一种2,4,5三氟苯甲酰乙酸乙酯的制备方法及纯化方法与流程
本发明涉及有机合成领域,特别涉及一种2,4,5三氟苯甲酰乙酸乙酯的制备方法及纯化方法。
背景技术:
德拉沙星是一种新型动物专用氟喹诺酮类抗菌药,德拉沙星合成路线已有文献报道,但是有关此中间体,即2,4,5三氟苯甲酰乙酸乙酯的合成报道较少。然而,现有文献及专利报道的合成方法,需要加入大量酸,产生大量酸性废水且反应液在一定的酸性条件下形成氢键发生室温自交联,难以进行分离纯化。不适合大规模的工业化生产。
如下式所示,wo2006/113509,2006,a2专利公开了制备2,4,5三氟苯甲酰乙酸乙酯(化合物1的办法,溶剂用量大,反应过程中需要加入盐酸调节ph-2-3,产生大量的含酸废水,且产物在一定的酸性条件下易形成氢键发生室温自交联,且易于反应体系中形成螯合物,难以进行分离纯化。后处理成本高,环境压力大不利于放大生产。
技术实现要素:
本发明旨在提供一种操作简单,选择性高,经济环保,转化率高,有利于工业生产的2,4,5三氟苯甲酰乙酸乙酯的制备方法及纯化方法。
为了实现上述目的,本发明具体采用如下技术方案:
一种2,4,5三氟苯甲酰乙酸乙酯的制备方法,包括如下步骤:
步骤(1):5℃~80℃温度下,式2所示的2,4,5三氟苯甲酸与酰氯在催化剂作用下反应制得式3所示的2,4,5三氟苯甲酰氯;
步骤(2):0℃~5℃温度下,式3所示的2,4,5三氟苯甲酰氯与乙酰乙酸乙酯在弱碱性溶液发生反应,得到式4所示的中间产物;
步骤(3):步骤(2)所得式4所示的中间产物升温至50℃~80℃脱除乙酰基,回收溶剂,得到式1所示的2,4,5三氟苯甲酰乙酸乙酯。
优选的,步骤(1)中所述酰氯为草酰氯,步骤(1)在有机溶剂条件下反应;或酰氯选自二氯亚砜,步骤(1)反应不添加溶剂。
优选的,步骤(2)中的碱选自氧化钙,碳酸钾,碳酸钠,氢氧化钠,碳酸氢钾或碳酸氢钠中的一种或多种。此处所述的碱,也就是弱碱性溶液中所需的碱。
优选的,步骤(2)中2,4,5三氟苯甲酰氯与碱的摩尔比为1∶0.1-0.25。
优选的,步骤(2)中2,4,5三氟苯甲酰氯与弱碱的摩尔比为1∶0.1-0.2。
优选的,所述步骤(2)中的弱碱性溶液的溶剂选自1,2二氯乙烷,四氢呋喃,乙腈中的一种或多种。
优选的,步骤(2)中所述的2,4,5三氟苯甲酰氯与溶剂的质量体积比为1∶10。所述的质量体积比指的是g/ml。
一种前述制备方法制备所得2,4,5三氟苯甲酰乙酸乙酯的纯化方法,包括如下步骤:2,4,5三氟苯甲酰乙酸乙酯粗品经活性炭脱色,在醇水体系中重结晶得高纯度产品,所述的醇水体系中醇的质量分数为15%~30%。此处所述的醇的质量分数是指的醇的质量/(醇+水的质量)。
优选的,所述的醇水体系中的醇选自甲醇,乙醇或异丙醇。
优选的,所述纯化的具体步骤为:
a:2,4,5三氟苯甲酰乙酸乙酯粗品溶于可溶溶剂中,经活性炭脱色,过滤,蒸除溶剂;
b:步骤b所得产品溶于醇水体系中,加热至50~80℃溶清;
c:步骤c所得溶液降温至室温,后置于冰浴中0℃-10℃析晶得纯品。
有益效果
本发明合成路线简单,后处理方便。
反应生成式3所示的化合物的过程中产生少量盐酸气,这也是其他发明所不可避免的,用尾气吸收装置处理。后续两步反应在弱碱性条件下进行,采取一锅法,易于操作,反应过程中只产生少量废渣,且有效避免了分子内氢键及螯合物结构,溶剂可回收套用。
粗品精制用一定比例的醇+水混合体系重结晶,意外的发现一定比例醇+水体系重结晶可以制得几乎全是酮式结构的化合物1,经济环保,转化率高,有利于工业生产。
附图说明
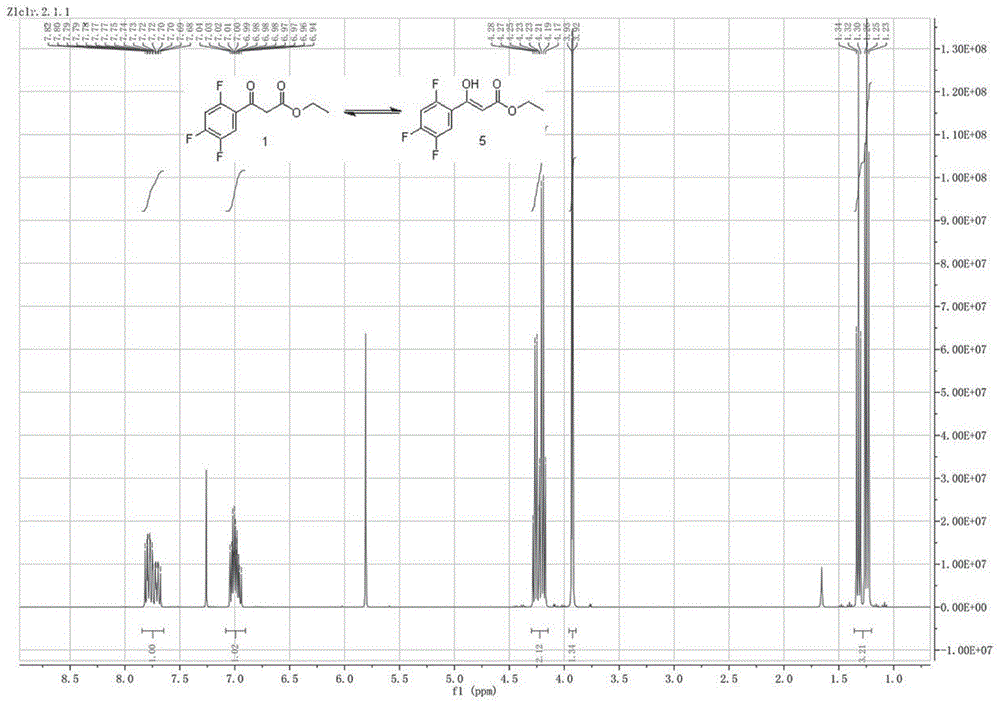
图1是本发明实施例3混合物1和5核磁图谱。
图2是本发明实施例3混合物1和5液相图谱。
图3是本发明实施例4混合物1和5液相图谱。
图4是本发明实施例5混合物1和5液相图谱。
图5是本发明实施例6混合物1和5液相图谱。
图6a、图6b是本发明实施例7混合物1和5液相图谱。
图7是本发明实施例8混合物1和5液相图谱。
图8是本发明实施例9混合物1和5液相图谱。
图9是本发明实施例10化合物1的液相图谱。
图10是本发明实施例11化合物1的液相图谱。
图11是本发明实施例12化合物1的液相图谱。
图12是本发明实施例13化合物1的液相图谱。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。以下对至少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使用的任何限制。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本申请的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
除非另外具体说明,否则在这些实施例中阐述的部件和步骤的相对布置、数字表达式和数值不限制本发明的范围。对于相关领域普通技术人员已知的技术、方法和设备可能不作详细讨论,但在适当情况下,所述技术、方法和设备应当被视为授权说明书的一部分。
概述本发明所用的的1h-nmr谱bruker-400核磁共振仪,化学位移是百万分之一,内标是四甲基硅烷。耦合常数(j)接近0.1hz。使用的缩略语如下说明:s,单重峰;d,双重峰;t,三重峰;q,四重峰;qu,五重峰;m,多重峰;br,宽峰。
液相色谱仪采用安捷伦1220infinityii液相色谱系统,具体分析方法如下:
取样品适量,加稀释剂(acn∶h2o=8∶2)溶解并稀释制成每1ml中约含1.0mg的溶液,作为供试品溶液;照高效液相色谱法(中国药典2015年版通则0512)试验,使用welchxtimatec184.6*150mm,3.5μm或其他等效色谱柱;以10mmk2hpo4(ph=6.8)水溶液为流动相a,乙腈为流动相b;柱温为35℃;流速为每分钟1.0ml;检测波长为240nm;按下表进行梯度洗脱:
精密量取供试品溶液20μl注入液相色谱仪,记录色谱图;按峰面积归一法计算并报告单杂和总杂含量。
下面结合附图对本发明进行详细说明,以方便本领域技术人员理解本发明。
实施例1化合物3的制备
2,4,5三氟苯甲酰氯(3)的合成:在配有回流冷凝管和磁力搅拌的三口烧瓶中,在冷凝管顶部安装一个干燥管,再连接一个尾气吸收装置,用5%氢氧化钠吸收尾气,然后依次加入100g(0.57mol)2,4,5,三氟苯甲酸,80ml二氯甲烷,加入1-2滴dmf作为催化剂,然后开始搅拌,并滴加79.31g(0.63mol)草酰氯,时间控制在30min左右,滴加完毕后,室温搅拌3h,溶液变得澄清透明,取少量反应液加入甲醇淬灭,进行薄层色谱(tlc)监控(展开剂体系eto-ac/pe/hoac(体积比)=20/10/1)。反应结束后,反应液冷却至室温,然后在40℃,0.1mpa条件下旋蒸出溶剂和低沸点杂质,溶剂直接回收套用,得到110.4g淡黄色液体,摩尔收率100%.
实施例2化合物3的制备
2,4,5三氟苯甲酰氯3的合成:在配有回流冷凝管和磁力搅拌的三口烧瓶中,在冷凝管顶部安装一个干燥管,再连接一个尾气吸收装置,用5%氢氧化钠吸收尾气,然后依次加入100g(0.57mol)2,4,5,三氟苯甲酸,然后加入101.5g(0.85m01)二氯亚砜做溶剂,加入1-2滴dmf作为催化剂,回流搅拌3h,溶液变得澄清透明,取少量反应液加入甲醇淬灭,进行薄层色谱(tlc)监控(展开剂体系eto-ac/pe/hoac(体积比)=20/10/1)。反应结束后,反应液冷却至室温,然后在40℃,0.1mpa条件下旋蒸出二氯亚砜,得到109g淡黄色液体,收率98.6%
实施例3化合物1和5混合物的制备
1000ml三口烧瓶中依次加入500ml乙腈,7.8g(0.057mol)碳酸钾,冰浴控温0-5℃以加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h,然后升至室温,即得到式4所示的化合物,不经过任何后处理,继续加热至70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品136.6g,收率97.3%,hplc检测显示化合物5:化合物1的比例75∶24。粗品产物的核磁共振氢谱图如图1所示,粗品产物的液相色谱图如图2所示。核磁数据:
1h-nmr(400mhz,cdcl3),δ1.25-1.32(3h,m),3.92-3.93(1h,d,4.0hz),4.17-4.28(2h,m),6.98-7.04(1h,m),7.68-7.82(1h,m)。
实施例4化合物1和5混合物的制备
1000ml三口烧瓶中依次加入500ml乙腈,15.6g(0.114mol)碳酸钾,冰浴控温0℃-5℃,加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h,后升至室温,.即得到化合物4,不经过任何后处理,加热至70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品134.0g,收率92.6%,hplc检测化合物5:化合物1比例为79∶19。粗品产物的液相色谱图如图3所示。
实施例5化合物1和化合物5混合物的制备
1000ml三口烧瓶中依次加入500ml二氯乙烷,7.8g(0.057mol)碳酸钾,冰浴控温0℃-5℃加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控,0-5℃,30min加毕。5℃左右保温2h,后升至室温,即得到化合物4。后不经过任何后处理,加热至70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品135.8g,收率96.7%,hplc显示几乎全是化合物5,纯度>95%,粗品产物的液相色谱图如图4所示。
实施例6化合物1和化合物5混合物的制备
1000ml三口烧瓶中依次加入500ml二氯乙烷,15.6g(0.114mol)碳酸钾,冰浴控温0℃-5℃,加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h.后升至室温,即得到化合物4。不经过任何后处理,加热至70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品134.9g,收率96.1%,hplc检测化合物5:化合物1比例为79∶17。品产物的液相色谱图如图5所示。
实施例7化合物1和化合物5混合物的制备
1000ml三口烧瓶中依次加入500ml乙腈,6.1g(0.057mol)碳酸钠,冰浴控温0℃-5℃,加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h.后升至室温,即得到化合物4。不经过任何后处理,加热70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品136.8g,收率98.2%,hplc检测化合物5:化合物1比例为54∶44粗品产物的液相色谱图如图6a和图6b所示。
实施例8化合物1和化合物5混合物的制备
1000ml三口烧瓶中依次加入500ml乙腈,12.2g(0.114mol)碳酸钠,冰浴控温0℃-5℃,加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h.后升至室温,保温5h.即得到化合物4.反应体系不经过任何后处理,加热70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品138.6g,收率98.4%,hplc检测化合物5:化合物1比例为24∶75。粗品产物的液相色谱图如图7所示。
实施例9化合物1和化合物5混合物的制备
1000ml三口烧瓶中依次加入500ml四氢呋喃,15.3g(0.143mol)碳酸钠,冰浴控温0℃-5℃,加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h.后升至室温,保温5h.即得到化合物4。反应体系不经过任何后处理,加热70摄氏度,反应5h,取样tlc中控后,降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品130.9g,收率93.3%,hplc检测显示化合物5:化合物1比例为69∶20粗品产物的液相色谱图如图8所示。
实施例10化合物1和化合物5混合物的制备
1000ml三口烧瓶中依次加入500ml乙腈,2.28g(0.057mol)氢氧化钠,冰浴控温0℃-5℃加入81.25g(0.625mol)乙酰乙酸乙酯,搅拌1h,滴加按照实施例1方法制备得到的110.4g(0.57mol)2,4,5三氟苯甲酰氯,滴加过程控温0℃-5℃,30min加毕。5℃左右保温2h.后升至室温,保温5h.即得到化合物4.反应体系不经过任何后处理,加热70摄氏度,反应5h,取样tlc中控后,体系比较杂。降至室温,过滤除去残渣,回收溶剂,得到化合物1和化合物5烯醇异构混合物粗品100.0g,收率71.3%,hplc检测显示化合物5:化合物1比例77∶12。粗品产物的液相色谱图如图9所示。
实施例11
产品的精制
50g由实施例3制备的化合物1和化合物5的烯醇异构粗品,100ml甲苯溶解,加入5g活性炭脱色,过滤除去活性炭,滤液旋干,加入350ml20%乙醇加热70℃左右溶清,搅拌下自然降温,大约45℃析出晶体,自然降至室温后,放置冰浴中0℃-10℃析晶1h.过滤得白色晶体48.2g,收率96.4%,hplc纯度>99.5%,精制产物的液相色谱图如图10所示。
实施例12
产品的精制
50g由实施例3化合物1和化合物5的烯醇异构粗品,100ml甲苯溶解,加入5g活性炭脱色,过滤除去活性炭,滤液旋干,加入350ml20%甲醇加热70℃左右溶清,搅拌下自然降温,大约48℃析出晶体,自然降至室温后,放置冰浴中0℃-10℃析晶1h.过滤得白色晶体48.6g,收率97.2%,hplc纯度>99.5%,精制产物的液相色谱图如图11所示。
实施例13
产品的精制
50g由实施例4化合物1和化合物5的烯醇异构粗品,100ml乙酸乙酯溶解,加入5g活性炭脱色,过滤除去活性炭,滤液旋干,加入350ml30%异丙醇加热70℃左右溶清,搅拌下自然降温,大约40℃析出晶体,自然降至室温后,放置冰浴中0℃-10℃析晶1h.过滤得白色晶体47.9g,收率95.8%,hplc纯度>99.5%,精制产物的液相色谱图如图12所示。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!