金属有机配合物及其制备方法和应用与流程
本发明涉及光电材料技术领域,尤其是涉及一种金属有机配合物及其制备方法和应用。
背景技术:
有机发光二极管(organiclightemittingdiodes,简称:oleds)是一种利用有机化合物作为发光层材料的发光二极管,自技术问世以来,由于其具有自发光特性、高发光效率、全彩色能力、宽视角、高对比度、低功耗、低重量、潜在大面积彩色显示器和灵活性等诸多优点,而受到学术界和工业界的广泛关注,被认为是下一代大尺寸高分辨率显示器和固态照明面板的竞争对手,具有制造简单、生产成本低等优点。其中磷光有机发光二极管(phosphorescentorganiclightemittingdiodes,简称:pholeds)由于同时利用单线态(singlet,25%)和三线态(triplet,75%)发光使其内部理论发光量子效率达到100%,从而大幅降低器件功耗,因此在平面显示器及照明应用等方面显示出巨大的发展潜力。
尽管各种oleds产品现已实现商业化多年,高效、稳定、易于加工的发光器件仍有大量需求。目前,oleds仍有一些缺点需要改进。
有鉴于此,特提出本发明。
技术实现要素:
本发明的第一目的在于提供金属有机配合物,以解决现有技术中存在的发光效率低等技术问题。
本发明的第二目的在于提供金属有机配合物的制备方法,操作简单,条件温和。
本发明的第三目的在于提供金属有机配合物在有机发光二极管中的应用。
本发明的第四目的在于提供一种有机发光二极管。
为了实现本发明的上述目的,特采用以下技术方案:
金属有机配合物,具有如下结构通式:
其中,m选自pt和pd中的任一种;
r1~r9各自独立的选自h、卤素、-ora和c1~6烷基中的任一种;
ra选自h和c1~6烷基中的任一种。
在本发明的具体实施方式中,r1~r9均为h。
本发明还提供了上述金属有机配合物的制备方法,包括如下步骤:
铂盐或钯盐与化合物ⅰ在溶剂中回流反应;
其中,所述化合物ⅰ的结构式如下:
在本发明的具体实施方式中,化合物ⅰ的制备方法包括如下步骤:
化合物ⅱ与三溴化硼在溶剂中进行脱保护反应;
其中,所述化合物ⅱ的结构式如下:
在本发明的具体实施方式中,化合物ⅱ的制备方法包括如下步骤:
化合物ⅲ与化合物ⅳ在催化剂的作用下于溶剂中进行suzuki偶联反应;
其中,所述化合物ⅲ与化合物ⅳ的结构式分别如下:
在本发明的具体实施方式中,化合物ⅲ的制备方法包括如下步骤:
化合物ⅴ与化合物ⅵ在l-脯氨酸/cui和无机碱的作用下于溶剂中进行反应;
其中,所述化合物ⅴ与化合物ⅵ的结构式分别如下:
本发明还提供了上述任意一种所述金属有机配合物在有机发光二极管中的应用。
本发明还提供了有机发光二极管,包含上述任意一种所述金属有机配合物。
在本发明的具体实施方式中,所述有机发光二极管包括发射层,所述发射层包括上述任意一种所述金属有机配合物。
与现有技术相比,本发明的有益效果为:
(1)本发明的金属有机配合物具有高发光效率和高电子迁移率;
(2)本发明的金属有机配合物可作为发光中心用于有机发光二极管的发射层。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
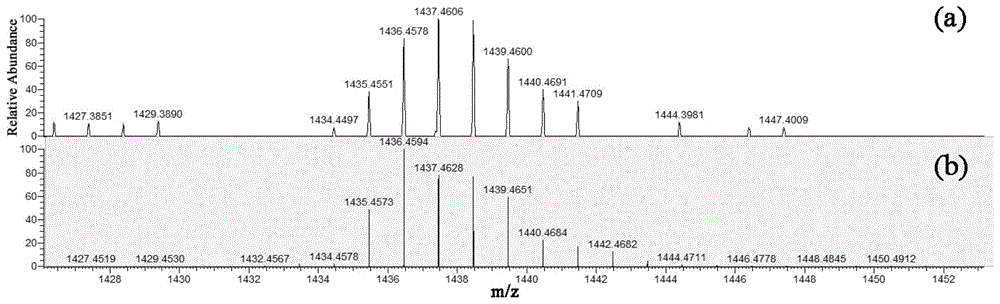
图1为本发明实施例1的金属有机配合物c1的高分辨质谱图;其中,(a)为正离子模式(2m+h+)分子离子峰;(b)为正离子模式(2m+h+)理论模拟峰;
图2为本发明实施例1的化合物ⅱ1的核磁氢谱图;
图3为本发明实施例1的金属有机配合物c1的核磁氢谱图;
图4为本发明实施例2的金属有机配合物c1的高分辨质谱图;其中,(a)为正离子模式(m+h+)分子离子峰;(b)为正离子模式(m+h+)理论模拟峰;
图5为本发明实施例1的金属有机配合物c1在溶液状态下的紫外-可见吸收图谱;
图6为本发明实施例2的金属有机配合物c2在溶液状态下的紫外-可见吸收图谱;
图7为本发明实施例1的金属有机配合物c1在固体状态下的发射图谱;
图8为本发明实施例2的金属有机配合物c2在固体状态下的发射图谱;
图9为本发明实施例1的金属有机配合物c1在pmma薄膜状态下的发射图谱;
图10为本发明实施例2的金属有机配合物c2在pmma薄膜状态下的发射图谱;
图11为本发明实施例1的金属有机配合物c1在溶液状态下的发射图谱;
图12为本发明实施例2的金属有机配合物c2在溶液状态下的发射图谱;
图13为本发明实施例1的金属有机配合物c1的tga分析图谱;
图14为本发明实施例2的金属有机配合物c2的tga分析图谱。
具体实施方式
下面将结合附图和具体实施方式对本发明的技术方案进行清楚、完整地描述,但是本领域技术人员将会理解,下列所描述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为限制本发明的范围。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
金属有机配合物,具有如下结构通式:
其中,m选自pt和pd中的任一种;
r1~r9各自独立的选自h、卤素、-ora和c1~6烷基中的任一种;
ra选自h和c1~6烷基中的任一种。
本发明提供了一类cnno四齿螯合金属有机配合物,属于磷光材料,具有优异的发光效率和高的电子迁移率以及较长的发光寿命。
在本发明的具体实施方式中,r1~r9均为h。
其中,c1~6烷基是指碳数为1~6的烷基,如可以为甲基、乙基、丙基、异丙基等等。
具体的,所述金属有机配合物的结构式可以分别如下:
本发明还提供了上述金属有机配合物的制备方法,包括如下步骤:
铂盐或钯盐与化合物ⅰ在溶剂中回流反应;
其中,所述化合物ⅰ的结构式如下:
在本发明的具体实施方式中,所述铂盐包括四氯铂酸钾。
在本发明的具体实施方式中,所述钯盐包括醋酸钯。
在本发明的具体实施方式中,所述溶剂包括冰醋酸。
在本发明的具体实施方式中,化合物ⅰ与铂盐或钯盐的摩尔比为1﹕(0.8~1.2),优选为1﹕(1~1.1),如可以为1﹕1。
在本发明的具体实施方式中,化合物ⅰ与溶剂的比例为1mmol﹕(300~500)ml,优选为1mmol﹕(320~450)ml。
在实际操作中,所述回流反应的温度可以为110~120℃条件下进行,所述回流反应的时间为20~30h,优选为22~26h,如可以为24h。
在本发明的具体实施方式中,所述回流反应后,还包括冷却至室温,进行固液分离,收集固体并采用冰醋酸对所述固体进行洗涤,干燥得到粗产物。进一步的,将所述粗产物进行柱层析分离,得到金属有机配合物。其中,进行柱层析分离时,可采用二氯甲烷和正己烷作为洗脱剂,具体的,二氯甲烷和正己烷的体积比为1﹕1。
在本发明的具体实施方式中,化合物ⅰ的制备方法包括如下步骤:
化合物ⅱ与三溴化硼在溶剂中进行脱保护反应;
其中,所述化合物ⅱ的结构式如下:
在本发明的具体实施方式中,所述脱保护反应中的溶剂包括二氯甲烷。进一步的,所述化合物ⅱ与溶剂的比例为1mmol﹕(55~65)ml,如可以为1mmol﹕60ml。
在本发明的具体实施方式中,所述化合物ⅱ与三溴化硼的摩尔比为1﹕(4~5),如可以为1﹕4。
在本发明的具体实施方式中,所述脱保护反应的温度为55~65℃,所述脱保护反应的时间为10~18h。
在本发明的具体实施方式中,所述脱保护反应后,冷却至室温,采用二氯甲烷对反应后的物料进行萃取,收集有机相;对所述有机相除水处理后,除去有机溶剂,得到粗产物。进一步的,将所述粗产物进行柱层析分离,得到化合物ⅰ。
在本发明的具体实施方式中,化合物ⅱ的制备方法包括如下步骤:
化合物ⅲ与化合物ⅳ在催化剂的作用下于溶剂中进行suzuki偶联反应;其中,所述化合物ⅲ与化合物ⅳ的结构式分别如下:
在本发明的具体实施方式中,所述催化剂包括四(三苯基膦)钯和碳酸钾,催化剂的用量为suzuki偶联反应中的常规用量。
在本发明的具体实施方式中,所述suzuki偶联反应的溶剂包括水和四氢呋喃。进一步的,所述水和四氢呋喃的体积比为1﹕1。所述化合物ⅲ与溶剂的比例为1g﹕(35~45)ml。如可以为1g﹕40ml。
在本发明的具体实施方式中,所述suzuki偶联反应的温度为75~85℃,所述suzuki偶联反应的时间为20~28h,如可以为24h。进一步的,在氮气保护条件下进行所述suzuki偶联反应。
在本发明的具体实施方式中,化合物ⅲ与化合物ⅳ的摩尔比为1﹕(1~5),如可以为1﹕(4~5)。
在本发明的具体实施方式中,所述suzuki偶联反应后,冷却至室温,固液分离,收集液体然后除去溶剂,通过柱层析分离的方式纯化,得到化合物ⅱ。
在本发明的具体实施方式中,化合物ⅲ的制备方法包括如下步骤:
化合物ⅴ与化合物ⅵ在l-脯氨酸/cui和无机碱的作用下于溶剂中进行反应;其中,所述化合物ⅴ与化合物ⅵ的结构式分别如下:
在本发明的具体实施方式中,所述无机碱包括碳酸钾。
在本发明的具体实施方式中,所述溶剂包括dmso。化合物ⅴ与溶剂的比例为1mmol﹕(3~5)ml,如可以为1mmol﹕4ml。
在本发明的具体实施方式中,所述反应的温度为135~145℃,所述反应的时间为20~28h,如可以为24h。进一步的,所述反应在氮气保护的条件下进行。
在本发明的具体实施方式中,所述反应完成后,冷却至室温,固液分离收集液体,除去所述液体中的溶剂,通过柱层析分离的方式纯化,得到化合物ⅲ。
在本发明的具体实施方式中,化合物ⅴ与化合物ⅵ的摩尔比为1﹕(0.8~1.2),优选为1﹕(1~1.1),如可以为1﹕1。
在本发明的具体实施方式中,化合物ⅴ、l-脯氨酸、cui和无机碱的摩尔比可以为1﹕(0.15~0.25)﹕(0.05~0.15)﹕(1.5~2.5),如可以为1﹕0.2﹕0.1﹕2。
具体的,由化合物ⅴ与化合物ⅵ制备金属有机配合物的合成路线如下:
本发明还提供了上述任意一种所述金属有机配合物在有机发光二极管中的应用。
本发明还提供了有机发光二极管,包含上述任意一种所述金属有机配合物。
在本发明的具体实施方式中,所述有机发光二极管包括发射层,所述发射层包括上述任意一种所述金属有机配合物。
实施例1
本实施例提供了金属有机配合物c1,其结构式如下:
金属有机配合物c1的合成路线如下:
具体的制备步骤如下:
(i)将6,6′-二溴-2,2′-联吡啶(3.14g,10.0mmol)、3,6-二叔丁基咔唑(2.79g,10.0mmol)、l-proline(0.230g,2.00mmol)、cui(0.190g,1.00mmol)和k2co3(2.76g,20.0mmol)溶于无水二甲基亚砜(40.0ml)中,然后将得到的混合物在氮气保护、140℃的条件下回流加热24h。反应结束后,将反应后混合物冷却至室温,过滤收集滤液,除去溶剂,将残余物通过硅胶柱纯化,得到纯化的化合物ⅲ1。
(ii)将化合物ⅲ1(1.00g,1.95mmol)、2-甲氧基苯基硼酸(1.22g,8.00mmol)、四(三苯基膦)钯(0.231g,0.200mmol)和碳酸钾(2.21g,16.0mmol)溶于水(20.0ml)和四氢呋喃(20.0ml)中,然后将得到的混合物在氮气保护、80℃的条件下回流加热24h。反应结束后,将反应后混合物冷却至室温,过滤收集滤液,除去溶剂,将残余物通过硅胶柱纯化,得到的白色固体为化合物ⅱ1。
(iii)将化合物ⅱ1(0.270mg,0.5mmol)和三溴化硼(0.501g,2.0mmol)溶于无水二氯甲烷(30.0ml)中,将混合得到的混合溶液在60℃下搅拌18h。反应结束后,将反应体系冷却至室温,用二氯甲烷萃取,收集的有机相中加无水硫酸镁进行除水,过滤除去硫酸镁,旋转蒸发除去二氯甲烷,得到粗产物,将粗产物经硅胶柱纯化后得到的白色固体为化合物ⅰ1。
(iv)将化合物ⅰ1(0.130g,0.247mmol)和四氯铂酸钾(0.103g,0.247mmol)溶于冰醋酸(80.0ml)中,得到的混合物在115℃下回流反应24h。反应结束后,冷却至室温,过滤,将固体用冰醋酸洗涤,干燥后得到的粗产物,将粗产物经硅胶柱纯化,得到的橙色固体为金属有机配合物c1;其中,洗脱剂为体积比为1﹕1的二氯甲烷和正己烷。
实施例2
本实施例提供了金属有机配合物c2,其结构式如下:
金属有机配合物c2的合成路线如下:
具体的制备步骤参考实施例1,区别仅在于,将步骤(iv)替换为步骤(v),步骤(v)具体如下:
(v)将化合物ⅰ1(0.0936g,0.178mmol)和醋酸钯(0.0400g,0.178mmol)溶于冰醋酸(80.0ml)中,得到的混合物在115℃下回流反应24h。反应结束后,冷却至室温,过滤,将固体用冰醋酸洗涤,干燥后得到的粗产物,将粗产物经硅胶柱纯化,得到的黄色固体为金属有机配合物c2;其中,洗脱剂为体积比为1﹕1的二氯甲烷和正己烷。
实验例1
本发明合成的金属有机配合物c1和金属有机配合物c2的结构表征数据如下:
图1为本发明实施例1的金属有机配合物c1的高分辨质谱图,图2为本发明实施例1的化合物ⅱ1的核磁氢谱图(氘代试剂为氘代氯仿),图3为本发明实施例1的金属有机配合物c1的核磁氢谱图(氘代试剂为氘代氯仿),图4为本发明实施例2的金属有机配合物c1的高分辨质谱图。由上述结构表征数据验证得出成功制备得到了金属有机配合物c1和金属有机配合物c2。
实验例2
图5和图6分别是本发明制得的金属有机配合物c1和金属有机配合物c2在溶液状态下(将金属有机配合物c1和金属有机配合物c2分别溶解于二氯甲烷中)于25℃的紫外-可见吸收图谱。
图7和图8分别为本发明制得的金属有机配合物c1和金属有机配合物c2在固体状态下于25℃的磷光发射图谱,激发波长分别为520nm和473nm。
图9和图10分别为本发明制得的金属有机配合物c1和金属有机配合物c2在pmma薄膜状态下于25℃的磷光发射图谱,激发波长分别为520nm和473nm。其中,pmma薄膜中金属有机配合物的添加量均为2wt%,其余为pmma(pmma薄膜采用常规方法制得,如:金属有机配合物样品与聚甲基丙烯酸甲酯按质量比1﹕49溶于二氯甲烷(acs级),将该溶液滴于石英玻璃表面,待溶剂挥发后即可)。
图11和图12分别为本发明制得的金属有机配合物c1和金属有机配合物c2在溶液状态下(将金属有机配合物c1和金属有机配合物c2分别溶解于二氯甲烷中)于25℃的磷光发射图谱,激发波长分别为520nm和473nm。
其中,图7~图12的发射图谱均为归一化后的结果,以方便对比。具体归一化的方法为常规归一化方法,如:将光谱数据同时除以最高峰值,使最高峰值变为1。
图13和图14分别为本发明制得的金属有机配合物c1和金属有机配合物c2在氮气条件下的tga分析图谱(升温速率为34.4℃/min)。
对本发明制得的金属有机配合物c1和金属有机配合物c2在上述不同状态下的发光性能进行汇总,具体见表1。
表1不同金属有机配合物在不同状态下的发光性能
备注:表格中“em”代表“发射波长”,“φ”代表“磷光量子产率”,“τ”代表“磷光寿命”,“solid”代表“固体状态”,“pmma”代表“pmma薄膜状态”,“liquid”代表“溶液状态”。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种Ni@C@g-C<sub>3</sub>N<sub>4</sub>纳米复合物及其制备方法和应用
- 一种石墨烯-三聚氰胺泡沫气凝胶及其制备方法
- 制备聚胺的方法
- 一种低温固化三聚氰胺甲醛树脂胶及其制备方法
- 一种生物质改性三聚氰胺甲醛树脂微球泡沫的制备方法
- 一种三羟甲基三聚氰胺的制备方法
- 一种Fe@C@g-C<sub>3</sub>N<sub>4</sub>纳米复合物及其制备方法和应用
- 一种g-C<sub>3</sub>N<sub>4</sub>/Bi<sub>3</sub>TaO<sub>7</sub>表面复合光催化剂的制备方法
- Ag<sub>2</sub>CrO<sub>4</sub>负载的g-C<sub>3</sub>N<sub>4</sub>复合光催化剂及其制备方法和应用
- 一种高聚合度的三聚氰胺聚磷酸盐的制备方法
- 一种羰基锝-99m标记的美法仑配合物及其制备方法与用图
- 一种2-羰基-3-苯基丙酸苯甲酰腙二苯基锡配合物及其制备方法和应用
- 一种新型金属有机配合物纤维及其衍生多孔碳纤维的制备方法
- 包含新的配体结构的金属配合物的制作方法
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用