根据本发明的可以用作粘合促进剂的化合物具有芴骨架,例如式(1a)或(1b)的结构,使得在保持粘合提高效果最大化的同时,由于芴骨架而产生分子间自由体积,并且填充密度不受影响。此外,由于包含更多芳族的结构特性,所述化合物可以提供不影响聚酰亚胺膜的耐热性和作为光学特性的厚度方向上的延迟值的高耐热性聚酰亚胺膜。
具有式(1a)的结构的粘合促进剂可以由包含芴结构的酸酐与氨基丙基四乙氧基硅烷的反应制备。
具有式(1b)的结构的粘合促进剂可以由包含芴结构的二胺与酸酐封端的四乙氧基硅烷的反应制备。
特别地,在式(1a)或(1b)的化合物中,烷氧基硅烷(si-or)部分可以通过水或水分转化为硅烷醇基(si-oh)。硅烷醇基可以与玻璃或金属发生缩合反应,使得其可以与玻璃或金属表面强烈结合。
根据一个实施方案,式(1a)的化合物可以选自下式(3a)至(3f)的化合物。
在上式(3a)至(3f)中,r1和r2与式(1a)中限定的相同。
根据一个实施方案,式(1b)的化合物可以选自下式(4a)至(4f)的化合物。
在上式(4a)至(4f)中,r3和r4与式(1b)中限定的相同。
本发明提供了聚酰亚胺共聚物,其通过使包含以下的聚酰亚胺前体组合物聚合和固化来制备:
作为聚合组分的酸二酐、二胺和二甲基硅氧烷(dms)-二苯基硅氧烷(dps)低聚物;
具有正分配系数(logp)的溶剂;和
作为粘合促进剂的式(1a)或(1b)的化合物。
根据一个实施方案,尺寸为50nm或更小的dms-dps低聚物的区域均匀地分布在聚酰亚胺基体中,并且dms-dps区域占据的体积可以为总体积的15体积%至30体积%。dms-dps区域的尺寸优选为1nm至50nm,或5nm至40nm,或10nm至30nm,以实现均匀分布。
根据一个实施方案,粘合促进剂可以以每摩尔酸二酐0.001摩尔至0.5摩尔的量包含在内。
此外,相对于100重量份的聚酰亚胺前体,粘合促进剂可以以0.1重量份至10重量份的量包含在内。
根据一个实施方案,粘合促进剂可以包含在聚酰亚胺前体组合物中以形成下式(5a)或(5b)的结构。
[式5a]
[式5b]
在式(5a)和(5b)中,r1、r2、r3、r4、a、b、n和m各自与式(1a)和(1b)中限定的相同,
z为衍生自四羧酸二酐的残基,以及
y为衍生自二胺的残基。
即,如上式(5a)或(5b)所示,根据一个实施方案的粘合促进剂与通过四羧酸二酐和二胺的反应形成的聚酰胺酸的重复单元的末端键合。因此,氨基不会暴露,并因此不与酸反应产生盐。并且可以提供这样的聚酰亚胺树脂:其具有增加的粘合强度,并且由于芴结构而没有增加厚度方向上的延迟。
根据一个实施方案,dms-dps低聚物可以具有下式(6)的结构:
[式6]
其中,p和q为摩尔分数,以及设p+q=100,则p为70至90且q为10至30。
具有式(6)的结构的二胺化合物的分子量可以为4000g/mol或更大,优选为4400g/mol或更大,更优选为5000g/mol或更大。在本文中,分子量意指重均分子量并且可以通过使用nmr分析或酸碱滴定计算胺当量来确定。
当具有式(6)的结构的二胺的分子量小于4000g/mol时,耐热性可能降低。例如,所得聚酰亚胺的玻璃化转变温度(tg)降低或者热膨胀系数过度增加。
根据一个实施方案,在本发明中可以使用一种或更多种二胺。式(6)的二胺可以以总二胺的1mol%至20mol%、优选1mol%至10mol%的量包含在内。
根据一个实施方案,基于聚酰亚胺共聚物的总固体含量(即,聚酰亚胺前体的固体含量的重量或聚合组分(二胺和酸二酐)的总重量),式(6)的二胺可以以10重量%至50重量%,优选10重量%至40重量%的量添加。如果相对于聚合物的总重量过量添加包含式(6)的结构的二胺,例如以50重量%或更大、或者40重量%或更大的量添加,则聚酰亚胺的机械特性(例如模量)可能降低并且膜强度可能降低,使得在过程中可能发生物理损坏,例如膜撕裂。如果过量添加具有式(6)的结构的二胺,则其具有源自具有硅氧烷结构的聚合物的玻璃化转变温度(tg)。由此,可能在350℃或更低的低过程温度下出现玻璃态。在无机膜沉积期间,由于聚合物的流动现象,可能在膜的表面上产生褶皱,并且无机膜可能开裂。
根据本发明,分布在聚酰亚胺基体中的dms-dps区域具有为纳米尺寸(例如1nm至50nm、或5nm至40nm、或10nm至30nm)的连续相,使得可以在保持耐热性和机械特性的同时使残余应力最小化。在不具有这样的连续相的情况下,虽然可以获得残余应力的减小效果,但耐热性和机械特性显著降低,难以用于加工。
在本文中,dms-dps区域是指具有dms-dps结构的聚合物的分布区域,并且其尺寸是指围绕该区域的圆的直径。
优选地,包含dms-dps结构的部分(区域)在聚酰亚胺基体中以连续相连接,其中连续相意指其中纳米尺寸的区域均匀分布的形状。
因此,在本发明中,即使dms-dps具有高分子量,其也可以均匀地分布在聚酰亚胺基体中而没有相分离,使得雾度特性降低并因此获得具有更透明的特性的聚酰亚胺。此外,由于dms-dps结构以连续相存在,因此可以更有效地改善聚酰亚胺的机械强度和应力松弛效果。根据这些特性,根据本发明的组合物可以提供热特性和光学特性以及在涂覆和固化之后具有减少的基底弯曲现象的平坦聚酰亚胺膜。
通过将包含硅氧烷结构的式(6)的结构引入聚酰亚胺结构中,本发明可以改善聚酰亚胺的模量强度并且可以使由外力引起的应力减轻。包含硅氧烷结构的聚酰亚胺可以表现出极性。并且由于与不包含硅氧烷结构的聚酰亚胺结构的极性差异,可能发生相分离,从而使得硅氧烷结构在整个聚酰亚胺结构中不均匀地分布。在这种情况下,由于硅氧烷结构而难以表现出聚酰亚胺的物理特性的改善效果,例如强度提高效果和应力松弛效果,并且由于由相分离引起的雾度增加,膜的透明度可能劣化。特别地,当包含硅氧烷结构的二胺具有高分子量时,由所述二胺制备的聚酰亚胺表现出更明显的极性,并且可能更明显地发生聚酰亚胺之间的相分离现象。然而,当使用具有低分子量结构的硅氧烷二胺时,应添加大量的硅氧烷二胺以表现出诸如应力松弛的效果。这可能引起工艺问题,例如低温下的tg,因此聚酰亚胺膜的物理特性可能劣化。因此,与添加低分子量硅氧烷二胺的情况相比,当添加高分子量硅氧烷二胺时,可以在分子中大量形成松弛链段,因此,即使在较低的量下也可以有效地表现出应力松弛效果。因此,本发明人研究了用于使具有高分子量硅氧烷结构的式(6)的二胺更均匀地分布在聚酰亚胺基体上而没有相分离的方法。
本发明通过使用包含具有高分子量的si结构的二胺在具有正分配系数(logp)的有机溶剂中聚合来制备聚酰亚胺,可以提供无色透明且具有优异的耐热性的聚酰亚胺膜。
具有正分配系数(logp)的溶剂可以为基于酰胺的溶剂,并且基于酰胺的溶剂可以为选自以下的至少一者:二甲基丙酰胺(dmpa)、二乙基丙酰胺(depa)、n,n-二乙基乙酰胺(deac)、n,n-二乙基甲酰胺(def)和n-乙基吡咯烷酮(nep)。
通过使用如上所述的有机溶剂,根据本发明的聚酰亚胺共聚物可以减少由于其中引入有式(6)的结构的柔性聚酰亚胺结构与其他聚酰亚胺结构之间的极性差异而引起的相分离。常规地,使用两种有机溶剂以解决相分离问题。然而,即使用一种有机溶剂,本发明也可以减少由于相分离引起的白浊,使得可以制备更透明的聚酰亚胺膜。
另一方面,存在将极性溶剂和非极性溶剂混合以解决白浊问题的方法。然而,由于极性溶剂具有高挥发性,其可能在制造过程期间预先挥发,这可能引起诸如过程再现性劣化的问题。此外,可能无法完全解决相分离的问题,导致所制备的聚酰亚胺膜的高雾度和低透明度。
在本发明中,为了使包含式(6)的结构的聚酰亚胺结构均匀地分布在整个聚酰亚胺基体中,使用具有正分配系数(logp)的溶剂,特别是具有正logp的基于酰胺的溶剂。更具体地,通过使用包含两亲性分子结构的溶剂,可以解决由于使用极性溶剂而引起的过程问题。此外,即使仅使用一种溶剂,由于两亲性分子结构,聚酰亚胺也可以均匀地分布,并且适合于解决由相分离引起的问题。因此,可以提供具有显著改善的雾度特性的聚酰亚胺。
根据一个实施方案,二酐可以选自在分子结构中包含下式(7a)至(7h)的四价有机基团的四羧酸二酐。
[式7a]
[式7b]
[式7c]
[式7d]
[式7e]
[式7f]
[式7g]
[式7h]
在式(7a)至(7h)中,r11至r24各自独立地为选自以下的取代基:选自-f、-cl、-br和-i的卤素原子;羟基(-oh);硫醇基(-sh);硝基(-no2);氰基;具有1至10个碳原子的烷基;具有1至4个碳原子的卤代烷氧基;具有1至10个碳原子的卤代烷基;和具有6至20个碳原子的芳基,
a1为0至2的整数,a2为0至4的整数,a3为0至8的整数,a4和a5各自独立地为0至3的整数,a7和a8各自独立地为0至3的整数,a10和a12各自独立地为0至3的整数,a11为0至4的整数,a15和a16各自独立地为0至4的整数,a17和a18各自独立地为0至4的整数,以及a6、a9、a13、a14、a19和a20各自独立地为0至3的整数,
n为1至3的整数,以及
a11至a16各自独立地选自-o-、-cr'r"-、-c(=o)-、-c(=o)o-、-c(=o)nh-、-s-、-so2-、亚苯基、及其组合,其中r'和r"各自独立地选自氢原子、具有1至10个碳原子的烷基和具有1至10个碳原子的氟烷基。
根据一个实施方案,基于总二胺含量,二胺可以以80mol%至99mol%的量包含在分子结构中含有下式(8)的二价有机基团的二胺。
[式8]
在式(8)中,
r31和r32各自独立地为选自以下的取代基:选自-f、-cl、-br和-i的卤素原子;羟基(-oh);硫醇基(-sh);硝基(-no2);氰基;具有1至10个碳原子的烷基;具有1至4个碳原子的卤代烷氧基;具有1至10个碳原子的卤代烷基;和具有6至20个碳原子的芳基,优选为选自以下的取代基:卤素原子;卤代烷基;烷基;芳基;和氰基。例如,卤素原子可以为氟(-f);卤代烷基可以为包含氟原子的具有1至10个碳原子的氟烷基,例如选自氟甲基、全氟乙基和三氟甲基;烷基可以选自甲基、乙基、丙基、异丙基、叔丁基、戊基和己基;芳基可以选自苯基和萘基。更优选地,它们可以被氟原子或包含氟原子的取代基(例如氟烷基)取代。
q可以选自单键、-o-、-cr'r"-、-c(=o)-、-c(=o)o-、-c(=o)nh-、-s-、-so2-、亚苯基、及其组合,其中r'和r"各自独立地选自氢原子、具有1至10个碳原子的烷基和具有1至10个碳原子的氟烷基。
在本文中,本发明的“基于氟的取代基”意指“氟原子取代基”以及“包含氟原子的取代基”。
式(8)的二胺可以选自由下式(8a)至(8d)表示的化合物。
在式(8a)至(8d)中,q与上述相同。
根据一个实施方案,在总四羧酸二酐中,四羧酸二酐可以以20mol%至80mol%,优选30mol%至80mol%,更优选30mol%至70mol%的量包含具有由下式(9)表示的结构的四羧酸二酐。
[式9]
根据一个实施方案,在总四羧酸二酐中,四羧酸二酐可以以20mol%至80mol%,优选20mol%至60mol%,更优选20mol%至50mol%的量包含具有由下式(10)表示的结构的四羧酸二酐。
[式10]
在式(10)中,
q1和q2各自独立地选自单键、-o-、-c(=o)-、-c(=o)o-、-c(=o)nh-、-s-、-so2-、亚苯基、及其组合。
根据一个实施方案,式(10)的化合物可以为下式(10a)至(10e)的化合物。
通过在聚酰亚胺结构中包含芴结构,可以减小膜的厚度方向上的延迟。
在本发明中,可以一起使用选自包含下式(11a)至(11r)的四价有机基团的四羧酸二酐中的至少一者。
在式(11l)中,a2可以选自单键、-o-、-c(=o)-、-c(=o)nh-、-s-、-so2-、亚苯基、及其组合,v为0或1的整数;在式(11r)中,x为1至10的整数。
式(11a)至(11r)的四价有机基团中存在的至少一个氢原子可以被选自以下的取代基取代:选自-f、-cl、-br和-i的卤素原子;羟基(-oh);硫醇基(-sh);硝基(-no2);氰基;具有1至10个碳原子的烷基;具有1至4个碳原子的卤代烷氧基;具有1至10个碳原子的卤代烷基;和具有6至20个碳原子的芳基。
或者,在本发明中,式(9)和(10)的四羧酸二酐可以一起使用。当式(9)和(10)的四羧酸二酐一起使用时,相对于四羧酸二酐的总含量,式(10)的四羧酸二酐的含量可以在10mol%至30mol%,优选10mol%至25mol%,更优选15mol%至25mol%的范围内。通过将包含芴结构的式(10)的化合物与式(9)的化合物一起用于制备聚酰亚胺,减轻了由于热引起的平面方向上的收缩。因此,可以改善耐热性例如玻璃化转变温度和在加热步骤之后的冷却步骤期间出现的膜的收缩现象。
根据本发明的一个实施方案,四羧酸二酐的总含量和二胺的含量可以为1:1.1至1.1:1的摩尔比。为了改善反应性和可加工性,优选地,四羧酸二酐的总含量相对于二胺过量,或者二胺的含量相对于四羧酸二酐的总含量过量。
根据本发明的一个实施方案,优选地,四羧酸二酐的总含量与二胺的含量的摩尔比为1:0.99至0.99:1,优选为1:0.98至0.98:1。
可以在聚合反应中使用的有机溶剂可以具有在25℃下的正分配系数(logp值)和180℃或更低的沸点。更具体地,分配系数logp值可以为0.01至3、或0.01至2、或0.1至2。
分配系数可以使用来自acd/labs的acd/percepta平台的acd/logp模块来计算。acd/logp模块使用基于使用2d分子结构的qspr(quantitativestructure-propertyrelationship,定量结构-特性关系)方法的算法。
正分配系数值意味着溶剂的极性为疏水的。根据本发明人的研究,如果使用具有正分配系数(logp)值的特定溶剂来制备聚酰亚胺前体组合物,则可以改善溶液的反润湿现象。此外,通过使用具有正logp值的溶剂,可以在不使用用于控制涂覆膜的表面张力或光滑度的添加剂例如流平剂的情况下控制溶液的反润湿现象。由于不使用另外的材料例如添加剂,因此可以消除品质和过程的问题,例如最终产品中包含低分子物质,并且可以更有效地形成具有均匀特性的聚酰亚胺膜。
例如,在将聚酰亚胺前体组合物涂覆在玻璃基底上的过程中,在固化或在湿度条件下放置涂覆溶液期间,由于涂层的收缩而可能发生溶液的反润湿。涂覆溶液的这种反润湿现象引起膜厚度的变化,导致膜的抗弯性不足。因此,可能发生膜断裂或在切割时可能出现边缘开裂。即,可能存在可加工性差和产率降低的问题。
如果向其上涂覆有包含具有负logp的极性溶剂的聚酰亚胺前体溶液的基底中引入极性的细小异物,则异物的极性可能在存在异物的部分周围引起散发性的涂层裂纹或厚度变化。相比之下,当使用具有正logp的疏水性溶剂时,即使在引入极性的细小异物时,也可以减少或抑制涂层裂纹、厚度变化等。
具体地,包含具有正logp的溶剂的聚酰亚胺前体组合物的由以下方程式(1)所定义的反润湿率可以为0%至0.1%或更小:
[方程式1]
反润湿率(%)=[(a-b)/a]×100
在方程式1中,
a:当将聚酰亚胺前体组合物完全涂覆在基底(100mm×100mm)上时测量的面积,
b:从涂覆有聚酰亚胺前体组合物或pi膜的基底的边缘末端发生反润湿现象之后测量的面积。
聚酰亚胺前体组合物和膜的反润湿现象可能在涂覆聚酰亚胺前体组合物的溶液之后的30分钟内发生。特别地,随着从边缘开始反润湿,边缘变厚。
在用根据本发明的聚酰亚胺前体组合物涂覆基底,然后在湿度条件下放置10分钟或更长时间,例如10分钟或更长时间,例如40分钟或更长时间之后,反润湿率为0.1%或更小。例如,即使在20℃至30℃的温度下以及在40%或更大的湿度条件(更具体地,40%至80%,即40%、50%、60%、70%、80%的湿度条件,例如50%的湿度条件)下放置10分钟至50分钟之后,也可以表现出0.1%或更小,优选0.05%,更优选接近0%的非常低的反润湿率。
上述反润湿率即使在固化之后也得以保持。例如,在将聚酰亚胺前体组合物涂覆在基底上,然后例如在20℃至30℃的温度下以及在40%或更大的湿度条件(更具体地,40%至80%,即40%、50%、60%、70%、80%的湿度条件)下放置10分钟或更长时间之后,例如在50%的湿度条件下放置10分钟至50分钟之后,固化的聚酰亚胺膜的反润湿率可以为0.1%或更小,即,即使在通过热处理进行的固化过程中反润湿也可以几乎不发生或者可以消失,并且具体地为0.05%,更优选接近0%。
根据本发明的聚酰亚胺前体组合物可以解决该反润湿现象,从而使得可以获得具有更均匀的特性的聚酰亚胺膜并进一步改善制备过程的产率。
此外,根据本发明的溶剂的通过标准astmd1475所测量的密度可以为1g/cm3或更小。如果密度大于1g/cm3,则相对粘度可能增加并且过程的效率可能降低。
四羧酸二酐和二胺的反应可以通过聚酰亚胺前体的常规聚合方法例如溶液聚合来进行。具体地,将二胺溶解在有机溶剂中,然后通过添加四羧酸二酐来进行聚合反应。
聚合反应可以在惰性气体或氮气流中进行,并且可以在无水条件下进行。
聚合反应期间的反应温度可以为-20℃至80℃,优选为0℃至80℃。如果反应温度太高,则反应性可能变高,分子量可能变大,并且前体组合物的粘度可能增加,这在过程中可能是不利的。
优选地,考虑到成膜步骤期间的涂覆特性等,聚酰亚胺前体组合物以使得组合物具有合适粘度的量包含固体含量。根据一个实施方案,可以调节组合物的含量使得聚酰亚胺前体的总含量为8重量%至25重量%,优选为10重量%至25重量%,更优选为10重量%至20重量%或更小。
或者,可以将聚酰亚胺前体组合物调节为具有3,000cp或更大、或者4,000cp或更大的粘度。聚酰亚胺前体组合物的粘度为10,000cp或更小,优选为9,000cp或更小,更优选为8,000cp或更小。当聚酰亚胺前体组合物的粘度超过10,000cp时,在加工聚酰亚胺膜期间消泡的效率降低。这不仅导致过程的效率降低,而且还导致所制备的膜的表面粗糙度由于气泡产生而劣化。这可能引起电特性、光学特性和机械特性劣化。
根据本发明的聚酰亚胺的重均分子量可以为10,000g/mol至200,000g/mol、或20,000g/mol至100,000g/mol、或30,000g/mol至100,000g/mol。根据本发明的聚酰亚胺的分子量分布(mw/mn)优选为1.1至2.5。当聚酰亚胺的重均分子量或分子量分布在上述范围之外时,可能难以形成膜,或者聚酰亚胺膜的特性(例如透射率、耐热性和机械特性)可能劣化。
然后,可以使作为聚合反应结果获得的聚酰亚胺前体酰亚胺化以制备透明聚酰亚胺膜。此时,酰亚胺化过程可以具体为化学酰亚胺化或热酰亚胺化过程。
例如,在将脱水剂和酰亚胺化催化剂添加到聚合的聚酰亚胺前体组合物中之后,将聚合的聚酰亚胺前体组合物在50℃至100℃的温度下加热并通过化学反应酰亚胺化,或者使溶液回流以除去醇并使其酰亚胺化以获得聚酰亚胺。
在化学酰亚胺化方法中,可以使用吡啶、三乙胺、皮考啉或喹啉作为酰亚胺化催化剂。此外,可以使用经取代或未经取代的含氮杂环化合物、含氮杂环化合物的n-氧化物化合物、经取代或未经取代的氨基酸化合物、具有羟基的芳族烃化合物或芳族杂环化合物,特别是低级烷基咪唑,例如1,2-二甲基咪唑、n-甲基咪唑、n-苄基-2-甲基咪唑、2-甲基咪唑、2-乙基-4-甲基咪唑和5-甲基苯并咪唑;异喹啉;经取代的吡啶,例如3,5-二甲基吡啶、3,4-二甲基吡啶、2,5-二甲基吡啶、2,4-二甲基吡啶和4-正丙基吡啶;和对甲苯磺酸。
作为脱水剂,可以使用酸酐,例如乙酸酐。
或者,可以将聚酰亚胺前体组合物涂覆在基底上并进行热处理以进行酰亚胺化。
聚酰亚胺前体组合物可以为其中聚酰亚胺前体溶解在有机溶剂中的溶液的形式。例如,当在有机溶剂中合成聚酰亚胺前体时,溶液可以为所获得的反应溶液或者可以通过用另一种溶剂稀释该反应溶液而获得。当聚酰亚胺前体作为固体粉末获得时,可以将其溶解在有机溶剂中以制备溶液。
根据本发明的用于由聚酰亚胺前体溶液制备膜的方法包括以下步骤:
将聚酰亚胺前体溶液施加到基底上;以及
对施加的聚酰亚胺前体溶液进行热处理。
作为基底,可以没有任何特别限制地使用玻璃基底、金属基底、塑料基底等。其中,可以优选玻璃基底,其在聚酰亚胺前体的酰亚胺化和固化过程期间具有优异的热稳定性和化学稳定性,并且即使不用另外的脱模剂进行任何处理也可以容易地分离,同时不损坏在固化之后形成的聚酰亚胺膜。
施加步骤可以根据常规施加方法进行。具体地,可以使用旋涂法、棒涂法、辊涂法、气刀法、凹版印刷法、逆辊法、接触辊法、刮刀法、喷涂法、浸渍法、刷涂法等。其中,更优选通过允许连续过程并能够提高聚酰亚胺的酰亚胺化率的流延法进行。
此外,可以将聚酰亚胺前体组合物以使得最终制备的聚酰亚胺膜具有适合于显示器基底的厚度的厚度范围施加在基底上。
具体地,其可以以使得厚度为10μm至30μm的量施加。在施加聚酰亚胺前体组合物之后,还可以在固化过程之前任选地进行用于除去残留在聚酰亚胺前体组合物中的溶剂的干燥过程。
干燥过程可以根据常规方法进行。具体地,干燥过程可以在140℃或更低、或80℃至140℃的温度下进行。如果干燥温度低于80℃,则干燥过程变长。如果干燥温度超过140℃,则酰亚胺化快速进行,使得难以形成具有均匀厚度的聚酰亚胺膜。
然后,将聚酰亚胺前体组合物施加在基底上并在ir烘箱中、在热风烘箱中或在热板上进行热处理。热处理温度的范围可以为300℃至500℃,优选为320℃至480℃。热处理可以在上述温度范围内以多步加热过程进行。热处理过程可以进行20分钟至70分钟,优选20分钟至60分钟。
此后,可以根据常规方法将形成在基底上的聚酰亚胺膜从基底上剥离,得到聚酰亚胺膜。
本发明的聚酰亚胺前体组合物中包含的有机溶剂可以与聚合反应中使用的有机溶剂相同。
在本发明中,可以添加硅烷偶联剂、可交联化合物、用于有效地促进酰亚胺化的酰亚胺化促进剂等,只要不损害效果即可。
此外,基于聚酰亚胺的膜的雾度可以为2或更小,优选为1或更小,或者0.9或更小,从而提供具有改善的透明度的聚酰亚胺膜。此时,聚酰亚胺膜的厚度可以为8μm至15μm,优选为10μm至12μm。
此外,聚酰亚胺膜可以为这样的透明无色的聚酰亚胺膜,其对380nm至760nm波长的光的透射率为80%或更大,并且黄度指数(yi)为约15或更小,优选为约10或更小,更优选为约8或更小。通过具有如上所述的优异的透光率和黄度,可以表现出显著改善的透明度和光学特性。
根据本发明的聚酰亚胺膜的玻璃化转变温度(tg)可以为350℃或更高,优选为360℃或更高,更优选为370℃或更高。
根据本发明的聚酰亚胺膜可以具有优异的根据温度变化的热稳定性。例如,其在100℃至400℃的温度范围内进行n+1次加热和冷却过程之后的热膨胀系数可以为-10ppm/℃至100ppm/℃,优选为-7ppm/℃至90ppm/℃,更优选为80ppm/℃或更小。
此外,根据本发明的式(1a)或(1b)的化合物通过将芴结构引入其结构中而可以在保持聚酰亚胺膜的特性的同时提供膜的减小的延迟值。例如,包含上述化合物作为粘合促进剂的聚酰亚胺膜的面内延迟(rin)可以为约0nm至100nm,厚度方向上的延迟值(rth)可以为约-1000nm至1000nm,或-700nm至700nm,优选为-600nm至600nm,更优选为-500nm至500nm,或-200nm至200nm。在厚度方向上的延迟的所述范围内,可以表现出适合于显示器的可视性。当厚度方向上的延迟为1000nm或者-1000nm或更大时,在聚酰亚胺膜中产生相位差,并且光被扭曲,使得可视性可能显著降低。
根据一个实施方案,包含粘合促进剂的聚酰亚胺膜与载体基底的粘合力可以为至少5gf/in,优选为至少10gf/in。
本发明提供了可用作用于聚酰亚胺树脂的粘合促进剂的新的化合物,以提供新的聚酰亚胺膜,所述新的聚酰亚胺膜在保持现有特性例如高透明度、耐热性、机械特性和低残余应力的同时,即使在高温过程中也具有优异的与载体基底的粘合。
在本发明的另一个实施方案中,提供了包含聚酰亚胺共聚物的模制品。
根据本发明的聚酰亚胺共聚物可以用于电路基底用保护膜、电路基底用基膜、电路基底用绝缘层、半导体用层间绝缘膜、阻焊剂、柔性电路基底或柔性显示器基底。特别地,其适用于需要高温过程的使用低温多晶硅(lowtemperaturepolysilicon,ltps)的oled设备,但不限于此。
发明实施方式
在下文中,将详细地描述本发明的实施方案,使得本领域技术人员可以容易地实施本发明。然而,本发明可以以许多不同的形式实施,并且不应被解释为限于本文阐述的实施方案。
<合成例1>
通过反应方案(1)制备具有式(20)的结构的化合物。
[式20]
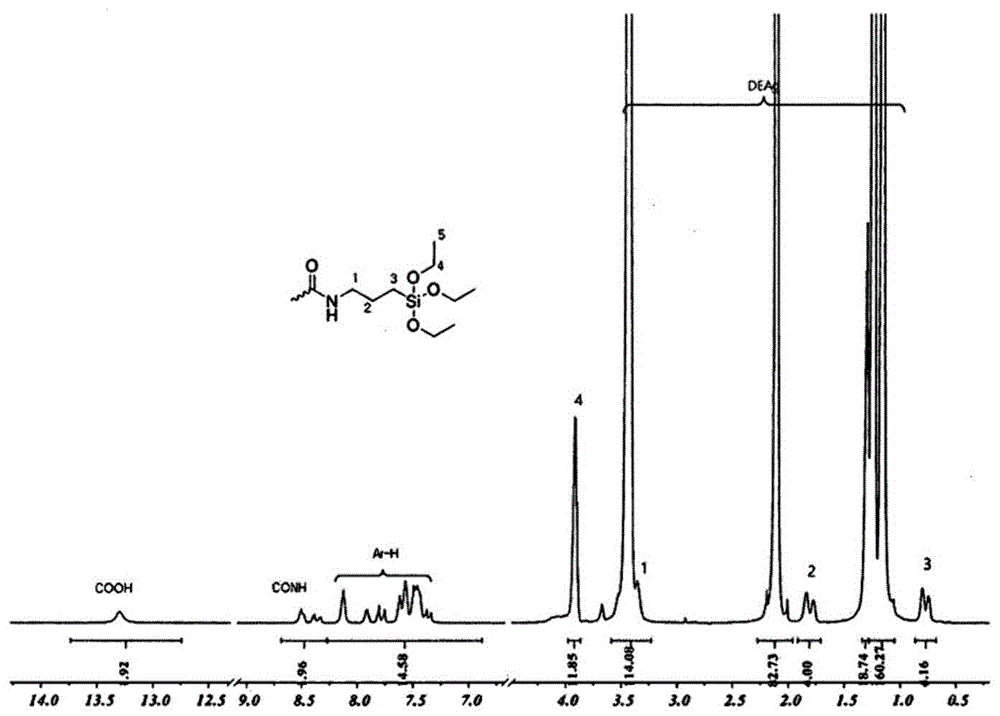
更具体地,在氮气流经的反应器中加入1463gdeac(二乙基乙酰胺),然后在将反应器的温度保持在25℃的同时添加并溶解0.916mol单端胺改性的aptes(3-氨基丙基三乙氧基硅烷)。在相同温度下,向添加有aptes的溶液中添加0.458molbpaf(9,9'-双(3,4-二羧基苯基)芴二酐),并搅拌24小时。合成的化合物的1h-nmr示于图1和图2中。图3示出了式(20)的化合物和aptes的1h-nmr峰的比较。
[反应方案1]
<nmr测量方法>
使用填充有丙酮-d6的插入管,用bruker700mhznmr测量未稀释溶液的nmr谱。
从图1至图3中的1hnmr谱的测量结果(其中由aptes单体与bpaf反应产生的(co)nch2峰出现在3.4ppm附近)可以看出,发现进行了合成反应。
<合成例2>
以与合成例1中相同的方式根据反应方案2进行合成,不同之处在于使用bpda(3,3',4,4'-联苯四羧酸二酐)代替bpaf。
[反应方案2]
<聚合例1>
向氮气流经的反应器中加入124gn,n-二乙基乙酰胺(deac)(分配系数:0.32),然后在将反应器的温度保持在25℃的同时添加并溶解0.0010mol两端胺改性的dms-dps(分子量:5700g/mol,p=73.3,q=26.7)和0.0390moltfmb(2,2'-双(三氟甲基)联苯胺)。在相同温度下,向添加有dms-dps和tfmb的溶液中添加0.032molpmda和0.008molbpaf(9,9'-双(3,4-二羧基苯基)芴二酐),并搅拌3小时,然后在80℃下搅拌4小时。
dms-dps的结构如下:
其中,p和q为摩尔分数,以及设p+q=100,则p为70至90且q为10至30。
<实施例1>
向聚合例1中制备的聚酰亚胺前体溶液中,基于100重量份的聚酰胺酸,添加0.5重量份的合成例1中获得的化合物。
<比较例1>
向聚合例1中制备的聚酰亚胺前体溶液中,基于100重量份的聚酰胺酸,添加0.5重量份的合成例2中获得的化合物。
<实验例>
将实施例1和比较例1中制备的各聚酰亚胺前体溶液旋涂在玻璃基底上。将涂覆有聚酰亚胺前体溶液的玻璃基底放入烘箱中,以5℃/分钟的速率加热,使其在80℃下固化30分钟并在400℃下固化30分钟,以制备聚酰亚胺膜。
测量聚酰亚胺膜的yi、rth和tg,结果示于下表1中。
<黄度指数(yi)>
用coloreye7000a测量黄度指数(yi)。
<厚度方向上的延迟>
用axoscan测量厚度方向上的延迟(rth)。将膜切割成一定尺寸并测量厚度。然后,用axoscan测量延迟值。为了补偿延迟值,将在c板方向上校正时测量的厚度输入到axoscan。
<玻璃化转变温度(tg)>
将膜切割成5mm×20mm以制备样品,然后使用附件装载样品。实际测量的膜的长度等于16mm。牵拉力设定为0.02n。从100℃至400℃以5℃/分钟的加热速率进行第一升温步骤,然后从400℃至100℃以4℃/分钟的冷却速率进行冷却,并且从100℃至450℃以5℃/分钟的加热速率进行第二升温步骤。用tma(q400,ta公司)测量热膨胀的变化。
此时,将在第二升温步骤期间的升温区间中示出的拐点定义为tg。
<剥离强度>
如上所述制备的聚酰亚胺膜的剥离强度(粘合力)通过使用剥离强度分析仪(ta-xtplus,织构分析仪)以10mm/秒90°地剥离膜宽度为2.54cm且测量长度为10mm的样品来测量。
[表1]
从表1的结果可以看出,发现包含根据本发明的粘合促进剂的实施例1的聚酰亚胺膜在保持高剥离强度的同时将rth保持在低水平,但是在比较例1的情况下,rth增加。
因此,可以看出,根据本发明的用于聚酰亚胺树脂的粘合促进剂可以提供具有改善的粘合力和高耐热性的聚酰亚胺。
虽然已经参照本发明的具体实施方案具体示出和描述了本发明,但对于本领域技术人员来说显而易见的是,该具体描述仅仅是优选实施方案,并且本发明的范围不限于此。因此,本发明的范围旨在由所附权利要求及其等同方案限定。
- 还没有人留言评论。精彩留言会获得点赞!