二芳基丙烷二聚体类衍生物及其药物组合物和其应用的制作方法
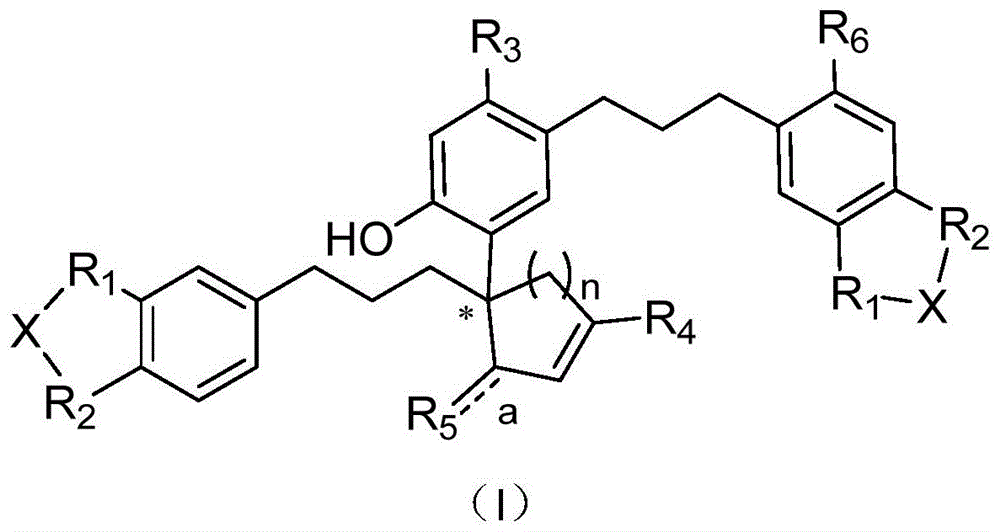
本发明属于药物技术领域,具体地,涉及二芳基丙烷二聚体类衍生物,以其为活性成分的药物组合物,其制备方法,以及它们在制备治疗或预防帕金森综合症及多巴胺相关疾病药物中的应用。
背景技术:
帕金森综合症(parkinson’sdisease,pd)是严重威胁老年人生活质量与身体健康的神经退行性疾病,尤其60岁以上老年人患病几率最大,主要影响患者的行动能力,部分患者也会表现出痴呆症状。我国人口老龄化严重,60岁以上人口有2.5亿,受到pd的极大威胁,其防治工作已经成为我国亟需解决的重大问题。而目前市售的pd药物中,没有一种可以治愈pd,虽然小部分药物可以达到缓解症状与延缓恶化的作用,但是这远不能减轻未来我国治疗pd患者的压力,因此开发更多高效的pd候选药物意义重大。
pd确切的发病机制尚不明确,众多研究表明pd患者存在大脑基底核细胞死亡现象,其直接影响了近70%基底核中黑质致密部的多巴胺分泌神经元。现有最高效的治疗药物为l-dopa与多巴脱羧酶抑制剂联合用药,后者能有效避免l-dopa在外周被转化为多巴胺而无法通过血脑屏障,增加了进入大脑中的l-dopa,达到治疗pd的作用。目前报道的多巴脱羧酶抑制剂活性不高(甲基多巴为临床用药,ic50=1200μm),亟需开发高效的新型抑制剂。
风吹楠属植物在东南亚被广泛用作治疗各种紊乱性疾病,本发明首次从该属植物大叶风吹楠中分离获得具有多巴脱羧酶抑制活性的新型天然二芳基丙烷二聚体类先导化合物1-4,并通过有机化学手段合成其衍生物,制备它们的药物组合物,进行多巴脱羧酶抑制活性评价。目前为止,尚未有关新型天然二芳基丙烷二聚体类先导化合物1-4及其衍生物、其药学上可接受的盐的报道,也没有其制备方法、含有这类化合物及衍生物的药物组合物及植物提取物,及它们在治疗和预防帕金森综合症及多巴胺相关疾病药物中应用的报道。
技术实现要素:
本发明的目的在于:提供一类新型天然二芳基丙烷二聚体类衍生物,以其为活性成分的药物组合物,其制备方法,以及二芳基丙烷二聚体类及其药物组合物在制备预防或治疗帕金森综合症及多巴胺相关疾病的药物中的应用。
本发明的上述目的是通过如下的技术方案得以实现的:
式(i)所示的二芳基丙烷二聚体类衍生物或其药用盐,
式中小写字母a代表独立的双键或单键,用
其中x选自饱和或不饱和碳链(数量为0-2),r1和r2相同或不相同分别选自o和n;x不存在时,r1和r2相同或不相同分别选自h、oh、sh、sme、some、so2me、ome、卤素(f、cl、br、i)、nhc=or(r=me、et、npr、ipr、tbu、o-tbu、o-ally、obn)、nhr(r=me、et、npr、ipr、tbu、ally、bn)、nrr′(r和r′相同或不同选自me、et、npr、ipr)、五或六元环状二级胺(环上可以取代o、n等杂原子);
r3选自h、oh、ome;
r4选自h、卤素、or(r=me、et)、cho、co2h、co2r(r=me、et、npr、ipr、tbu、ally、bn、nhr′、nr′r′,其中r′选自me、et、npr、ipr);
a为单键时,r5选自h、oh、ome,a为双键时,r5选自o、s、nor(r=me、et、npr、ipr、tbu、tbu、ally、bn);
r6选自h、卤素、oh、ome。
如所示式(i)所示的二芳基丙烷二聚体类衍生物,为如下化合物或其药用盐,
所述的二芳基丙烷二聚体类衍生物或其药用盐,其中所述的药用盐是指与酸或碱形成的药学上可接受的盐,所述的酸为酒石酸、柠檬酸、甲酸、乙酸、乙二酸、丙酸、丁酸、己酸、草酸、马来酸、己二酸、天冬氨酸、苯磺酸、樟脑酸、樟脑磺酸、十二烷基硫酸、延胡索酸、乳酸、盐酸、氢溴酸、氢碘酸、硫酸、磷酸等,所述的碱为氨水、碳酸氢钠、碳酸钠、碳酸氢钾、碳酸钾等。
制备权利要求式(i)所示的二芳基丙烷二聚体类衍生物的方法,方法i:取风吹楠属植物大叶风吹楠枝叶(10kg),经干燥、粉碎后,用70%丙酮/水(20l)提取4次(每次2天)。提取物浓缩后剩余水相(6l)加乙酸乙酯(6l)萃取,收集乙酸乙酯层,并用乙酸乙酯(18l)萃取水相3次(每次6l),合并有机层,浓缩得到乙酸乙酯提取物376g。将该提取物经硅胶柱(100-200目)以氯仿:丙酮=1:0、8:2、5:5、2:8、0:1梯度洗脱,每个梯度洗脱溶剂5l,每个馏分接取250ml,得到约20个馏分,根据每个馏分tlc(氯仿:丙酮=1:1和氯仿:甲醇=8:1)分析的主斑点情况进行合并,最终得到5段(fr.a-e)。fr.b(28g)经凝胶(sephadexlh-20)以氯仿:甲醇=3:2(500ml)洗脱,每个馏分接取25ml,得到约20个馏分,用tlc检测(氯仿:甲醇=15:1)合并获得4段(fr.b1-b4)。fr.b2(320mg)经半制备高效液相色谱分离[甲醇:水=48:52或者乙腈:水=35:65,体积比,流速3ml/min,色谱柱:zorbaxxdb-c18(9.4mm×25cm)]得到化合物1(3.7mg)和3(3.3mg)外消旋体,经手性拆分[甲醇:水=95:5,体积比,流速:3ml/min,色谱柱:ultimatecellu-dr(10mm×25cm)column]分别得到光学纯化合物1(1.1mg)、2(1.3mg)、3(1.0mg)、4(1.2mg)。
方法ii:应用化学合成制备权利1所示化合物,其特征在于,将uniti和unitii偶联(coupling)合成或将unitii与化合物i偶联生成化合物ii后再与化合物iii在碱性条件(base)下偶联合成。其中偶联(coupling)的条件为金属pd催化剂催化、光催化、电化学促进等,碱性条件(base)为双(三甲基硅基)氨基锂(lihmds)、双(三甲基硅基)氨基钾(khmds)、双(三甲基硅基)氨基钠(nahmds)、二异丙基氨基锂(lda)、叔丁基锂、仲丁基锂、氢化钠、叔丁醇钾等。
药物组合物,其中含有治疗有效量的权利要求1或2或所示的二芳基丙烷二聚体类衍生物或其药用盐和药学上可接受的载体。
所述药物组合物,特征在于药物组合物为片剂、胶囊、丸剂、注射剂、缓控释制剂、喷雾剂、微粒(纳米)给药体系。
式(i)所示的二芳基丙烷二聚体类衍生物或其药用盐在制备治疗或预防帕金森综合症的药物中的应用。
式(i)所示的二芳基丙烷二聚体类衍生物或其药用盐在制备治疗或预防多巴胺相关疾病的药物中的应用。
式(i)所示的二芳基丙烷二聚体类衍生物或其药用盐在制备多巴脱羧酶抑制剂的药物中的应用。
本发明同时还提供了上面所示的二芳基丙烷二聚体类衍生物或其药用盐在制备治疗或预防帕金森综合症及多巴胺相关疾病的药物中的应用。
如所述的应用,其中所述的多巴胺相关疾病为早老年痴呆、高血压及心血管并发症、精神性疾病、认知障碍、神经内分泌失调等。
本发明的二芳基丙烷二聚体类衍生物或其药物组合物可经口服、鼻吸入、直肠或肠胃外给药,给药量因药物不同而各有不同,对成人来说,每天1-1000mg比较合适。
二芳基丙烷二聚体类衍生物可从风吹楠属同属植物中提取分离得到,而片剂、胶囊、口服液、针剂、注射用冻干粉或粉针剂等药物剂型的制备也是本领域的常规知识,因此二芳基丙烷二聚体类衍生物与相应载体制备的药物剂型也能够由本领域的技术人员实现。
本发明的典型化合物包括(但不限于)下表1中所列化合物。
表1典型化合物
出于示例的目的(而非限制)提供下列实施例。
附图说明:
图1为本发明二芳基丙烷二聚体类衍生物结构示意图。
具体实施方式:
下面结合附图,用本发明的实施例来进一步说明本发明的实质性内容,但并不以此来限定本发明。
下面的试验实施例和制剂实施例可更详细地说明本发明,但不以任何形式限制本发明。根据本发明实质对本发明的改进或补充均属于本发明范围。本公开的某些化合物可以根据下列方法和本领域技术人员的知识制备。所有从植物中分离的化合物至少需要核磁(1hnmr和13cnmr)、旋光、高分辨质谱hrms表征,所有合成的化合物至少需要核磁(1hnmr和13cnmr)高分辨质谱hrms表征。
实施例1
二芳基丙烷二聚体类衍生物1-4的提取分离与鉴定:
取风吹楠属植物大叶风吹楠枝叶(10kg),经干燥、粉碎后,用70%丙酮/水(20l)提取4次(每次2天)。提取物浓缩后剩余水相(6l)加乙酸乙酯(6l)萃取,收集乙酸乙酯层,并用乙酸乙酯(18l)萃取水相3次(每次6l),合并有机层,浓缩得到乙酸乙酯提取物376g。将该提取物经硅胶柱(100-200目)以氯仿:丙酮=1:0、8:2、5:5、2:8、0:1梯度洗脱,每个梯度洗脱溶剂5l,每个馏分接取250ml,得到约20个馏分,根据每个馏分tlc(氯仿:丙酮=1:1和氯仿:甲醇=8:1)分析的主斑点情况进行合并,最终得到5段(fr.a-e)。fr.b(28g)经凝胶(sephadexlh-20)以氯仿:甲醇=3:2(500ml)洗脱,每个馏分接取25ml,得到约20个馏分,用tlc检测(氯仿:甲醇=15:1)合并获得4段(fr.b1-b4)。fr.b2(320mg)经半制备高效液相色谱分离[甲醇:水=48:52或者乙腈:水=35:65,体积比,流速3ml/min,色谱柱:zorbaxxdb-c18(9.4mm×25cm)]得到化合物1(3.7mg)和3(3.3mg)外消旋体,经手性拆分[甲醇:水=95:5,体积比,流速:3ml/min,色谱柱:ultimatecellu-dr(10mm×25cm)column]分别得到光学纯化合物1(1.1mg)、2(1.3mg)、3(1.0mg)、4(1.2mg)。
化合物1:棕色油状物;uv(meoh)λmax(logε)204(4.05),226(3.60),288(3.30)nm;
化合物2:棕色油状物;uv(meoh)λmax(logε)204(4.05),226(3.60),288(3.30)nm;
化合物3:棕色油状物;uv(meoh)λmax(logε)204(3.78),226(3.33),292(3.05)nm;
化合物4:棕色油状物;uv(meoh)λmax(logε)204(3.78),226(3.33),292(3.05)nm;
表2化合物1-4的1hnmr数据(δinppm,jinhz;800mhz,methanol-d4)
表3化合物1-4的13cnmr数据(δinppm,jinhz;200mhz,methanol-d4)
实施例2
二芳基丙烷二聚体类衍生物1和2的化学合成:
向干燥的250ml两口瓶中加入化合物unitii(4.30g,8.09mmol)、pd(oac)2(0.36g,1.62mmol)、(±)-binap(1.21g,1.94mmol),氩气抽换气3次,加入1,4-二氧六环(100ml)溶解并搅拌30min。加入nahmds的四氢呋喃溶液(2.0m,10.11ml,20.22mmol)和化合物uniti(2.20g,7.28mmol)的1,4-二氧六环溶液(50ml),升温至90℃反应至tlc检测到uniti消耗完全,约20h。加入水(10ml)猝灭反应,减压蒸除溶剂,剩余加水(50ml)和乙酸乙酯(80ml)分层,用乙酸乙酯萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(石油醚:乙酸乙酯=3:1洗脱,300ml)得到粗产物为黄色油状物(2.20g)。将该粗产物溶于乙酸乙酯:甲醇=5:1(200ml),加入pd/c(200mg),连接氢气球,置换氢气3次,室温反应至tlc检测到粗产物消耗完全,约16h。反应液经硅藻土过滤,减压浓缩得到棕色油状物,经硅胶柱分离纯化(氯仿:甲醇=20:1)后得到目标产物1和2为外消旋体混合物(1.54g,32%收率),再经手性制备型hplc[甲醇:水=95:5,体积比,流速:3ml/min,色谱柱:ultimatecellu-dr(10mm×25cm)column]分别得到光学纯化合物1(0.78g)和2(0.74g)。1hnmr(500mhz,cd3od)δ6.73(s,1h),6.66(dd,j=7.8,4.7hz,2h),6.62(s,1h),6.61(dd,j=7.9,1.5hz,1h),6.59(s,1h),6.25(s,1h),5.88(s,2h),5.87(s,2h),5.32(s,1h),3.75(s,3h),3.74(s,3h),2.66(dt,j=13.8,6.1hz,1h),2.57–2.48(m,4h),2.48–2.42(m,1h),2.21(d,j=12.8hz,1h),2.14(td,j=13.9,3.9hz,1h),2.07(d,j=12.8hz,1h),2.00–1.94(m,1h),1.71(d,j=7.0hz,2h),1.51–1.45(m,1h),1.42(tdd,j=12.4,8.3,4.3hz,1h);13cnmr(125mhz,cd3od)δ201.3,171.7,157.1,153.7,153.4,149.0,147.4,147.0,137.3,128.1,124.5,124.2,122.3,113.4,110.6,109.8,109.0,104.1,102.0,102.0,101.9,95.8,94.8,57.1,56.9,48.7,39.1,36.88,31.9,31.8,30.8,30.4,26.8;hresimsm/z571.1961[m-h]-(calcdforc33h32o9,571.1974)。
实施例3
二芳基丙烷二聚体类衍生物3和4的化学合成:
向干燥的250ml两口瓶中加入化合物unitii(2.50g,4.46mmol)、pd(oac)2(0.20g,0.89mmol)、(±)-binap(0.71g,1.07mmol),氩气抽换气3次,加入1,4-二氧六环(100ml)溶解并搅拌30min。加入nahmds的四氢呋喃溶液(2.0m,5.58ml,11.15mmol)和化合物uniti(1.21g,4.01mmol)的1,4-二氧六环溶液(50ml),升温至90℃反应至tlc检测到uniti消耗完全,约20h。加入水(10ml)猝灭反应,减压蒸除溶剂,剩余加水(50ml)和乙酸乙酯(80ml)分层,用乙酸乙酯萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(石油醚:乙酸乙酯=5:1洗脱,300ml)得到粗产物为黄色油状物(1.15g)。将该粗产物溶于乙酸乙酯:甲醇=5:1(100ml),加入pd/c(100mg),连接氢气球,置换氢气3次,室温反应至tlc检测到粗产物消耗完全,约12h。反应液经硅藻土过滤,减压浓缩得到棕色油状物,经硅胶柱分离纯化(氯仿:甲醇=40:1)后得到目标产物3和4为外消旋体混合物(1.07g,40%收率),再经手性制备型hplc[甲醇:水=95:5,体积比,流速:3ml/min,色谱柱:ultimatecellu-dr(10mm×25cm)column]分别得到光学纯化合物3(0.50g)和4(0.52g)。1hnmr(500mhz,cd3od)δ6.69(d,j=7.9hz,1h),6.68(s,1h),6.66(d,j=1.4hz,1h),6.66(d,j=1.4hz,1h),6.66(dd,j=4.6,3.2hz,2h),6.66(dd,j=4.6,3.2hz,2h),6.65(d,j=1.4hz,1h),6.61(d,j=1.5hz,1h),6.61(d,j=1.6hz,2h),6.61(d,j=1.6hz,1h),6.60(d,j=1.5hz,1h),6.60(d,j=1.5hz,1h),6.23(s,1h),5.89(s,2h),5.86(dd,j=4.9,1.2hz,2h),5.30(s,1h),3.73(s,3h),2.68–2.63(m,1h),2.55–2.50(m,1h),2.48(d,j=7.4hz,2h),2.46(td,j=7.5,3.3hz,3h),2.20(d,j=12.8hz,1h),2.14–2.08(m,1h),2.05(d,j=12.8hz,1h),2.00–1.94(m,1h),1.75(dd,j=13.6,6.8hz,2h),1.47(qd,j=12.8,5.9hz,1h),1.39(ddd,j=12.5,8.7,4.4hz,1h);13cnmr(125mhz,cd3od)δ201.4,171.8,157.2,153.5,149.1,149.1,147.2,147.02,137.9,137.4,128.3,124.0,122.4,122.4,113.5,109.9,109.8,109.1,109.0,104.3,102.1,102.0,102.0,94.9,57.2,48.8,39.1,37.0,36.3,33.2,32.0,30.3,26.9;hresimsm/z601.2069[m-h]-(calcdforc34h34o10,601.2079).
实施例4
二芳基丙烷二聚体类衍生物5的化学合成:
向干燥的25ml单口瓶中加入化合物1(0.10g,0.17mmol)和二氯甲烷(10ml)在-78℃冷阱中搅拌10min。加入selectfluor(0.06g,0.18mmol),继续搅拌反应至tlc检测到化合物1消耗完全,约2h。加入水(1ml)猝灭反应,加水(10ml)分层,用二氯甲烷萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(石油醚:乙酸乙酯=3:1洗脱,300ml)得到目标产物为黄色油状物(23mg,21%收率)。1hnmr(500mhz,cd3od):δ7.19(1h,s),6.97(1h,s),6.93(1h,s),6.69(1h,dd,j=8.4,1.5hz),6.64(1h,dd,j=2.8,1.5hz),6.55(1h,dd,j=8.4,2.8hz),6.34(1h,s),6.22(1h,s),5.95(2h,s),5.93(2h,s),3.79(3h,s),2.99(1h,d,j=12.5hz),2.75(1h,d,j=12.5hz),2.71(2h,t,j=7.4hz),2.55(2h,t,j=7.7hz),2.40(2h,t,j=7.3hz),1.85(2h,t,j=7.0hz),1.71(2h,m),1.59(2h,m);13cnmr(125mhz,cd3od)δ209.8,172.3,163.2,157.0,154.7,147.9,147.6,147.5,147.2,136.6,135.2,129.2,128.3,122.4,121.3,121.1,120.3,111.0,108.4,108.4,103.0,101.5,101.5,100.6,51.9,42.5,42.4,35.4,35.1,32.6,30.0,29.9,26.0;hresimsm/z589.1959[m-h]-(calcdforc33h30fo9,589.1952)。
实施例5
二芳基丙烷二聚体类衍生物6的化学合成:
向干燥的25ml单口瓶中加入化合物1(0.10g,0.17mmol)和二氯甲烷(10ml)在-40℃冷阱中搅拌10min。加入ncs(0.06g,0.18mmol),继续搅拌反应至tlc检测到化合物1消耗完全,约0.5h。加入水(1ml)猝灭反应,加水(10ml)分层,用二氯甲烷萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(石油醚:乙酸乙酯=3:1洗脱,300ml)得到目标产物为黄色油状物(34mg,33%收率)。1hnmr(500mhz,cd3od):δ7.17(1h,s),6.95(1h,s),6.93(1h,s),6.72(1h,dd,j=8.4,1.5hz),6.60(1h,dd,j=2.8,1.5hz),6.58(1h,dd,j=8.4,2.8hz),6.30(1h,s),6.28(1h,s),5.93(2h,s),5.91(2h,s),3.78(3h,s),2.92(1h,d,j=12.5hz),2.71(1h,d,j=12.5hz),2.67(2h,t,j=7.4hz),2.52(2h,t,j=7.7hz),2.44(2h,t,j=7.3hz),1.88(2h,t,j=7.0hz),1.76(2h,m),1.54(2h,m);13cnmr(125mhz,cd3od)δ206.3,172.1,163.8,156.1,155.5,148.3,147.1,147.4,147.3,136.5,135.8,129.3,127.9,122.8,121.5,121.4,121.0,111.2,108.8,108.8,102.8,101.1,101.1,100.5,50.4,42.2,42.3,35.1,35.1,32.2,30.6,29.4,26.8;hresimsm/z605.1571[m-h]-(calcdforc33h30clo9,605.1578)。
实施例6
二芳基丙烷二聚体类衍生物7的化学合成:
向干燥的25ml单口瓶中加入化合物1(0.10g,0.17mmol)和四氢呋喃:水=9:1,(10ml),加入naoh(6.8mg,0.17mmol),在40℃下继续搅拌反应至tlc检测到化合物1消耗完全,约16h。减压蒸除溶剂,加水(10ml)和氯仿(10ml)分层,用氯仿萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(氯仿:甲醇:乙酸=40:1:0.05洗脱,200ml)得到目标产物为棕色油状物(61mg,65%收率)。1hnmr(500mhz,cd3od):δ7.18(1h,s),6.93(1h,s),6.70(1h,s),6.69(1h,s),6.56(1h,d,j=8.4hz),6.54(1h,d,j=8.4hz),6.34(1h,s),6.28(1h,s),5.93(4h,s),2.98(2h,dd,j=12.5hz),2.54(2h,t,j=7.6hz),2.41(4h,t,j=7.4hz),1.81(2h,t,j=7.0hz),1.73(2h,m),1.58(1h,m);13cnmr(125mhz,cd3od)δ209.4,175.2,163.2,154.7,147.9,147.9,147.5,147.5,136.6,136.6,135.2,129.2,128.3,122.4,121.1,121.1,120.3,108.4,108.4,108.4,108.4,101.5,100.6,42.5,42.4,35.4,35.1,35.1,33.6,29.9,26.0,26.8;hresimsm/z557.1808[m-h]-(calcdforc32h29o9,557.1812)。
实施例7
二芳基丙烷二聚体类衍生物8的化学合成:
向干燥的250ml两口瓶中加入化合物unitii(1.80g,3.40mmol)、pd(oac)2(0.15g,0.68mmol)、(±)-binap(0.51g,0.81mmol),氩气抽换气3次,加入1,4-二氧六环(70ml)溶解并搅拌30min。加入nahmds的四氢呋喃溶液(2.0m,10.11ml,20.22mmol)和化合物uniti(0.87g,3.06mmol)的1,4-二氧六环溶液(50ml),升温至90℃反应至tlc检测到uniti消耗完全,约20h。加入水(10ml)猝灭反应,减压蒸除溶剂,剩余加水(50ml)和乙酸乙酯(80ml)分层,用乙酸乙酯萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(石油醚:乙酸乙酯=3:1洗脱,300ml)得到粗产物为黄色油状物(1.15g)。将该粗产物溶于乙酸乙酯:甲醇=5:1(100ml),加入pd/c(100mg),连接氢气球,置换氢气3次,室温反应至tlc检测到粗产物消耗完全,约12h。反应液经硅藻土过滤,减压浓缩得到橙色油状物,经硅胶柱分离纯化(氯仿:甲醇=30:1)后得到目标产物8为外消旋体混合物(0.86g,48%收率),再经手性制备型hplc[甲醇:水=95:5,体积比,流速:3ml/min,色谱柱:ultimatecellu-dr(10mm×25cm)column]分别得到光学纯化合物8(0.42g,23%收率)。1hnmr(500mhz,cd3od)δ6.93(s,1h),6.70(s,1h),6.68(s,1h),6.64(d,j=2.8hz,1h),6.63(s,1h),6.56(d,j=8.4hz,1h),6.55(d,j=8.4hz,1h),6.34(s,1h),6.04(s,1h),5.93(s,2h),5.92(s,2h),3.99(s,3h),2.54(t,j=13.7,7.6,hz,2h),2.48–2.39(m,6h),2.26(m,1h),2.08-1.99(m,2h),1.83(t,j=7.0hz,2h),1.77–1.70(m,4h);13cnmr(125mhz,cd3od)δ201.1,163.2,158.8,154.7,147.9,147.9,147.5,147.5,136.6,136.6,135.2,122.4,121.1,121.1,120.3,108.4,108.4,108.4,108.4,106.0,101.6,101.5,100.8,54.5,53.1,35.4,35.3,35.1,35.1,33.6,29.9,27.3,26.0;hresimsm/z545.2185[m-h]-(calcdforc33h33o8,545.2175)。
实施例8
二芳基丙烷二聚体类衍生物9的化学合成:
向干燥的25ml单口瓶中加入化合物7(80.0mg,0.14mmol)、cocl2(2.0mg,0.014mmol)、cs2co3(45.0mg,0.14mmol),氩气抽换气3次,加入1,2-二甲氧基乙烷(8ml)和水(0.1ml)溶解,将反应用34w的led蓝光灯照射。室温反应至tlc检测到7消耗完全,约30h。减压蒸除溶剂,剩余物经快速柱色谱(石油醚:乙酸乙酯=6:1洗脱,100ml)得到粗产物为黄色油状物(60mg)。将该粗产物溶于乙酸乙酯(10ml),加入pd/c(10mg),连接氢气球,置换氢气3次,室温反应至tlc检测到粗产物消耗完全,约12h。反应液经硅藻土过滤,减压浓缩得到黄色油状物(50mg)。将该粗产物溶于甲醇(10ml),加入o-甲基盐酸羟胺(12mg,0.14mmol),室温反应至tlc检测到粗产物消耗完全,约24h。经硅胶柱分离纯化(氯仿:甲醇=40:1)后得到目标产物9(20mg,26%收率)。1hnmr(500mhz,cd3od)δ6.96(s,1h),6.76(s,1h),6.68(s,1h),6.66(d,j=2.8hz,1h),6.62(brs,1h),6.53(d,j=8.4hz,1h),6.51(d,j=8.4hz,1h),6.36(s,1h),6.04(s,1h),5.93(s,2h),5.92(s,2h),3.80(s,3h),2.70–2.61(m,2h),2.54(t,j=13.5,5.4,hz,2h),2.40(m,4h),2.00-1.94(m,2h),1.90(t,j=13.5,5.4,hz,2h),1.85-1.70(m,4h),1.54(m,2h);13cnmr(125mhz,cd3od)δ164.5,154.7,153.8,147.7,147.7,147.5,147.5,135.8,135.6,135.2,124.4,122.2,122.3,121.6,109.2,109.0,107.2,107.4,107.0,103.1,101.2,100.1,61.9,42.5,35.4,35.3,35.1,33.9,33.6,30.4,28.1,26.2;hresimsm/z546.2482[m+h]+(calcdforc32h36no7,546.2492)。
实施例9
二芳基丙烷二聚体类衍生物10的化学合成:
向干燥的25ml单口瓶中加入化合物1(0.10g,0.17mmol)和tbscl(63.0mg,0.43mmol),加入二氯甲烷(10ml)溶解,接着加入咪唑(35.3mg,0.43mmol),在室温下继续搅拌反应至tlc检测到化合物1消耗完全,约0.5h。减压蒸除溶剂,加水(10ml)和氯仿(10ml)分层,用氯仿萃取3次,合并有机相,干燥(na2so4)。减压浓缩有机相,剩余物经快速柱色谱(石油醚:乙酸乙酯=10:1洗脱,100ml)得到无色油状物(140mg)。将该无色油状物溶解在二氯甲烷(10ml)中,加入n,n-二甲基甲酰胺(15μl,0.20mmol)和三氯氧磷(25μl,0.20mmol),在室温下继续搅拌反应至tlc检测到该无色油状物消耗完全,约2h。加水加热至回流反应0.5h,加入水(10ml)分层,水层用二氯甲烷(10ml)萃取3次,合并有机相,干燥(na2so4),减压蒸除溶剂,得到淡黄色油状物(100mg)。然后将该油状物溶解在二氯甲烷(10ml)中,加入碳酸氢钠(16.9mg,0.20mmol)和间氯过氧苯甲酸(75%,46mg,0.20mmol),封管加热至60℃反应8h。冷却至0℃,加入盐酸水溶液(1n,1ml)和水(5ml)分层,水层用氯仿:甲醇=5:1(20ml)萃取多次,合并有机相,干燥(na2so4),减压蒸除溶剂,得到棕色油状物,经快速硅胶柱色谱分离(氯仿:甲醇=20:1,200ml)得到目标产物为棕色油状物(23mg,23%收率)。1hnmr(500mhz,cd3od)δ6.72(d,j=7.9hz,1h),6.70(s,1h),6.66(d,j=1.4hz,1h),6.64(d,j=1.4hz,1h),6.62(dd,j=4.6,3.2hz,2h),6.60(dd,j=4.6,3.2hz,2h),6.59(d,j=1.4hz,1h),6.54(d,j=1.5hz,1h),6.52(d,j=1.6hz,2h),6.50(d,j=1.6hz,1h),6.44(d,j=1.5hz,1h),6.42(d,j=1.5hz,1h),6.24(s,1h),5.90(s,2h),5.88(dd,j=4.9,1.2hz,2h),5.28(s,1h),2.65–2.61(m,1h),2.58–2.54(m,1h),2.48(d,j=7.4hz,2h),2.45(td,j=7.5,3.3hz,3h),2.22(d,j=12.8hz,1h),2.16–2.10(m,1h),2.07(d,j=12.8hz,1h),2.02–1.93(m,1h),1.76(dd,j=13.6,6.8hz,2h),1.49(qd,j=12.8,5.9hz,1h),1.31(ddd,j=12.5,8.7,4.4hz,1h);13cnmr(125mhz,cd3od)δ202.1,170.5,155.2,154.5,148.3,148.6,147.2,147.0,138.1,136.6,126.1,125.1,123.2,122.1,114.4,108.9,108.8,108.5,108.0,106.2,104.5,101.9,102.0,97.4,49.2,38.5,37.6,36.1,33.5,32.6,30.8,27.2;hresimsm/z587.1922[m-h]-(calcdforc33h31o10,587.1917).
实施例10
二芳基丙烷二聚体类衍生物11和12的化学合成:
向干燥的25ml单口瓶中加入化合物1(0.10g,0.17mmol),加入二氯甲烷(10ml)溶解并置于-78℃冷阱搅拌10min,加入三溴化硼(35μl,0.17mmol),在-78℃下继续搅拌反应至tlc检测到化合物1消耗完全,约0.5h。加水(10ml)和氯仿(10ml)分层,用氯仿:甲醇=5:1(30ml)萃取多次,合并有机相,干燥(na2so4)。减压蒸除溶剂,得到棕色油状物,经快速硅胶柱色谱分离(氯仿:甲醇=20:1–5:1,200ml)得到目标产物12为棕色油状物(57mg,60%收率),得到化合物11为棕色油状物(18mg,20%收率)。化合物11:1hnmr(500mhz,cd3od)δ6.65(s,1h),6.64(s,1h),6.62(s,1h),6.61(s,1h),6.57(s,1h),6.28(s,1h),5.38(s,1h),3.75(s,3h),2.64(dt,j=13.4,5.8hz,1h),2.62–2.56(m,4h),2.51–2.46(m,1h),2.25(d,j=12.5hz,1h),2.18(td,j=13.4,3.6hz,1h),2.12(d,j=12.5hz,1h),2.10–1.88(m,1h),1.72(d,j=7.0hz,2h),1.58–1.47(m,1h),1.45(tdd,j=12.3,8.5,4.1hz,1h);13cnmr(125mhz,cd3od)δ202.6,172.1,157.1,155.2,155.1,152.2,148.2,148.0,139.2,130.4,123.2,123.1,121.7,116.2,113.2,110.2,109.1,106.7,105.2,103.3,97.6,95.1,56.8,56.2,49.0,38.2,34.8,32.5,32.0,31.1,30.9,27.0;hresimsm/z547.1961[m-h]-(calcdforc31h31o9,547.1968)。化合物12:1hnmr(500mhz,cd3od)δ6.68(s,1h),6.66(s,1h),6.65(s,1h),6.64(s,1h),6.58(s,1h),6.34(s,1h),5.89(s,2h),5.35(s,1h),3.76(s,3h),2.65(dt,j=13.4,5.8hz,1h),2.61–2.55(m,4h),2.55–2.49(m,1h),2.28(d,j=12.7hz,1h),2.14(td,j=13.4,3.6hz,1h),2.11(d,j=12.7hz,1h),2.12–1.90(m,1h),1.76(d,j=7.0hz,2h),1.56–1.46(m,1h),1.44(tdd,j=12.6,8.2,4.6hz,1h);13cnmr(125mhz,cd3od)δ201.8,171.4,157.5,155.8,151.4,151.0,149.1,148.7,138.6,131.9,125.1,124.5,122.7,118.2,115.1,113.2,108.4,107.6,106.4,101.9,98.4,96.3,57.6,56.6,48.4,38.4,35.6,32.3,32.1,31.6,31.0,26.5;hresimsm/z559.1959[m-h]-(calcdforc32h31o9,559.1968)。
实施例11:
本发明的化合物二芳基丙烷二聚体类衍生物具有明显的多巴脱羧酶抑制活性,实验方法和结果如下:
一、实验方法:
1.将待测试二芳基丙烷二聚体类衍生物样品分别用dmso溶解配制为浓度10mm的溶液,储存备用。
2.配制缓冲液:取dw适量,加热煮沸30min,通入n2自然冷却至室温。称取0.203克mgcl2·6h2o,加入1ml的dw溶解。称取0.39克的trisbase、0.19克nacl以及0.002克bsa溶解于45ml的dw中,向该溶液依次加入mgcl2水溶液(1m,250ml)、浓盐酸(160ml)、β-巯基乙醇(25ml)定容至500.00ml。
3.配制酶反应体系:
1)酶反应混合液:0.1mnadh(3.8ml)+6.59μmpepc25.0ml+0.1mplp(0.5ml)+ddc(4.6ml)+dw(466.1ml)
2)底物反应混合液:0.5mpppa(10.0ml)+mdase(2.0ml)+l-dopa(1m,0.75ml)+dw(487.0ml)
3)空白对照反应液:0.5mpppa(5.0ml)+mdase(1.0ml)+dw(244.0ml)
4.各组分分别加样到384孔板:dmso/待测样品(1.0ml),酶反应混合液(24.5ml),底物反应液/空白对照(24.5ml),共计50.0ml。采用封板膜封板,立刻至biotek酶标仪(340nm)读取数值a;37℃放置1h后,再读取数值b。
5.数据处理
酶活性=数据a-数据b;数据越高表示酶活性越高。
二、实验结果:(见表4)。本发明对一系列二芳基丙烷二聚体类衍生物进行了体外多巴脱羧酶抑制活性评价,结果显示该类化合物对多巴脱羧酶具有较明显地抑制作用,多数衍生物的活性远远强于阳性对照甲基多巴,是一类治疗和预防帕金森综合症及多巴胺相关疾病的潜力化合物。
表4.二芳基丙烷二聚体类衍生物对人源多巴脱羧酶的抑制活性
a甲基多巴(mdopa)为阳性对照.
实施例12:
本发明的二芳基丙烷二聚体类衍生物,与赋形剂重量比1:1的比例加入赋形剂,制粒压片。
实施例13:
本发明的二芳基丙烷二聚体类衍生物或其药用盐,按常规胶囊制剂方法制成胶囊。
实施例14:
按下述配方制成片剂
实施例15:
胶囊剂:二芳基丙烷二聚体类衍生物或其药用盐100mg
淀粉适量
硬脂酸镁适量
制备方法:将二芳基丙烷二聚体类衍生物或其药用盐,与助剂混合,过筛,在合适的容器中均匀混合,把得到的混合物装入硬明胶胶囊。
实施例16:
安瓿剂:二芳基丙烷二聚体类衍生物或其药用盐2mg,氯化钠10mg;
制备方法:将二芳基丙烷二聚体类衍生物或其药用盐和氯化钠溶解于适量的注射用水中,过滤所得溶液,在无菌条件下装入安瓿瓶中。
- 还没有人留言评论。精彩留言会获得点赞!