一种渐变式聚酯的合成方法
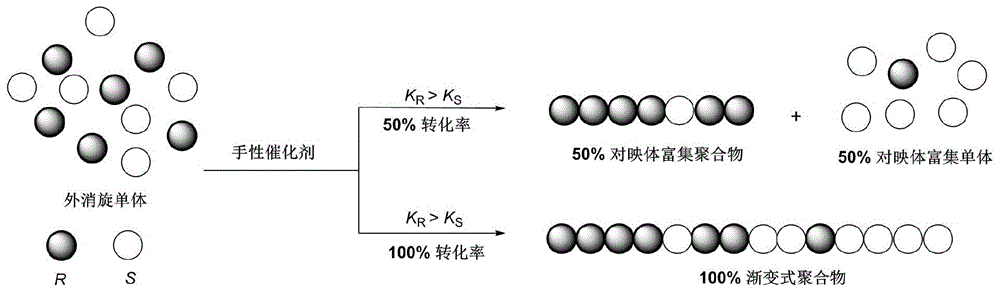
本发明属于高分子合成材料领域,具体涉及一种渐变式聚酯的合成方法。
背景技术:
:手性是自然界的基本属性,在自然界以及生命体中蕴藏着大量的手性分子。手性的研究在生命科学、制药以及材料科学等领域发挥着重要的作用。在材料科学领域,手性聚合物作为一种特殊的高分子材料,聚合物链的手性显著影响所得高分子材料的性能,比如空间排布、材料强度、介电性能以及材料强度等;这些特殊性能使得合成的聚合物具有不同于普通聚合物的独特应用,如手性分离材料、手性液晶材料、手性传感材料和手性电磁材料等,因此对于手性聚合物的研究具有重要的研究和应用价值。目前手性聚合物的研究大多集中在天然手性聚合物以及少数人工合成手性聚合物。人工合成手性聚合物的发展较为缓慢,主要是因为如何精确控制聚合过程的立体化学以及如何精确表征聚合物的整体手性是制约其发展的关键科学问题。对于人工合成手性聚合物的研究,其中渐变式聚合物作为一种具有特殊结构的新型手性聚合物,近年来引起了科学家的注意,这种新型聚合物的特征是聚合物链的手性渐变式变化,从聚合物链的一端到另一端,一种手性单体逐渐减少,另一种手性单体逐渐增加。这种结构聚合物目前有一些成功的研究报道,但是目前这种结构聚合物的报道多是关于聚丙交酯及其衍生物,其它不同单体类型的渐变式聚合物的研究有待进一步发展。以聚ε-己内酯为例,作为一类重要的高分子材料,它具有优异的生物相容性、生物可降解性和机械性能,被广泛应用于食品包装、医疗器械、医用高分子等领域。催化ε-己内酯单体开环聚合是合成聚ε-己内酯材料最重要的方法。通过在ε-己内酯单体中引入官能团来对聚合物结构进行修饰,可进一步提高聚ε-己内酯材料的综合性能,获得功能高分子材料从而拓宽材料应用范围。取代基的引入也将手性引入ε-己内酯单体,但聚合物链中手性中心的绝对构型并没有受到足够重视,对于手性聚ε-己内酯的研究鲜有报道,所得聚ε-己内酯材料多为无规聚合物。开发新型的具有手性功能特征结构的渐变式聚酯材料,突破制约手性聚合物发展的“合成难”和“表征难”等瓶颈问题,对于拓展结构多样、种类多样的聚酯材料在各领域的应用潜能,推动手性聚合物研究的进一步发展具有重要意义。该领域的研究目前具有很大的研究缺口,尚未有成功实现的案例,需要进一步探究。技术实现要素:本发明为解决现有技术缺少对于聚合物链手性的研究以及手性聚合物研究存在“合成难”与“表征难”的技术问题,而提供了一种渐变式聚酯的合成方法。本发明的一种渐变式聚酯的合成方法按以下步骤进行:在常压、惰性气体保护下,在有机溶剂中,以外消旋取代内酯为聚合单体,以手性磷酸(cpa)为催化剂,以醇类化合物为引发剂,利用聚合单体中两种对映异构体反应速率的差异实现开环拆分聚合,获得渐变式聚酯。本发明的反应过程如下所示:进一步限定,所述外消旋取代内酯的结构为以下结构中的任意一种:其中r为烷基或芳基。进一步限定,所述外消旋取代内酯的结构具体为cl-1~cl-8及la中的任意一种:进一步限定,所述手性磷酸(cpa)的结构为以下结构中的任意一种:其中r代表烷基或芳基。进一步限定,所述手性磷酸(cpa)的结构具体为cpa-1~cpa-4中的任意一种:进一步限定,所述醇类化合物为苄醇或异丙醇。进一步限定,所述有机溶剂为甲苯、氯苯、四氢呋喃或二氯甲烷中的一种。进一步限定,所述醇类化合物、手性磷酸和外消旋取代内酯的摩尔比为(1~2):(1~5):(20~500)。进一步限定,所述醇类化合物、手性磷酸和外消旋取代内酯的摩尔比为(1~2):1:50。进一步限定,所述外消旋取代内酯在有机溶剂中的浓度为0.1mol/l~5mol/l。进一步限定,所述外消旋取代内酯在有机溶剂中的浓度为0.17mol/l~0.4mol/l。进一步限定,所述开环拆分聚合的反应条件如下:反应温度为0℃~100℃,反应时间为1min~7d。进一步限定,所述开环拆分聚合的反应条件如下:反应温度为25℃~100℃,反应时间为9h~7d。本发明采用手性磷酸催化剂对外消旋取代内酯的不对称拆分聚合方法,利用催化剂对两种不同手性单体的选择性不同来控制聚合物立体化学,手性催化剂催化聚合某一手性单体的反应速率优于催化另一手性单体的反应速率,因此聚合在50%转化率时,获得对映体富集的手性聚合物和另一手性单体;单体聚合结束时,获得渐变式聚合物,聚合物的手性在整个链中渐变式变化,反应过程示意图如图1所示。本发明与现有技术相比具有的显著效果:1)本发明对外消旋取代内酯的不对称拆分聚合的方法,可获得新型具有渐变式结构的聚酯,拓展了聚酯材料的类型及应用领域,推动了手性高分子功能材料的开发。2)本发明的方法操作简便,条件温和,聚合活性可控,生成的聚合物无金属残留。3)应用本发明的方法,可成功实现开环聚合过程中聚合物立体化学的精确控制,本方法也为所得聚合物材料的光学活性及微观结构的表征提供了新的手段,对手性聚合物的研究具有重要理论指导意义。附图说明图1为本发明不对称拆分聚合方法获得渐变式聚合物的反应过程示意图;图2为实施例2中cl-1聚合获得的聚ε-己内酯的1hnmr图;图3为实施例6中cl-2聚合获得的聚ε-己内酯的1hnmr图;图4为实施例7中cl-2聚合获得的聚ε-己内酯的hplc图;图5为实施例17~27中cl-5聚合的动力学曲线。具体实施方式实施例1:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1ml甲苯,ε-己内酯(cl-1)单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于90℃下搅拌反应9h,获得渐变式聚ε-己内酯。实施例2:本实施例与实施例1不同的是:反应时间为32h。其他步骤及参数与实施例1相同。实施例3:本实施例与实施例1不同的是:催化剂为cpa-2,反应时间为16h。其他步骤及参数与实施例1相同。实施例4:本实施例与实施例3不同的是:反应时间为34h。其他步骤及参数与实施例3相同。对实施例1~4过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。具体结果见表1。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi),具体结果见表1。从表1的数据可以看出,对于cl-1单体的开环聚合,应用不同的手性磷酸催化剂,均能获得1.9左右的拆分常数,即催化剂对两种不同手性单体具有1.9倍的催化聚合速率差,显示成功获得了渐变式聚ε-己内酯,实施例2所得聚ε-己内酯的核磁共振氢谱(1hnmr)如图2所示。此外,从表1还可以看出,cpa-1比cpa-2具有更强的酸性,故其催化速率更快。对获得聚合物进行凝胶渗透色谱(gpc)检测结果表明,获得的聚合物分子量符合理论值且分子量分布较窄,显示该聚合过程为可控活性聚合。表1.取代ε-己内酯cl-1的开环聚合实施例5:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1ml甲苯,ε-己内酯(cl-2)单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于90℃下搅拌反应34h,获得渐变式聚ε-己内酯。实施例6:本实施例与实施例5不同的是:反应时间为108h。其他步骤及参数与实施例5相同。实施例7:本实施例与实施例5不同的是:催化剂为cpa-2,反应时间为46h。其他步骤及参数与实施例5相同。实施例8:本实施例与实施例7不同的是:反应时间为132h。其他步骤及参数与实施例7相同。对实施例5~8过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。具体结果见表2。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi),具体结果见表2。从表2的数据可以看出:①对于cl-2单体的开环聚合,获得的拆分常数均大于1,可知应用手性磷酸催化剂cpa-1与cpa-2均能成功获得渐变式聚ε-己内酯,实施例6所得聚合物的核磁共振氢谱(1hnmr)如图3所示。所得聚合物分子量分布窄,聚合活性可控。cpa-2催化剂具有更好的拆分聚合效果,聚合开始时拆分常数最高可达3.7,随着聚合反应的继续进行,拆分常数逐渐降低,存在手性减小的现象,这可能是因为cl-2单体开环后形成的二级醇手性链端对聚合反应的影响不可忽视,在聚合过程中,聚合物链的空间位阻增大,导致了聚合物链端手性匹配的困难从而使得拆分效果有所下降。②图4给出了实施例7中cl-2聚合获得的聚ε-己内酯的高效液相色谱(hplc)谱图,其峰信息见表3。从谱图中可获得剩余单体的对映体过量值(ee)。作为一种可降解聚合物,反应时间的延长使得聚合物有所降解,从而分子量略低于理论值,分子量分布略微增大。表2.取代ε-己内酯cl-2的开环聚合表3.峰信息表峰号保留时间面积高度面积(%)113.72025255679145250162.930220.0451487699059120637.070总计401326692043707100.00实施例9:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入设定体积的甲苯(详见表3),ε-己内酯(cl-3)单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入设定当量比(详见表3)的苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于50℃下搅拌反应90h,获得渐变式聚ε-己内酯。实施例10:本实施例与实施例9不同的是:于70℃下搅拌反应50h。其他步骤及参数与实施例9相同。实施例11:本实施例与实施例9不同的是:于90℃下搅拌反应24h。其他步骤及参数与实施例9相同。实施例12:本实施例与实施例9不同的是:催化剂为cpa-3,于90℃下搅拌反应22h。其他步骤及参数与实施例9相同。实施例13:本实施例与实施例9不同的是:催化剂为cpa-2,于90℃下搅拌反应24h。其他步骤及参数与实施例9相同。实施例14:本实施例与实施例13不同的是:ε-己内酯(cl-3)单体浓度为0.05mol/l,反应时间为66h。其他步骤及参数与实施例13相同。实施例15:本实施例与实施例13不同的是:苄醇的当量比不同(详见表4),反应时间为13h。其他步骤及参数与实施例13相同。对实施例9~15过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。具体结果见表4。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi),具体结果见表4。从表4的数据可以看出,对于cl-3单体的开环聚合,系统探究了初始单体的浓度、引发剂当量比、催化剂类型以及反应温度对聚合效果的影响。表4中给出了不同反应条件下在50%左右转化率获得的实验结果,具体如下:①当使用cpa-1作为催化剂时,50℃下催化聚合,52%的转化率可获得24%的ee值,计算拆分常数为1.9,这显示该催化剂可催化cl-3不对称拆分聚合获得渐变式聚ε-己内酯。②升高温度可提高催化速率但对选择性影响不大。当使用cpa-2与cpa-3作为催化剂时,与cpa-1相比,所得聚合物分子量分布略宽,可能是因为催化剂酸性的增强使得酯交换反应增加。③应用cpa-3催化剂所得的拆分常数为1.5略低于其他催化剂,催化剂中吸电子基团的引入降低了选择性。④相比之下,cpa-2催化剂催化聚合的拆分效果最好,拆分常数最高可达2.7,降低初始单体浓度反应速率降低,选择性略微下降。⑤增加苄醇引发剂的当量比使得催化速率显著提高,不过对选择性影响不大。不同反应条件下均能成功获得渐变式聚己内酯。表4.取代ε-己内酯cl-3的开环聚合a初始单体浓度为0.25mol/l,b初始单体浓度为0.05mol/l。实施例16:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1ml甲苯,ε-己内酯(cl-4)单体(250μmol,50equiv.),cpa-2催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于90℃下搅拌反应7天,获得渐变式聚ε-己内酯。对实施例16过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi)。本实施例中,cl-4单体由于具有大位阻的叔丁基基团,聚合活性较低,在cpa-2催化剂催化下,经过7天的反应,聚合转化率达到51%,高效液相色谱(hplc)检测剩余手性单体的ee值为31%,计算可得拆分常数s为2.4,表明cpa-2对两种不同手性单体催化聚合具有2.4倍的速率差,这证明该取代ε-己内酯单体也可成功获得渐变式聚ε-己内酯。实施例17:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1.5ml甲苯,ε-己内酯(cl-5)单体(250μmol,50equiv.),cpa-2催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于90℃下搅拌反应4h,获得渐变式聚ε-己内酯。实施例18:本实施例与实施例17不同的是:反应时间为7h。其他步骤及参数与实施例17相同。实施例19:本实施例与实施例17不同的是:反应时间为9h。其他步骤及参数与实施例17相同。实施例20:本实施例与实施例17不同的是:反应时间为12h。其他步骤及参数与实施例17相同。实施例21:本实施例与实施例17不同的是:反应时间为17h。其他步骤及参数与实施例17相同。实施例22:本实施例与实施例17不同的是:反应时间为21h。其他步骤及参数与实施例17相同。实施例23:本实施例与实施例17不同的是:反应时间为32h。其他步骤及参数与实施例17相同。实施例24:本实施例与实施例17不同的是:反应时间为38h。其他步骤及参数与实施例17相同。实施例25:本实施例与实施例17不同的是:反应时间为48h。其他步骤及参数与实施例17相同。实施例26:本实施例与实施例17不同的是:反应时间为60h。其他步骤及参数与实施例17相同。实施例27:本实施例与实施例17不同的是:反应时间为72h。其他步骤及参数与实施例17相同。实施例28:本实施例与实施例17不同的是:反应时间为108h。其他步骤及参数与实施例17相同。对实施例17~28过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。具体结果见表5。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi),具体结果见表5。从表5的数据可以看出,cpa-2催化cl-5单体的开环聚合,聚合过程中最高拆分常数可达4.1,催化剂对两种不同手性单体具有不同的聚合速率,从而成功获得cl-5单体的渐变式聚合物。图5为实施例17~27聚合过程的动力学曲线,聚合在50%转化率处反应速率有明显的转折点,这显示聚合开始阶段优先聚合反应速率快的对映体,聚合后半段主要聚合另一手性的对映体,进一步证明了该发明方法获得渐变式聚合物的可行性。聚合过程中分子量随转化率呈线性增长且符合理论值,分子量分布较窄,显示该单体聚合也表现出可控活性聚合特征。表5.取代ε-己内酯cl-5的开环聚合实施例29:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1ml甲苯,ε-己内酯(cl-6)单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于40℃下搅拌反应11h,获得渐变式聚ε-己内酯。对实施例29过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi)。本实施例中,cl-6单体由于具有小位阻的甲基基团,聚合活性高,在cpa-1催化剂催化下,经过11小时的反应,聚合转化率达到57%,分子量(mn)为2900g/mol及分子量分布(pdi)为1.19。高效液相色谱(hplc)检测剩余手性单体的ee值为18%,计算可得拆分常数s为1.5,表明cpa-2对两种不同手性单体催化聚合具有1.5倍的速率差,这证明该取代ε-己内酯单体可成功获得渐变式聚ε-己内酯。实施例30:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1ml甲苯,ε-己内酯(cl-7)单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于25℃下搅拌反应24h,获得渐变式聚ε-己内酯。实施例31:本实施例与实施例30不同的是:催化剂为cpa-4,反应时间为5d。其他步骤及参数与实施例30相同。对实施例30~31过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。具体结果见表6。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi),具体结果见表6。从表6的数据可以看出,cl-7单体由于6号位点具有小位阻的甲基基团,聚合活性较高,在cpa-1催化剂催化下,经过24小时的反应,聚合转化率达到48%,高效液相色谱(hplc)检测剩余手性单体的ee值为45%,计算可得拆分常数s为4.4,表明cpa-1对两种不同手性单体催化聚合具有4.4倍的速率差,这证明该取代ε-己内酯单体也可成功获得渐变式聚ε-己内酯。表6.取代ε-己内酯cl-7的开环聚合实施例32:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入1ml甲苯,ε-己内酯(cl-8)单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于50℃下搅拌反应3天,获得渐变式聚ε-己内酯。对实施例32过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi)。本实施例中,cl-8单体由于6号位点具有乙基基团,聚合活性明显降低,在cpa-1催化剂催化下,反应3天,聚合转化率达到57%,测得聚合物的分子量(mn)为2000g/mol及分子量分布(pdi)为1.19。高效液相色谱(hplc)检测剩余手性单体的ee值为37%,计算可得拆分常数s为2.5,表明cpa-1对两种不同手性单体催化聚合具有2.5倍的速率差,这证明该取代ε-己内酯单体也可成功获得渐变式聚ε-己内酯。实施例33:本实施例的一种渐变式聚酯的合成方法按以下步骤进行:取5ml的schlenk管,抽烤并置换氩气后,于手套箱中加入0.5ml二氯甲烷,rac-la单体(250μmol,50equiv.),cpa-1催化剂(5μmol,1equiv.),然后加入5μl苄醇的甲苯溶液,所述苄醇的甲苯溶液中苄醇的浓度为1mol/l,将反应管移出手套箱,于25℃下搅拌反应26h,获得渐变式聚ε-己内酯。对实施例33过程中以及反映结束后的相关参数进行如下检测,具体检测过程如下:检测1:催化剂对两种手性单体的选择性不同采用拆分常数s进行评估,对拆分常数进行分析从而获得渐变式聚酯聚合物的光学活性特征,具体过程如下:在反应过程中,通过针管抽取0.1ml反应体系,加入三乙胺淬灭反应后真空去除溶剂,通过核磁共振氢谱(1hnmr)检测聚合反应t时间的转化率(ct);通过薄层色谱(tlc)收集未反应的手性单体,通过高效液相色谱检测t时间两种手性单体的对映体过量值(eet);通过kagan方程计算拆分常数s=ln[(1-ct)(1-eet)]/ln[(1-ct)(1+eet)]。检测2:单体聚合结束后,真空去除溶剂,冰甲醇洗涤分离出聚合物,并真空干燥至恒重。所得聚合物通过凝胶渗透色谱(gpc)测得聚合物的分子量(mn)及分子量分布(pdi)。本实施例中,rac-la单体,在cpa-1催化剂催化下,反应26小时,聚合转化率达到49%,高效液相色谱(hplc)检测剩余手性单体的ee值为29%,计算可得拆分常数s为2.4,表明cpa-1对两种不同手性单体催化聚合具有2.4倍的速率差,这证明rac-la单体也可成功获得渐变式聚丙交酯。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1